

Abstract
Huntingtin interacting protein 1-related (Hip1r) is an F-actin- and clathrin-binding protein involved in vesicular trafficking that is crucial for parietal cell function and epithelial cell homeostasis in the stomach. Gastric parietal cells in Hip1r-deficient mice are lost by apoptotic cell death, which leads to a progressive epithelial cell derangement, including glandular hypertrophy, zymogenic cell loss and expansion of a metaplastic mucous cell lineage known as spasmolytic polypeptide expressing metaplasia (SPEM). The epithelial cell changes are associated with infiltration of inflammatory cells. Since inflammatory mediators, such as IFNγ, have been shown to contribute to the development of the gastric epithelial cell metaplasia after Helicobacter infection, we tested whether IFNγ played a role in the spontaneous progressive epithelial metaplasia observed in Hip1r-deficient mice. Hip1r-deficient mice were crossed with IFNγ-deficient mice and single and double mutant mice were analyzed at 3 and 12 months of age. Histopathology scoring showed that loss of IFNγ tempered the spontaneous development of metaplastic lesions in Hip1r-deficient mice. Loss of IFNγ was observed to abrogate the glandular hypertrophy evident in Hip1r mutant stomach, although increased epithelial cell proliferation and elevated gastrin levels were not affected by the presence or absence of this pro-inflammatory cytokine. Analysis of cell lineage markers in the double mutant mice demonstrated that IFNγ specifically affected the development of metaplastic mucous cells in the neck region, while the parietal cell, surface mucous cell and zymogenic cell alterations remained similar to the histopathology in the Hip1r mutant. Morphometric analysis showed that IFNγ was required for the mucous cell hypertrophy and hyperplasia observed in Hip1r-deficient mice. Together, these findings demonstrate that IFNγ is critical for the development of the gastric epithelial cell metaplasia that results from parietal cell atrophy in the Hip1r-deficient mice.
Keywords: atrophic gastritis, gastric cancer, mucous neck cell, parietal cell, spasmolytic polypeptide-expressing metaplasia (SPEM), stomach
Gastric cancer is the second most common cause of cancer-related death worldwide (1). Human gastric epithelial cell transformation and eventual gastric cancer development are thought to result from a chronic inflammatory condition initiated by Helicobacter pylori infection (2). The current pathway for gastric cancer development, as proposed by Correa and others (3, 4), is a progression from inflammation-induced changes in the gastric mucosa, to chronic and then atrophic gastritis associated with the loss of parietal cells, with subsequent metaplastic changes including the formation of spasmolytic polypeptide-expressing metaplasia (SPEM) and/or intestinal metaplasia. Parietal cells in particular are considered to play a critical role in gastric epithelial cell homeostasis, as evidenced by disturbed epithelial cell differentiation in mouse models of parietal cell loss, including reduced numbers of zymogenic cells and expansion of an aberrant mucous cell population termed SPEM that emerges from cells of the zymogenic lineage (5, 6). These characteristic epithelial cell changes have been observed in numerous mouse models of parietal cell loss, including those exhibiting parietal cell death induced by toxins (6, 7) or gene mutation (8), as well as those with progressive cell loss resulting from activation of complex inflammatory pathways, including autoimmune gastritis (9, 10) or infection with gastric pathogenic Helicobacter sp. (11). The common cellular derangement observed in all of these divergent pathological processes strongly suggests that parietal cell function is necessary for gastric epithelial cell homeostasis.
We have recently described a mouse mutant with spontaneous parietal cell apoptosis that serves as a useful model to study the complex gastric epithelial cell changes initiated by parietal cell loss. Huntingtin interacting protein 1 related (Hip1r) is an F-actin- and clathrin-binding protein involved in the dynamic vesicular trafficking associated with parietal cell acid secretion (8). Loss of Hip1r results in parietal cell apoptosis, with subsequent spontaneous development of multifaceted gastric epithelial cell changes, including glandular hypertrophy, expansion of surface mucous cells and disruption of the zymogenic lineage characterized by loss of zymogenic chief cells, expansion of cells that co-stain for chief and mucous neck cell markers and emergence of metaplastic mucous cells (8, 12). The loss of zymogenic cells accompanied by the expansion of metaplastic TFF2-expressing mucous cells is diagnostic for SPEM. Importantly, Hip1r-deficient mice have an associated gastric inflammatory cell infiltration, possibly resulting from low gastric acid levels creating conditions permissive for bacterial overgrowth (8). However, it is still unknown if and how inflammation might play a role in the multi-lineage epithelial cell derangement associated with parietal cell loss.
It is well established that chronic inflammation is crucial for the initiation and progression of epithelial cell metaplasia and gastric cancer development. However, the roles of specific inflammatory mediators in this complex process are not clear. Clinical studies have shown that specific gene polymorphisms for interleukin 1β (IL-1β) are associated with increased cytokine levels and increased incidence of gastric cancer (13, 14). Indeed, expression of IL-1β in the gastric mucosa of transgenic mice triggered spontaneous gastric inflammation and gastric cancer development, which was accelerated after Helicobacter felis infection (15). Together these studies suggest that IL-1β can induce gastric neoplasia, but roles for other pro-inflammatory cytokines remain to be described.
Analysis of cytokine profiles in mice after Helicobacter pylori infection suggested that mucosal inflammation is Th1-mediated, characterized by increased expression of the pro-inflammatory cytokine interferon gamma (IFNγ) (16, 17). Further support for the potential importance of this cytokine was provided by the observation of increased expression of direct IFNγ target genes in mouse gastric epithelial cell populations after Helicobacter pylori infection (18) and by the observation of reduced metaplasia in Helicobacter infected IFNγ-deficient mouse models (16, 17, 19, 20). Furthermore, treatment of C57BL/6 mice with IFNγ induced the expansion of mucous cells with features similar to SPEM (21). In addition, characterization of responses to IFNγ in the human gastric cancer cell line NCI-N87 showed increased expression of the mucous neck cell markers TFF2 and Muc6, which are also upregulated in SPEM (21). Together these data suggested that IFNγ might play a role in the development of SPEM in response to gastric inflammation.
