

Abstract
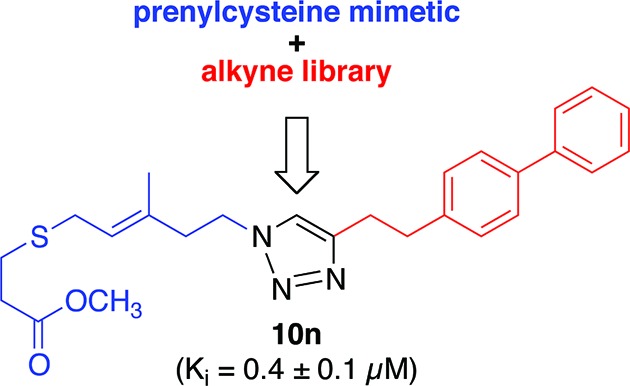
We report the design and synthesis of novel FTPA-triazole compounds as potent inhibitors of isoprenylcysteine carboxyl methyltransferase (Icmt), through a focus on thioether and isoprenoid mimetics. These mimetics were coupled utilizing a copper-assisted cycloaddition to assemble the potential inhibitors. Using the resulting triazole from the coupling as an isoprenyl mimetic resulted in the biphenyl-substituted FTPA triazole 10n. This lipid-modified analogue is a potent inhibitor of Icmt (IC50 = 0.8 ± 0.1 μM; calculated Ki = 0.4 μM).
Keywords: Isoprenylcysteine carboxyl methyltransferase (Icmt), Ras, prenylcysteine, dipolar cycloaddition, S-farnesyl-thiopropionic acid (FTPA), triazole
The post-translational processing of members of the Ras protein superfamily is under active investigation because approximately 20% of human cancers result from mutated Ras proteins.1 These proteins contain a −CaaX motif that is first isoprenylated on the cysteine thiol with a farnesyl group by farnesyltransferase (FTase) or a geranylgeranyl group by geranylgeranyltransferase-1 (GGTase-1). Following lipidation, two critical modifications occur sequentially at the endoplasmic recticulum (ER) by membrane-associated enzymes. First, the endoprotease Ras converting enzyme-1 (Rce-1) cleaves the terminal −aaX residues, resulting in a newly exposed prenylcysteine at the C terminus of the protein. Second, isoprenylcysteine carboxylmethyltransferase (Icmt) methyl esterifies the carboxylate using S-adenosyl-methionine (SAM) as the methyl donor. The increased hydrophobicity imparted by these modifications is thought to allow for proper localization of the Ras proteins to the plasma membrane and thus their function.2
Because blocking Ras localization should prevent its functioning in tumor progression, this post-translational modification pathway has been the target for the development of therapeutics against mutant Ras-based tumors. To this end, potent FTase inhibitors were developed that showed promise against some tumors but proved ineffective against K-Ras driven tumors,3 due in part to alternative geranylgeranylation of K-Ras. We chose to focus on developing inhibitors of Icmt, the only known enzyme that catalyzes the methyl esterification of the terminal prenylcysteine.4 Genetic knockout studies have provided support for the importance of Icmt-mediated methylation in Ras signaling.5
Substrate specificity studies revealed several key elements necessary for recognition by Icmt, including a C15 or C20 isoprenyl lipid and a requirement for a thioether bridge between the lipid and the carboxylate motifs.6,7 Early work by Rando et al. identified the minimal substrate for Icmt as farnesyl thiopropionic acid (FTPA, 1).8 Using this information as a starting point, inhibitors of Icmt were developed from studies of rotationally restricted prenylcysteines,9 library screening efforts,10 and amide-11,12 and isoprenyl-modified13 prenylcysteines. In recent studies, we have established the prenylcysteine analogue POP-3MB as a low micromolar inhibitor of human Icmt.12
We have now utilized FTPA as a starting point for nonamino acid Icmt inhibitor scaffolds, incorporating a triazole as a thioether or isoprene mimetic. This approach allowed us to utilize the synthetic advantages of copper-assisted dipolar cycloaddition (click chemistry) for rapid and facile analogue assembly.14 Herein, we report the synthesis and evaluation of FTPA analogues assembled utilizing click chemistry. Triazole 10n represents our most potent Icmt inhibitor to date, with an in vitro IC50 of 0.8 ± 0.1 μM.
In the initial design, a 1,4-disubstituted 1,2,3-triazole was positioned as the thioether replacement and cysteine backbone modifier that would join the lipid and carboxylate motifs of the FTPA analogues (Scheme 1). This approach had two key advantages: (a) It replaced the potentially labile allylic thioether, and (b) the building blocks needed for the reaction were readily available. Azido-esters 3a–d underwent cycloaddition with alkynes 4a–c,15,15b followed by saponification to afford 5a–g in acceptable yields. This synthetic sequence generated analogues with varying flexibility between the carboxylate, the triazole, and the lipid motifs.
Scheme 1. Synthesis of Triazole for Sulfur Analogues.

Reagents and conditions: (a) Sodium ascorbate, cupric sulfate, tBuOH/H2O, room temperature. (b) LiOH, MeOH; 20–80% over two steps. The number in parentheses indicates Icmt specific activity as percent of AFC control in the presence of 10 μM inhibitor.
These analogues (Scheme 1) were evaluated for activity using a vapor diffusion assay.16 Compounds 5a–g were modest inhibitors of Icmt but were not substrates. Structure–activity relationships (SAR) determined an optimal two-methylene bridge (5b) between the carboxylate and the triazole. Triazole-containing analogues based on the FTPA structural motif thus warranted further exploration, as this approach could generate diverse compounds quickly and easily.
Previous efforts examined isoprenyl-modified AFC analogues as Icmt inhibitors.13 These efforts demonstrated that the correct (E)-geometry of the first isoprene must be maintained as well as inclusion of the sulfur heteroatom.7 Furthermore, saturated lipids were also not mimetics,13 suggesting that specific “druglike” lipid structural motifs will interact with the active site of Icmt. We also took compounds developed previously as ligands for other isoprenyl binding pockets into consideration when designing our analogues. Aryl mimetics of isoprenoids have been utilized in the development of nanomolar squalene synthase17 and FTase ligands,18−19a and thus, we focused on this approach in our subsequent set of potential Icmt inhibitors.
The synthesis of the FTPA-triazoles began from iodide 6, which underwent Zr-assisted carboalumination,20 affording the desired trisubstituted olefin 7 (Scheme 2). Corey-Kim chlorination21 and alkylation with methyl 3-mercaptopropionate resulted in ester 8. Sodium azide displaced the primary iodide, affording the desired cycloaddition partner 9. Copper-mediated cycloaddition with a variety of terminal alkynes (with a focus on aromatic moieties) provided the FTPA-triazole analogues with good yields. These compounds were evaluated as both methyl esters (10a–m) and carboxylates (11a–m) following saponification.
Scheme 2. Synthesis of Triazole Substitutions in FTPA.

Reagents and conditions: (a) Me3Al, Cp2ZrCl2, (HCO)n, 0 °C–room temperature, 60%. (b) (i) N-Chlorosuccinimide, Me2S, DCM, −40 °C–room temperature; (ii) methyl thiopropionate, DIEA, DCM, 0 °C–room temperature, 55% over two steps. (c) NaN3, DMF, 80 °C, 78%. (d) Sodium ascorbate, cupric sulfate, tBuOH/H2O, room temperature, 35–97%. (e) LiOH, MeOH, room temperature, 40–90%. The number in parentheses indicates Icmt specific activity as percent of AFC control in the presence of 10 μM inhibitor.
These FTPA-triazole analogues were evaluated using the vapor diffusion assay. Neither the methyl esters 10a–m nor the carboxylates 11a–m were Icmt substrates. The alkyl analogues 10a–d and 11a–d were modest inhibitors, and the esters showed inhibitory activity comparable to the acids. However, aryl rings attached to the triazole (10e–i and 11e–i) resulted in both ester and acid compounds that were poor inhibitors of Icmt. These data suggest that neither saturated lipids nor rigid aryl groups directly joined to the triazole satisfied the structural requirements necessary for inhibition.
Natural isoprenylcysteine substrates have a high degree of flexibility, and the aryl analogues described above possessed an inflexible motif. Increased inhibition of Icmt was afforded with increased spacing between the triazole and the aryl motifs. Analogue 10k was the most potent analogue in this series, with an IC50 of 41 ± 5 μM. Alternatively, 11m, with both a polar and a nonaromatic tail, displayed poor Icmt inhibition (IC50 > 100 μM). These data suggest that incorporation of aryl groups within the isoprenyl region via a trizole must be accompanied by a concomitant introduction of flexibility.