Although IFNγ has been strongly implicated in the epithelial cell remodeling in response to Helicobacter infection, its role in mediating the gastric epithelial cell lineage changes in response to parietal cell loss have not been investigated. Thus, in this study we examined the role of IFNγ for the spontaneous development of metaplasia in Hip1r-deficient mice by crossing this mutant with IFNγ-deficient mice to generate Hip1r/IFNγ double mutant mice. We found that IFNγ deficiency markedly reduced the gastric histological lesions and reduced the mucous cell metaplasia normally observed in Hip1r mutant mice. Importantly, these studies establish IFNγ as a critical cytokine contributing to the formation of SPEM in Hip1r-deficient mice.
MATERIALS AND METHODS
Mice
Mice were generated by crossing Hip1r-deficient mice (22) with IFNγ-deficient mice (Jackson Lab #002287). Mice were on a mixed 129X1 and C57BL/6 strain background, and all groups were composed of littermates generated from intercrossing compound heterozygotes. Control, Hip1r-deficient, Hip1r/IFNγ double mutant mice and IFNγ-deficient mice of both sexes were examined at 3 months and 12 months of age. Hip1r and IFNγ heterozygotes (+/−) did not exhibit gastric phenotypes and thus were grouped with mice carrying wild type alleles (+/+). Thus the control group included homozygous wild type and heterozygotes at both loci, which all exhibited similar phenotypes. IFNγ-deficient mice did not differ from controls for the phenotypes examined in this study. Mice were housed in ventilated and automated watering cages under specific pathogen-free conditions. Mouse use was approved by the University of Michigan Committee on Use and Care of Animals.
Histological Analysis
Stomachs were removed after an overnight fast and fixed in 4% paraformaldehyde as described previously (10). The analysis focused on the corpus region from the greater curvature. Paraffin tissue sections (5 μm) were stained with hematoxylin and eosin (H&E) to evaluate general histology and with Periodic Acid Schiff (PAS) and Alcian blue to evaluate mucin type changes. Histological scores were blindly evaluated by an experienced veterinary pathologist based on the following six parameters: mucosal hypertrophy, mucous cell metaplasia, dilated glands, disorganized glands, granulocytes and mononuclear cell infiltrates. Lesions were scored on a semi-quantitative scale of 0 to 3. Lesion absence in all 6 categories was given a score of 0. For mucosal hypertrophy, detectable thickening was scored 1, up to 2X normal thickness was scored 2, and thickening greater than 2X was given a score of 3. For mucous cell metaplasia, the presence of a few “foamy” epithelial cells was scored 1, foamy cells in most fields but with visible normal tissue was scored 2, and sections where the glandular epithelium was replaced by foamy mucous cells was scored 3. For the presence of dilated glands, occasional dilated glands was scored 1, more than 4 dilated glands per section was scored 2, and dilated glands that were present in all fields was scored 3. Tissue with detectable disorganized glands was scored 1, prominent but not widespread gland disorganization was scored 2, and disorganized glands that were present in all fields was scored 3. For granulocytes and mononuclear inflammatory cells, the presence of occasional clusters or widely scattered cells was scored 1, the presence of many clusters or focal extensive infiltrates was scored 2, and inflammatory cells present in all fields was scored 3.
Specific gastric cell types were identified by immunostaining for H+, K+-ATPase α subunit (1:1000; Medical and Biological Laboratories, Aichi, Japan), mucin-5AC (Muc5AC 1:75; Novocastra, Newcastle, UK), gastric intrinsic factor (1:2000 rabbit anti-human intrinsic factor; gift from David Alpers, Washington University, St. Louis, MO), and interferon gamma receptor 2 (IFNGR2 1:10; rabbit polyclonal; Santa Cruz, CA), and visualized with appropriate secondary antibodies as previously described (10). Griffonia simplicifolia lectin II (GSII 1:1000; Invitrogen Alexa 488 conjugated) was used to stain for mucous cells and 4′, 6-diamidino-2-phenylindole dihydrochloride (DAPI) was used for counterstaining cell nuclei. Imaging by digital microscopy was performed as previously described (10).
Proliferating cells were analyzed by immunostaining for Ki67 (1:200; Thermo Scientific, Waltham, MA). Image J (1.34u by Wayne Rasband, NIH, USA (http://rsb.info.nig.gov/ij/)) was used to calculate the number of Ki67-positive cells per gland. To identify proliferating mucous cells, tissue sections were immunostained for Ki67 as above, rinsed with 0.01% Triton X-100 in PBS, and then stained with GSII (1:1000 Alexa 594) 1 hr at room temperature. Secondary antibody staining was performed with goat anti-rabbit IgG Alexa 488 (1:400; Invitrogen) and slides were mounted with Prolong Gold Antifade Reagent with DAPI. Images were collected on a confocal microscope (Zeiss LSM510, Carl Zeiss, Germany).