Because Icmt can also efficiently methylate geranylgeranylated substrates,6 we hypothesized that further extension into the region of the terminal isoprene could be utilized for enhanced binding of our FTPA-triazole analogues. To examine this hypothesis, a series of biphenyl alkynes were utilized, as we have previously shown that the biphenyl group is an effective isoprene mimetic.12,13,18 These analogues also included incorporation of increased spacing between the triazole and the biphenyl motifs to afford flexibility. Of these analogues, the most potent inhibitor of Icmt in vitro was 10n. In our single point assay, 10n (10 μM) decreased the specific activity of Icmt to 14% of the no inhibitor control (Scheme 3). We experimentally determined an in vitro IC50 of 0.8 ± 0.1 μM for 10n and a calculated Ki of 0.4 μM (Cheng and Prusoff method22). Further biochemical evaluation found 10n to be a competitive inhibitor. Because the methyl ester 10n was more potent than the corresponding carboxylate, 11n, modification of the carboxylate group may represent a new approach for inhibitor design.23
Scheme 3. Triazole Substitutions in FTPA: Use of Biphenyl Alkynes.

Reagents and conditions: (a) Sodium ascorbate, cupric sulfate, tBuOH/H2O, room temperature; 36–78%. (b) LiOH, MeOH, room temperature, 50–96%. The number in parentheses indicates Icmt specific activity as percent AFC control with 10 μM inhibitor.
One particularly attractive feature of 10n is its ease of synthesis; it was prepared in five linear steps from homopropargyl iodide 6 in an unoptimized 14% overall yield. This route facilitated rapid additional SAR efforts through the synthesis of modified biphenyl analogues. Analogues with alterations to the orientation of the biphenyl moiety (10o–p) decreased Icmt inhibition. The ester 10o was nearly 30 times less potent than 10n, with an IC50 of 29 ± 5 μM (Table 1), and the ortho-10p analogue possessed further reduced inhibitory activity, indicating the importance of the para-biphenyl motif. Furthermore, inclusion of an oxygen linker (10q) was also detrimental to inhibition. Compounds containing electron-withdrawing groups in the terminal aromatic ring (10s, 10t) were approximately twice as potent as the electron rich analogue 10u.
Table 1. Cellular Activities (IC50) of Select FTPA-Triazole Analogues.
| MEF |
||||
|---|---|---|---|---|
| compd | cell-free (μM) | Icmt–/– (μM)a,c | Icmt+/+ (μM)a,c | PaTu-8902 (μM)b,c |
| 10g | ND | >100 | 96 ± 0.01 | ND |
| 10n | 0.8 ± 0.1 | >100 | 33 ± 1 | 8 ± 3 |
| 10o | 29 ± 5 | >100 | 55 ± 2 | ND |
| FTS | 25.5 ± 2 | 14.3 ± 2.2 | 15.3 ± 1.8 | 7.8 ± 1.7 |
MEFs are plated in 96-well plates at a density of 1000 cell/well in DMEM supplemented with 10% FBS. After incubation for 24 h, the media are replaced with DMEM supplemented with 5% FBS and 10n (various concentrations) or DMSO (0.1%). The cell viability is determined after 5 days using MTT. Twenty microliters of MTT (5 mg/mL) is added to each well and incubated for 4 h at 37 °C. Afterward, the media are removed, and 150 μL of DMSO are added to each well. The absorbance is determined at 590 nm using a Molecular Devices VERSAmax microplate reader.
IC50 determinations were performed in triplicate and calculated with Graphpad Prism V4.
ND, not determined.
The lead compound (10n) was tested for efficacy in vitro in several cell line model systems. First, MEF cells derived from Icmt+/+ and Icmt–/– animals4 were treated with increasing concentrations of 10n for 24 h, and cell viability was measured using a standard MTT assay (Table 1). In MEF Icmt–/– cells, 10n demonstrated an IC50 of >100 μM, whereas an IC50 of 33 μM was observed for wild-type MEF Icmt+/+ cells. Two analogues that are structurally related to 10n but are not Icmt inhibitors in vitro were also examined in this assay. Analogues 10g and 10o had IC50 values in MEF Icmt+/+ cells of 96 and 55 μM, respectively, and did not affect viability in MEF Icmt–/– cells (Table 1). Furthermore, treatment with FTS (S-farnesylthiosalicylic acid), an Icmt inhibitor that also possesses other cellular effects25 and has clinical potential,26 showed equivalent activity in both cell lines. Finally, as activating point mutations in K-Ras can be found in approximately 90% of pancreatic cancers,27 the in vitro efficacy of 10n was examined in the highly metastatic and “K-Ras addicted”28 pancreatic cancer cell line PaTu-8902. An IC50 of 8 μM was determined for 10n, which is comparable to the known Icmt inhibitor FTS (Table 1).
Proper posttranslational processing is a prerequisite for Ras membrane localization and function. To determine the cellular effects of our most potent inhibitor 10n, the membrane localization of fluorescently tagged K-Ras was visualized in the presence and absence of 10n.24−29 Jurkat T-cells that were transfected with GFP-K-Ras and treated with either delivery vehicle alone, the statin drug simvastatin (which blocks K-Ras prenylation), or 10n were fixed, and GFP-K-Ras localization was visualized using fluorescent microscopy. After 24 h of treatment, GFP-K-Ras localization was categorized into three groups: normal plasma membrane localization, partial mislocalization, and complete mislocalization. This classification was determined via visual inspection of randomly selected fields containing 100 cells. The results demonstrated that 10n led to a decrease in normal membrane localization (Figure 1). These data suggest that 10n is taken up into mammalian cells and functions to prevent K-Ras membrane localization.
Figure 1.
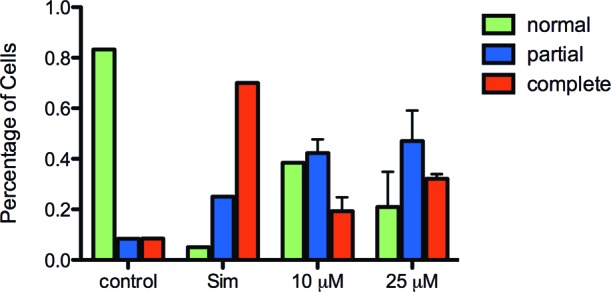
Mislocalization of GFP-KRas construct upon 10n administration. Jurkat T cells (E 6.1) were treated with vehicle, 25 μM Simvastatin (Sim), or 10n at indicated concentrations for 24 h. Cells were overlaid onto poly-l-lysine-coated coverslips (100 μg/mL), followed by fixation with 3.7% formaldehyde solution for 10 min. Subcellular localization of GFP-KRas was quantified using fluorescence microscopy (Olympus BH-2RFCA).
FTPA-triazole 10n was generated by removing the amide moiety present in other isoprenoid mimetics and by adding a biphenyl isoprenyl mimetic. Compound 10n was found to be a potent inhibitor of Icmt capable of inducing K-Ras mislocalization in a cellular model system. Furthermore, 10n appears to be selectively cytotoxic for Icmt+/+ MEF cells and has low micromolar activity against a metastatic pancreatic cancer cell line. Together, these data suggest, but do not prove, that 10n is targeting Icmt in cells. The lead triazole analogue 10n not only possesses higher potency than our best previous Icmt inhibitor (POP-3MB: IC50 = 2.5 μM),12 but it also possesses a lower molecular weight (450 vs 606), a lower CLog P (5.5 vs 8.8), and is much more easily assembled. To the best of our knowledge, 10n represents the first substrate-based compound to exhibit submicromolar inhibition against Icmt. Furthermore, 10n exhibits significantly improved ligand efficiency30 and is thus a superior starting point for drug development efforts.
Acknowledgments
We thank Mark Phillips (NYU) for the generous gift of GFP-KRas and Marietta Harrison (Purdue University) for helpful advice.
Supporting Information Available
Synthetic procedures, full experimental details, and details of biological assay. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding was provided by the NCI [R01CA112483 (R.A.G.) and P30CA21328 (Purdue University Center for Cancer Research CCSG)].