Confocal Analysis of Mucous Cells
Tissue sections were co-immunostained for E-cadherin (72 hrs, 4 °C at 1:1000; rat monoclonal from Invitrogen), rinsed with 0.01 % Triton X-100 in PBS, and then stained with GSII. Secondary antibody staining was performed with goat anti-rat IgG Alexa 488 (1:400; Invitrogen) and slides were mounted with Prolong Gold with DAPI. Confocal images were analyzed for mucous cell size and number using Image J software. For quantification of cell size, only cells with a distinctly visible cell membrane (E-cadherin-positive), positive GSII staining and a nucleus in the plane of section were counted (2–3 animals per group). For quantification of cell number, GSII-positive cells per gastric gland were counted from 2–3 representative fields per animal. Similar to the analysis above, only those cells with a visible cell membrane, positive GSII staining and a nucleus in the plane of section were included.
Measurement of Serum Gastrin
Radioimmunoassay (RIA) was used to measure serum gastrin levels as previously described (8). Briefly, blood was collected from fasted mice by cardiac puncture into heparinized tubes, and plasma was stored at −20°C until assayed. Samples were incubated with [125I] Tyr12-Gastrin I (human; Perkin Elmer Life and Analytical Sciences) and rabbit anti-gastrin antibody (1.27 μg/ml final concentration; Dako Cytomation) for 72h at 4°C, and gastrin was harvested by polyethylene glycol precipitation and centrifugation. Gastrin levels were calculated from the pellet radioactivity in comparison to a standard curve (2–500 pmol/l; human synthetic gastrin I diluted in heat-treated mouse serum; both from Sigma-Aldrich). The gastrin antibody recognizes both nonsulphated and sulphated forms of gastrin-17 and -34.
qRT-PCR Measurement of mRNA Abundance
Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) amplification of gastric epithelial specific markers was performed as previously described (12). Briefly, total RNA was isolated from corpus or antrum and reverse transcribed into cDNA using the iScript cDNA Synthesis Kit (BioRad, Hercules, CA). Triplicates for each sample were amplified by PCR to measure mRNA abundance of specific genes. Expression levels were normalized to the expression of the gene for glyceraldehyde 3-phosphate dehydrogenase (Gapdh), which remained the same in all groups. All primers used for qRT-PCR assays were validated via standard curve of a dilution series as previously described (8).
Statistics
GraphPad Prism software (v5.0, San Diego, CA) was used for statistical analysis and preparation of graphs. Histological scores were analyzed by non-parametric Mann–Whitney U test, and other data were analyzed by Student’s t-test or one-way ANOVA, with Bonferroni’s post-hoc test. Quantitative data were presented as mean ± SEM. P < 0.05 was considered significant.
RESULTS
IFNγ-deficiency Reduced Gastric Histological Lesions of Hip1r-deficient Mice
We determined whether increased IFNγ was a component of the inflammatory response that spontaneously develops in the stomachs of Hip1r-deficient mice by quantitative measurement of mRNA abundance. This analysis revealed increased expression of IFNγ in the acid-secreting corpus, but not the antrum of 3-month-old Hip1r-deficient mice in comparison to control mice (Figure 1).
Figure 1.
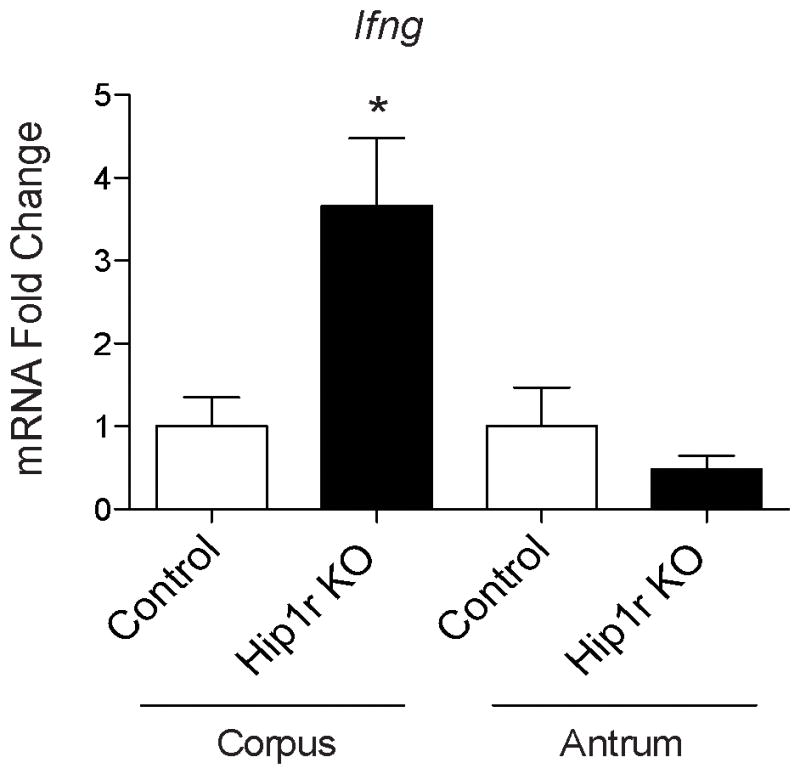
Increased IFNγ gene expression in Hip1r-deficient (KO) mice. Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) measurement of IFNγ (Ifng) gene expression from corpus and antrum of 3 month-old control (n=4) and Hip1r-KO (n=5) mice. Values were normalized to glyceraldehyde 3-phosphate dehydrogenase (Gapdh) levels and expressed as fold-change (mean ± s.e.m) relative to control values for each region and analyzed using Student’s t-test (*P < 0.05 vs. corpus control).