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bos J. L. ras oncogenes in human cancer: A review. Cancer Res. 1989, 49174682–4689. [PubMed] [Google Scholar]
- Michaelson D.; Ali W.; Chiu V. K.; Bergo M.; Silletti J.; Wright L.; Young S. G.; Philips M. Postprenylation CAAX Processing is Required for Proper Localization of Ras but not Rho GTPases. Mol. Cell. Biol. 2005, 16, 1606–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso A. D.; Kirschmeier P. T.; Bishop W. R. Farnesyl Transferase Inhibitors. J. Lipid Res. 2006, 47, 15–31. [DOI] [PubMed] [Google Scholar]
- Bergo M. O.; Leung G. K.; Ambroziak P.; Otto J. C.; Casey P. J.; Young S. G. Targeted Inactivation of the Isoprenylcysteine Carboxyl Methyltransferase Gene Causes Mislocalization of K-Ras in Mammalian Cells. J. Biol. Chem. 2000, 275, 17605–17610. [DOI] [PubMed] [Google Scholar]
- Bergo M. O.; Gavino B. J.; Hong C.; Beigneux A. P.; McMahon M.; Casey P. J.; Young S. G. Inactivation of Icmt inhibits transformation by oncogenic K-Ras and B-Raf. J. Clin. Invest. 2004, 113, 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan E. W.; Perez-Sala D.; Canada F. J.; Rando R. R. Identifying the Recognition Unit for G Protein Methylation. J. Biol. Chem. 1991, 266, 10719–10722. [PubMed] [Google Scholar]
- Henriksen B. S.; Anderson J. L.; Hrycyna C. A.; Gibbs R. A. Synthesis of desthio prenylcysteine analogs: Sulfur is important for biological activity. Bioorg. Med. Chem. Lett. 2005, 15, 5080–5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y. T.; Gilbert B. A.; Rando R. R. Farnesylcysteine Analogs to Probe Role of Prenylated Protein Methyltransferase. Methods Enzymol. 1995, 250, 226–234. [DOI] [PubMed] [Google Scholar]
- Marciano D.; Ben-Baruch G.; Marom M.; Egozi Y.; Haklai R.; Kloog Y. Farnesyl Derivatives of Rigid Carboxylic Acids-Inhibitors of ras-Dependent Cell Growth. J. Med. Chem. 1995, 38, 1267–1272. [DOI] [PubMed] [Google Scholar]
- Winter-Vann A. M.; Baron R.; Wong W.; de la Cruz J.; York J. D.; Gooden D. M.; Bergo M.; Young S. G.; Toone E. J.; Casey P. J. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 4336–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y. T.; Shi Y. Q.; Lim Y. H.; McGrail S. H.; Ware J. A.; Rando R. R. Mechanistic Studies on Human Platelet Isoprenylated Protein Methyltransferase: Farnesylcysteine Analogs Block Platelet Aggregation without Inhibiting the Methyltransferase. Biochemistry 1994, 33, 5414–5420. [DOI] [PubMed] [Google Scholar]
- Donelson J. L.; Hodges H. B.; Henriksen B. S.; Hrycyna C. A.; Gibbs R. A. Solid-Phase Synthesis of Prenylcysteine Analogs. J. Org. Chem. 2009, 74, 2975–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. L.; Henriksen B. S.; Gibbs R. A.; Hrycyna C. A. The Isoprenoid Substrate Specificity of Isoprenylcysteine Carboxylmethyltransferase: Development of Novel Inhibitors. J. Biol. Chem. 2005, 280, 29454–29461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem., Int. Ed. 2002, 41142596–2599. [DOI] [PubMed] [Google Scholar]
- Chan J.; Jamison T. F. Enantioselective synthesis of (-)-terpestacin and structural revision of siccanol using catalytic stereoselective fragment couplings and macrocyclizations. J. Am. Chem. Soc. 2004, 126, 91–92. [DOI] [PubMed] [Google Scholar]
- Lipshutz B. H.; Bulow G.; Fernandez-Lazaro F.; Kim S. K.; Lowe R.; Mollard P.; Stevens K. L. A convergent approach to coenzyme Q. J. Am. Chem. Soc. 1999, 121, 11673. [Google Scholar]