To study the specific function of IFNγ in the development of gastric metaplasia, Hip1r-deficient mice were crossed with IFNγ-deficient mice to generate Hip1r/IFNγ double mutant mice. Four genotypes were aged to 3 months and 12 months, including control, Hip1r-deficient, Hip1r/IFNγ double mutant and IFNγ-deficient, and histology was examined by H&E staining. As previously described (8, 12), Hip1r-deficient mice spontaneously developed hypertrophy and inflammatory cell infiltration at 3 months of age (Figure 2). In contrast, Hip1r/IFNγ double mutant mice had reduced hypertrophy and inflammation, although the cellular morphology was still altered in comparison to controls (described below). At 12 months of age, Hip1r-deficient mice exhibited more extensive hypertrophy and inflammation (arrows) as well as prominent mucous cell metaplasia (arrowheads), while Hip1r/IFNγ double mutants were spared these phenotypes (Figure 2). Histological scoring confirmed that Hip1r/IFNγ double mutant mice exhibited a reduction of histological lesions across multiple parameters, with statistically significant differences in: mucosal hypertrophy, disorganized glands, granulocytes and mononuclear cells (Figure 3). Together these results showed that IFNγ significantly contributes to the development of gastric gland pathology and inflammation in Hip1r-deficient mice.
Figure 2.

Spontaneous development of gastric metaplasia in Hip1r-deficient mice requires IFNγ. Paraffin sections from the corpus of control, Hip1r-KO, Hip1r/IFNγ-DKO, and IFNγ-KO mice at 3 and 12 months of age were stained with hematoxylin and eosin to evaluate general histology. Several aspects of the cellular changes that spontaneously developed in the Hip1r-KO, including inflammatory cell infiltrates (arrows) and mucous cell metaplasia (arrowheads), were not observed in the Hip1r/ IFNγ-DKO stomachs. Note that IFNγ-KO mice were indistinguishable from control. Scale bars: 100 μm.
Figure 3.

Histopathology in Hip1r KO mouse stomach was reduced by IFNγ deficiency. Histological scoring of H&E stained gastric paraffin sections from 12-month old control, Hip1r-KO, Hip1r/IFNγ-DKO and IFNγ-KO mice are displayed. Severity scores were determined for 6 different histological lesion categories and displayed as mean ± s.e.m., with n=6–10 as shown. Statistical comparisons were analyzed by Mann-Whitney U test; *P < 0.05, **P <0.01 Hip1r-KO vs. Hip1r/IFNγ-DKO in the same category.
Epithelial Cell Specific Effects of IFNγ
To define the cellular targets of IFNγ-mediated metaplasia, gastric epithelial cell-specific markers were examined. Examination of parietal cells by immunostaining for the alpha subunit of the H+, K+-ATPase proton pump showed that Hip1r/IFNγ double mutant mice had a reduction in parietal cells, similar to that observed in Hip1r-deficient mice (Figure 4a). Expression of this parietal cell marker was quantified by qRT-PCR measurement of mRNA abundance, confirming a similar reduction in expression in Hip1r-deficient and Hip1r/IFNγ double mutant mice (Figure 4b). These results suggest that the intrinsic defect in Hip1r-deficient parietal cells that results in apoptotic cell death (8) was not affected by the absence of the pro-inflammatory cytokine IFNγ.
Figure 4.

Parietal cell loss in Hip1r-deficient mice is not IFNγ-dependent. (a) Paraffin sections from the corpus of control, Hip1r-KO and Hip1r/IFNγ-DKO mice at 12 months of age were immunostained for the parietal cell specific marker H+, K+-ATPase alpha subunit. Scale bars: 100 μm. (b) Corpus mRNA abundance for the H+, K+-ATPase alpha subunit gene (Atp4a) in 3-month (n=4–7) and 12-month old (n=3–5) mice was measured by qRT-PCR. Values were normalized to Gapdh and expressed as fold-change (mean ± s.e.m.) relative to control, with statistical analysis by one-way ANOVA with Bonferroni’s post-hoc test (*P < 0.05 vs. control).
Our previously published studies had shown significant mucous cell alterations in the Hip1r mutant stomach, including increased numbers of surface mucous cells as well as the development of an aberrant mucous neck/zymogenic cell lineage termed SPEM that occupied the lower half of the affected glands (see Figure 2, arrowheads) (8, 12). This was confirmed by PAS/Alcian blue staining of gastric sections from 12 month old Hip1r-deficient mice, which showed both increased surface staining and extensive staining in the lower portion of the gastric glands in comparison to controls (Figure 5a). The expanded surface mucous cells were examined further by immunostaining for the surface mucous cell marker Muc5AC (Figure 5b). This staining demonstrated that the Hip1r/IFNγ double mutant exhibited a marked expansion of surface mucous cells similar to the Hip1r mutant, suggesting that IFNγ is not critical for the surface cell expansion.
Figure 5.

Expansion of surface mucous cells in Hip1r-deficient mice is not IFNγ-dependent. (a) To identify gastric mucins, paraffin sections from the corpus of control, Hip1r-KO and Hip1r/IFNγ-DKO mice at 12 months of age were stained with Periodic Acid Schiff/Alcian blue (PAS/AB). (b) Immunohistochemical staining for the surface mucous cell marker Muc5AC demonstrated that Hip1r/IFNγ-DKO mice exhibited similar increases in numbers of surface cells as observed in Hip1r-KO mice. Scale bars: 50 μm.
Further examination of the PAS/Alcian blue-stained gastric sections in the different groups showed that the Hip1r/IFNγ double mutant stomach differed markedly from the Hip1r mutant. In contrast to Hip1r-deficient mice, the double mutant did not exhibit the extensive neutral (red) and acidic (blue) mucin accumulation in the lower half of the glands (Figure 5a). This result suggested that IFNγ contributes to the development of mucous cell metaplasia. Specific staining for mucous cells with GSII lectin showed that Hip1r/IFNγ double mutant mice had a reduction in mucous cells in comparison to Hip1r-deficient mice at both 3 months and 12 months of age, suggesting that IFNγ is required to develop the histopathologic condition termed SPEM (Figure 6a and Figure 2 arrowheads).
Figure 6.

IFNγ-deficiency tempers the mucous cell metaplasia but does not affect the zymogenic cell loss observed in Hip1r-KO mice. (a) Paraffin sections from the corpus of control, Hip1r-KO and Hip1r/IFNγ-DKO mice at 3 and 12 months of age were co-stained with an antibody to the zymogenic cell marker gastric intrinsic factor (red) and the mucous cell specific Griffonia simplicifolia (GSII) lectin (green), with DAPI (blue) nuclear stain. Scale bar: 100 μm. (b) Corpus mRNA abundance for gastric intrinsic factor gene (Gif) expression in 3-month (n=4–7) and 12-month old (n=3–5) mice was measured by qRT-PCR. Values were normalized to Gapdh and expressed as fold-change (mean ± s.e.m.) relative to control with statistical analysis by one-way ANOVA with Bonferroni’s post-hoc test (*P < 0.05 and **P < 0.01 vs. control).
SPEM development normally includes loss of differentiated zymogenic cells (5) and thus we examined this cell type by immunostaining for the specific marker gastric intrinsic factor. Both Hip1r-deficient mice and Hip1r/IFNγ double mutant mice exhibited similar levels of zymogenic cell loss, shown by reduced immunostaining (Figure 6a, red), as well as reduced levels of gastric intrinsic factor mRNA (Gif) (Figure 6b). Furthermore, both Hip1r-deficient mice and Hip1r/IFNγ double mutant mice contained cells at the base of the glands that co-stained for zymogenic and mucous neck cell markers, which is a common feature of SPEM (Figure 6a). These data suggest that IFNγ is not responsible for the extensive zymogenic cell changes in Hip1r-deficient mice. Importantly, this analysis suggested that the metaplastic mucous cell expansion, observed by PAS/Alcian blue and by GSII staining, is not always reflected by the extent of zymogenic cell changes since the double mutant had an apparent tempering of mucous cell metaplasia in spite of marked zymogenic cell loss and expansion of co-stained cells. This finding suggested that IFNγ might directly target the zymogenic lineage to affect the emergence of metaplastic mucous cells.
IFNγ Receptor Expression in Mucous Neck Cells
To study whether IFNγ might directly target the zymogenic lineage, the gastric expression of IFNγ receptor was studied. Immunostaining for the non-ligand-binding beta chain, IFNGR2, showed expression in the mid- and base-regions of the gastric glands, consistent with localization in both mucous neck and zymogenic chief cells (Figure 7). Co-staining for mucous neck cells with GSII demonstrated that expression in the mid-region of the gland corresponded to mucous neck cells (Figure 7a). In contrast, co-staining with antibodies to IFNGR2 and H+, K+ ATPase demonstrated no co-localization in parietal cells (Figure 7b). These results suggest that IFNγ may directly target the zymogenic lineage to contribute to the development of mucous cell metaplasia in Hip1r-deficient mice.
Figure 7.

Interferon gamma receptor 2 (Ifngr2) is abundantly expressed in mucous neck cells. Paraffin sections from the corpus of control mice were co-stained with: (a) an antibody directed against Ifngr2 (red) and GSII lectin (green) to examine mucous neck cells, or (b) antibodies to Ifngr2 (red) and H+, K+-ATPase (green) to examine parietal cells. Mucous neck/zymogenic lineage cells stained strongly for Ifngr2 with no obvious staining in parietal cells. DAPI (blue) nuclear stain. Scale bars: 100μm.
Cell Proliferation and Glandular Hypertrophy
The gastric mucosal hypertrophy in Hip1r-deficient mice had been previously demonstrated to result from increased levels of the hormone gastrin in the blood (8). Since the Hip1r/IFNγ double mutant mice did not show this glandular hypertrophy (Figure 2 and 3), we examined the cellular basis for this effect by measuring cellular proliferation and circulating gastrin levels. Immunostaining for the cell proliferation marker Ki67 and quantification by morphometric analysis confirmed increased numbers of proliferating cells in Hip1r-deficient mice compared to control (Figure 8). Proliferating cells were observed to extend to the bottom of the gland in the region occupied by metaplastic mucous cells in the Hip1r-deficient mice. Co-staining for GSII and Ki67 confirmed proliferating mucous cells in this mutant (Figure 8c). In light of the differences in gland height, it was surprising to find that the Hip1r/IFNγ double mutant mice showed similarly increased proliferation as did the Hip1r-deficient mice. However, proliferating mucous cells were rarely observed in the double mutant (Figure 8c). In contrast, proliferating GSII-positive mucous cells were not observed in control mice (not shown).
Figure 8.

Increased epithelial cell proliferation in Hip1r-deficient mice is not IFNγ-dependent. (a) Paraffin sections from the corpus of control, Hip1r-KO, Hip1r/IFNγ-DKO, and IFNγ-KO mice at 12 months of age were stained for the cell proliferation marker Ki67 with hematoxylin counterstain. Scale bar: 100 μm. (b) Proliferation was quantified by morphometric counting of Ki67 positive cells per gastric gland in 3-month (n=3–5) and 12-month old (n=3–5) mice. Data are shown as mean ± s.e.m., with statistical analysis by one-way ANOVA with Bonferroni’s post-hoc test (*P < 0.05 vs. control). (c) Proliferating mucous cells in Hip1r-KO and Hip1r/IFNγ-DKO mice were assessed by co-immunostaining for Ki67 (green) and GSII lectin (red), and confocal microscopy was used to examine Ki67/GSII double-positive cells (arrows). Scale bar: 20 μm.