- Hrycyna C. A.; Wait S. J.; Backlund P. S.; Michaelis S. Yeast STE14 methyltransferase, expressed as TrpE-STE14 fusion protein in Escherichia coli, for in vitro carboxylmethylation of prenylated polypeptides. Methods Enzymol. 1995, 250, 251–266. [DOI] [PubMed] [Google Scholar]
- Biller S. A.; Abt J. W.; Pudzianowski A. T.; Rich L. C.; Slusarchyk D. A.; C. P. Ciosek J. Aromatic Isosteres as Conformational Probes for an Isoprenyl Subunit: Application to Inhibitors of Squalene Synthase. Bioorg. Med. Chem. Lett. 1993, 3, 595–600. [Google Scholar]
- Zhou C.; Shao Y.; Gibbs R. A. Aromatic Farnesyl Diphosphate Analogues: Vinyl Triflate-Mediated Synthesis and Preliminary Evaluation. Bioorg. Med. Chem. Lett. 2002, 12, 1417–1420. [DOI] [PubMed] [Google Scholar]
- Turek-Etienne T. C.; Strickland C. L.; Distefano M. D. Biochemical and Structural Studies with Prenyl Diphosphate Analogues Provide Insights into Isoprenoid Recognition by Protein Farnesyl Transferase. Biochemistry 2003, 42, 3716–3724. [DOI] [PubMed] [Google Scholar]
- Roberts M. J.; Troutman J. M.; Chehade K. A.; Cha H. C.; Kao J. P.; Huang X.; Zhan C. G.; Peterson Y. K.; Subramanian T.; Kamalakkannan S.; Andres D. A.; Spielmann H. P. Hydrophilic Anilinogeranyl Diphosphate Prenyl Analogues Are Ras Function Inhibitors. Biochemistry 2006, 455115862–15872. [DOI] [PubMed] [Google Scholar]
- Negishi E.; Horn D. E. V.; Yoshida T. Controlled carbometalation. 20. Carbometalation reaction of alkynes with organoalene-zirconocene derivatives as a route to stereo-and regiodefined trisubstituted alkenes. J. Am. Chem. Soc. 1985, 107, 6639–6647. [Google Scholar]
- Corey E. J.; Kim C. U.; Takeda M. A Method for Selective Conversion of Allylic and Benzylic Alcohols to Halides Under Neutral Conditions. Tetrahedron Lett. 1972, 4339–4342. [Google Scholar]
- Cheng Y.; Prusoff W. H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 108. [DOI] [PubMed] [Google Scholar]
- Gilbert B. A.; Tan E. W.; Perezsala D.; Rando R. R. Structure activity studies on the retinal rod outer segment isoprenylated protein methyltransferase. J. Am. Chem. Soc. 1992, 114103966–3973. [Google Scholar]
- Krzysiak A. J.; Rawat D. S.; Scott S. A.; Pais J. E.; Handley M.; Harrison M. L.; Fierke C. A.; Gibbs R. A. Combinatorial modulation of protein prenylation. ACS Chem. Biol. 2007, 26385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz A.; Haklai R.; Elad-Sfadia G.; Ballan E.; Kloog Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene 2001, 20, 7486–7493. [DOI] [PubMed] [Google Scholar]
- Tsimberidou A. M.; Rudek M. A.; Hong D.; Ng N. S.; Blair J.; Goldsweig H.; Kurzrock R. Phase 1 First-in-human clinical study of S-trans, trans-farnesylthiosalicylic acid (salirasib) in patients with solid tumors. Cancer Chemother. Pharmacol. 2010, 65, 235–241. [DOI] [PubMed] [Google Scholar]
- Downward J. Targeting ras signalling pathways in cancer therapy. Nature Rev. Cancer 2003, 3111–22. [DOI] [PubMed] [Google Scholar]
- Singh A.; Greninger P.; Rhodes D.; Koopman L.; Violette S.; Bardeesy N.; Settleman J. A Gene Expression Signature Associated with “K-Ras Addiction” Reveals Regulators of EMT and Tumor Cell Survival. Cancer Cell 2009, 156489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy E.; Philips M. Methods Enzymol., Part F 2001, 332, 50–62. [DOI] [PubMed] [Google Scholar]
- Reynolds C. H.; Bembenek S. D.; Tounge B. A. The role of molecular size in ligand efficiency. Bioorg. Med. Chem. Lett. 2007, 17, 4258–4261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.