To test whether IFNγ affected circulating levels of gastrin, serum gastrin was quantified by RIA. This analysis showed that both the Hip1r mutant and the Hip1r/IFNγ double mutant mice were hypergastrinemic (Figure 9), which is the likely explanation for the increased proliferation observed in both groups. Together, these results showed that IFNγ did not contribute to the hypergastrinemia and increased proliferation in Hip1r-deficient mice. Furthermore, the data showed that increased gastrin levels are associated with increased proliferation.
Figure 9.

Increased serum gastrin in Hip1r-deficient mice is not IFNγ-dependent. Serum gastrin levels were measured by radioimmunoassay in 3-month (n=6–9) and 12-month old (n=3–5) mice. Data are shown as mean ± s.e.m., with statistical analysis by one-way ANOVA with Bonferroni’s post-hoc test (*P < 0.05 and ***P < 0.001 vs. control or as shown).
Since loss of IFNγ affected GSII-positive mucous cell proliferation, we further examined this cell lineage to determine if alterations in these cells might contribute to the glandular hypertrophy. We hypothesized that expansion of metaplastic mucous cells in Hip1r-deficient mice led to increased gland height. We measured cell size and number after co-immunostaining for the cell membrane marker E-cadherin and GSII lectin. Analysis of confocal images from each group of animals revealed that mucous cells from Hip1r-deficient mice were significantly larger than control mice, and that the loss of IFNγ in the double mutant mice abrogated this phenotype (Figure 10). Mucous cell sizes in IFNγ-deficient mice were similar to control. Furthermore, Hip1r-deficient mice had significantly more mucous cells compared to control, and loss of IFNγ in the double mutant normalized mucous cell number. These results allude to the possibility that the elevation in IFNγ, acting through its receptor on zymogenic lineage cells, can induce gastric gland hypertrophy via increasing mucous cell size and number.
Figure 10.

Increased mucous cell number and size in Hip1r-deficient mice. (a) Paraffin sections from the corpus of control, Hip1r-KO, Hip1r/IFNγ-DKO and IFNγ-KO mice at 12 months of age were co-stained with an antibody to E-cadherin (green) and GSII lectin (red) and images collected by confocal microscopy. Arrows point to some mucous cells with insets showing higher powered views. (b) Quantification of cell size in control (n=101), Hip1r-KO (n=81), Hip1r/IFNγ-DKO (n=63) and IFNγ-KO (n=70) mucous cells. Data are shown as mean ± s.e.m., with statistical analysis by one-way ANOVA with Bonferroni’s post-hoc test (***P < 0.001 vs. all groups). (c) Quantification of mucous cell number in control (n=6), Hip1r-KO (n=10), Hip1r/IFNγ-DKO (n=4), and IFNγ-KO (n=3) mice. Data are shown as mean ± s.e.m., with statistical analysis by one-way ANOVA with Bonferroni’s post-hoc test (*P < 0.05 vs. control).
DISCUSSION
Chronic inflammation has long been considered a critical factor that disrupts gastric epithelial cell homeostasis and contributes to the development of gastric cancer. How immune cells and pro-inflammatory cytokines interact with the complicated gastric epithelial cell architecture and signaling network is an intriguing yet undefined question. In this study, we utilized a unique animal model, Hip1r-deficient mice, which develop progressive gastric epithelial cell lineage alterations due to intrinsic parietal cell loss, to understand the complex events associated with the multifaceted and progressive epithelial cell metaplasia termed SPEM (8, 12). In this study we focused on the contribution of the cytokine IFNγ by intercrossing Hip1r-deficient mice with IFNγ-deficient mice. The loss of IFNγ abrogated specific aspects of the zymogenic lineage metaplasia, but not the parietal cell or surface mucous cell changes (Table 1). Similar to the Hip1r-deficient mice, the double mutant had decreased numbers of zymogenic chief cells and increased numbers of cells expressing both chief and mucous neck cell markers. However, in contrast to the Hip1r-deficient mice, the double mutant did not develop metaplastic mucous cells. Thus, IFNγ was necessary for the development of some but not all aspects of SPEM. We also observed that IFNγ was not important for increased epithelial cell proliferation or elevated serum gastrin levels in Hip1r-deficient mice. Thus IFNγ appears to play a specific role in the complex cellular changes initiated by parietal cell loss.
Table 1.
Summary of gastric epithelial cell features in Hip1r-deficient mice and Hip1r/IFNγ-double mutant mice.
| Cell Type | Marker | Hip1r-KO | Hip1r/IFNγ DKO |
|---|---|---|---|
| Surface mucous | Muc5AC | Hyperplastic | Hyperplastic |
| Proliferative progenitor | Ki67 | Increased | Increased |
| Parietal | H+, K+-ATPase | Decreased | Decreased |
| Zymogenic lineage | |||
| Mucous neck | GSII lectin | Hyperplastic/hypertrophied | Normala |
| Transition | GSII and GIF | Increased | Increased |
| Zymogenic chief | GIF | Decreased | Decreased |
Cellular features, described in comparison to wild type, were determined by mRNA abundance and immunohistochemical analysis of cell specific markers in 12-month Huntingtin interacting protein 1-related (Hip1r)-deficient mice and Hip1r/IFNγ-double mutant mice. Abbreviations: KO, knockout; DKO, double knockout; Muc5AC, mucin 5AC; GSII, Griffonia simplicifolia; GIF, gastric intrinsic factor.
Loss of IFNγ protects against the spontaneous development of the mucous cell changes observed in the neck region of Hip1r-deficient glands.
Our studies suggest that IFNγ targets the zymogenic lineage through a mechanism that affects mucous cell number and size. This finding agrees with the results of a previously published study that showed mucous cell expansion after in vivo administration of IFNγ to C57BL/6 mice (21). Further in vitro analysis showed that IFNγ stimulation of NCI-N87 gastric cancer cells induced increased expression of the mucous neck cell markers Muc6 and TFF2, and, importantly, induced a significant increase in cell size similar to what we observed in the Hip1r-deficient mice (21). These studies together with our observation of abundant IFNGR2 expression in the zymogenic lineage is consistent with the notion that IFNγ can directly and preferentially target these gastric epithelial cells. Whether IFNγ is sufficient to trigger gastric metaplasia has been recently addressed by specific transgenic expression of this cytokine in the mouse stomach (23, 24). Although a low level of expression of this cytokine was not observed to induce gastric epithelial cell changes (23), higher levels triggered a profound and rapid mucous cell metaplasia (24).
The expansion of a metaplastic mucous cell population is a common characteristic of the pre-neoplastic metaplasia known as SPEM. Hip1r-deficient mice develop SPEM as a likely consequence of spontaneous apoptotic parietal cell death that occurs in this mutant (8, 12). Acute administration of the drug DMP-777, which rapidly induces parietal cell loss, has been shown to cause SPEM development without obvious inflammation in both rats (25) and mice (6). This finding indicates that parietal cell loss itself is sufficient to induce SPEM. However, use of the drug L-635, which induces rapid parietal cell loss accompanied by the induction of a prominent inflammatory cell infiltrate, resulted in a more aggressive development of mucous cell metaplasia, supporting the conclusion that inflammatory mediators play a critical role in SPEM development (6).
In our study, parietal cell loss in the absence of IFNγ does not appear to induce the complete SPEM phenotype, even when Hip1r/IFNγ double mutant mice were aged to 12 months old. Aspects of the SPEM phenotype that were preserved in the absence of IFNγ include the loss of zymogenic cells and the expansion of cells at the base of the gland that co-express mucous neck cell (GSII) and zymogenic chief cell (intrinsic factor) markers. Parietal cells are critically important for zymogenic chief cell differentiation and loss of parietal cells is associated with the rapid loss of zymogenic cells, reportedly by both apoptotic cell death (12) and trans-differentiation into the SPEM lineage (6). How parietal cells mediate this effect is unknown, although it has been suggested that parietal cells might secrete an essential growth factor required for zymogenic cell differentiation (26, 27).
In summary, our studies show that the gastritis that develops upon apoptotic loss of parietal cells in the Hip1r-deficient mouse includes increased expression of the pro-inflammatory cytokine IFNγ. IFNγ plays a unique role in the metaplastic lineage changes that occur in the gastric mucosa, specifically targeting hypertrophy and hyperplasia of mucous cells in the zymogenic lineage. While our studies support a possible direct effect of IFNγ to contribute to the pre-neoplastic SPEM phenotype, it is likely that these cells might be sensitive to a number of inflammatory mediators and thus the effects of IFNγ could also be indirect. Furthermore, our study demonstrates that different aspects of the SPEM phenotype are mediated by distinct mechanisms, with loss of zymogenic cells likely due to the loss of an essential, and as yet undefined, parietal cell factor, while pro-inflammatory cytokines, including IFNγ, contribute to the development of mucous cell metaplasia. Future studies should be directed towards understanding how inflammatory mediators might directly regulate the zymogenic lineage during metaplastic transformation.
Supplementary Material
Acknowledgments
Sources of Support: This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant RO1-DK-078926 (L.C. Samuelson). Core support was provided by NIDDK Gastrointestinal Hormone Research Center Grant P30-DK-34933.
We acknowledge Jianhua Ren for initiating this analysis, Allison Hoch for maintenance of the mouse colonies, and the morphology core of the University of Michigan Gastrointestinal Peptide Center for assistance with confocal microscopy. This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant RO1-DK-078926 (L.C. Samuelson). Core support was provided by NIDDK Gastrointestinal Hormone Research Center Grant P30-DK-34933.
Abbreviations
- DAPI
4′, 6-diamidino-2-phenylindole dihydrochloride
- Gapdh
Glyceraldehyde 3-phosphate dehydrogenase
- GSII
Griffonia simplicifolia lectin II
- H&E
Hematoxylin and eosin
- Hip1r
Huntingtin interacting protein 1-related
- H+, K+ - ATPase
Hydrogen potassium ATPase
- IFNγ
Interferon gamma
- Ifngr2
Interferon gamma receptor 2
- IL-1β
Interleukin-1 beta
- Muc5AC
Mucin-5AC
- PAS
Periodic Acid Schiff
- qRT-PCR
Quantitative reverse transcriptase polymerase chain reaction
- RIA
Radioimmunoassay
- SPEM
Spasmolytic polypeptide expressing metaplasia
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Polk DB, Peek RM., Jr Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer. 2010;10(6):403–414. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Correa P. A human model of gastric carcinogenesis. Cancer Res. 1988;48(13):3554–3560. [PubMed] [Google Scholar]
- 4.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007;117(1):60–69. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldenring JR, Nam KT, Mills JC. The origin of pre-neoplastic metaplasia in the stomach: Chief cells emerge from the Mist. Exp Cell Res. 2011;317(19):2759–2764. doi: 10.1016/j.yexcr.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nam KT, Lee HJ, Sousa JF, et al. Mature Chief Cells Are Cryptic Progenitors for Metaplasia in the Stomach. Gastroenterology. 2010;139(6):2028–U2324. doi: 10.1053/j.gastro.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nomura S, Yamaguchi H, Ogawa M, Wang TC, Lee JR, Goldenring JR. Alterations in gastric mucosal lineages induced by acute oxyntic atrophy in wild-type and gastrin-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2005;288(2):G362–375. doi: 10.1152/ajpgi.00160.2004. [DOI] [PubMed] [Google Scholar]
- 8.Jain RN, Al-Menhali AA, Keeley TM, et al. Hip1r is expressed in gastric parietal cells and is required for tubulovesicle formation and cell survival in mice. J Clin Invest. 2008;118(7):2459–2470. doi: 10.1172/JCI33569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Judd LM, Gleeson PA, Toh BH, van Driel IR. Autoimmune gastritis results in disruption of gastric epithelial cell development. Am J Physiol. 1999;277(1 Pt 1):G209–218. doi: 10.1152/ajpgi.1999.277.1.G209. [DOI] [PubMed] [Google Scholar]
- 10.Lopez-Diaz L, Hinkle KL, Jain RN, et al. Parietal cell hyperstimulation and autoimmune gastritis in cholera toxin transgenic mice. Am J Physiol Gastrointest Liver Physiol. 2006;290(5):G970–979. doi: 10.1152/ajpgi.00461.2005. [DOI] [PubMed] [Google Scholar]
- 11.Wang TC, Goldenring JR, Dangler C, et al. Mice lacking secretory phospholipase A2 show altered apoptosis and differentiation with Helicobacter felis infection. Gastroenterology. 1998;114(4):675–689. doi: 10.1016/s0016-5085(98)70581-5. [DOI] [PubMed] [Google Scholar]
- 12.Keeley TM, Samuelson LC. Cytodifferentiation of the postnatal mouse stomach in normal and Huntingtin-interacting protein 1-related-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2010;299(6):G1241–1251. doi: 10.1152/ajpgi.00239.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El-Omar EM, Carrington M, Chow WH, et al. The role of interleukin-1 polymorphisms in the pathogenesis of gastric cancer. Nature. 2001;412(6842):99. doi: 10.1038/35083631. [DOI] [PubMed] [Google Scholar]
- 14.Figueiredo C, Machado JC, Pharoah P, et al. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst. 2002;94(22):1680–1687. doi: 10.1093/jnci/94.22.1680. [DOI] [PubMed] [Google Scholar]
- 15.Tu S, Bhagat G, Cui G, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14(5):408–419. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawai N, Kita M, Kodama T, et al. Role of gamma interferon in Helicobacter pylori-induced gastric inflammatory responses in a mouse model. Infect Immun. 1999;67(1):279–285. doi: 10.1128/iai.67.1.279-285.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smythies LE, Waites KB, Lindsey JR, Harris PR, Ghiara P, Smith PD. Helicobacter pylori-induced mucosal inflammation is Th1 mediated and exacerbated in IL-4, but not IFN-gamma, gene-deficient mice. J Immunol. 2000;165(2):1022–1029. doi: 10.4049/jimmunol.165.2.1022. [DOI] [PubMed] [Google Scholar]
- 18.Mueller A, Merrell DS, Grimm J, Falkow S. Profiling of microdissected gastric epithelial cells reveals a cell type-specific response to Helicobacter pylori infection. Gastroenterology. 2004;127(5):1446–1462. doi: 10.1053/j.gastro.2004.08.054. [DOI] [PubMed] [Google Scholar]
- 19.Eaton KA, Mefford M, Thevenot T. The role of T cell subsets and cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J Immunol. 2001;166(12):7456–7461. doi: 10.4049/jimmunol.166.12.7456. [DOI] [PubMed] [Google Scholar]
- 20.Sayi A, Kohler E, Hitzler I, et al. The CD4+ T cell-mediated IFN-gamma response to Helicobacter infection is essential for clearance and determines gastric cancer risk. J Immunol. 2009;182(11):7085–7101. doi: 10.4049/jimmunol.0803293. [DOI] [PubMed] [Google Scholar]
- 21.Kang W, Rathinavelu S, Samuelson LC, Merchant JL. Interferon gamma induction of gastric mucous neck cell hypertrophy. Lab Invest. 2005;85(5):702–715. doi: 10.1038/labinvest.3700260. [DOI] [PubMed] [Google Scholar]
- 22.Hyun TS, Li L, Oravecz-Wilson KI, et al. Hip1-related mutant mice grow and develop normally but have accelerated spinal abnormalities and dwarfism in the absence of HIP1. Mol Cell Biol. 2004;24(10):4329–4340. doi: 10.1128/MCB.24.10.4329-4340.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tu SP, Quante M, Bhagat G, et al. IFN-gamma inhibits gastric carcinogenesis by inducing epithelial cell autophagy and T-cell apoptosis. Cancer Res. 2011;71(12):4247–4259. doi: 10.1158/0008-5472.CAN-10-4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Syu L-J, Ferris J, Qiao X, et al. Transgenic Expression of Interferon Gamma in the Gastric Corpus Leads to Inflammation, Progenitor Cells Expansion and Mucous Metaplasia. Gastroenterology. 2010;138(Suppl 1 5):S83. [Google Scholar]
- 25.Goldenring JR, Ray GS, Coffey RJ, et al. Reversible drug-induced oxyntic atrophy in rats. Gastroenterology. 2000;118(6):1080–1093. doi: 10.1016/s0016-5085(00)70361-1. [DOI] [PubMed] [Google Scholar]
- 26.Capoccia BJ, Huh WJ, Mills JC. How form follows functional genomics: gene expression profiling gastric epithelial cells with a particular discourse on the parietal cell. Physiol Genomics. 2009;37(2):67–78. doi: 10.1152/physiolgenomics.90408.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jain RN, Brunkan CS, Chew CS, Samuelson LC. Gene expression profiling of gastrin target genes in parietal cells. Physiol Genomics. 2006;24(2):124–132. doi: 10.1152/physiolgenomics.00133.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.