

1. Introduction: Goals of Multichromophore Studies
Multichromophoric systems have gained intense interest in recent decades.1 The arraying of chromophores at close proximity to one another leads to interesting and novel properties, fueling the design of novel materials for many applications. Examples of multichromophoric systems include conjugated polymers,2-5 dendrimers,6-8 photonic wires,9-12 functionalized polypeptides,13-16 oligoamides,17 viral nanoparticles18 as well as multichromophores on DNA, the topic of this review. These designed systems are under investigation for applications as synthetic photosystems and light-harvesting antennae,19-22 synthetic ion channels,23-25 photovoltaic solar cells,26-29 organic semiconductors2, 30 and new supramolecular structures in materials science.31
Some of the earliest developments of multichromophoric systems arose when researchers arrayed multiple chromophores such as porphyrins to synthesize artificial light-harvesting systems.32, 33 This mimics the most efficient light harvesting system present in nature - the photosynthetic system of photosynthetic plants, algae and bacteria.34-38
Multichromophoric systems have been shown to exhibit highly efficient excitation energy transfer and electron transfer properties with applications in semiconductor and organic electronic materials research.39, 40 For instance, perylene or pyrene chromophores can stack on one another with substantial overlap in their π-orbitals, allowing one-dimensional transport of charge carriers.41, 42 These columnar structures have potential applications in optical and molecular electronic devices such as organic field effect transistors and solar cells.
Conjugated polymers have been shown to be highly suited for the development of sensors due to their sensory signal amplification.43 Because of local disruptions of π-conjugation, conjugated polymers commonly act as multichromophoric systems. Early experiments by Zhou and Swager using poly(phenylene ethynylene) polymers with cyclophane receptors showed strongly enhanced quenching, which is attributed to the facile energy migration throughout the molecular wire, termed the “molecular wire effect”.44, 45 This “superquenching” effect has been harnessed in highly sensitive sensors in multiple applications.46-50
1.1. Advantages of multichromophoric systems
Arraying multiple chromophores at close proximity with one another gives the system many advantages over single chromophores. First, the collective absorption is higher. This is particularly useful in light harvesting applications in solar cell research.26, 28, 51 Second, new photophysical properties that are distinct from the monomeric components are observed.52 Interactions between chromophores in the ground state and excited state result in the formation of new complexes such as excited-state dimers (excimers) and exciplexes. These complexes typically exhibit broadened absorptions and red-shifted emission spectra as a result of proximity. In the ground state, the formation of J-aggregates and H-aggregates from multiple chromophores results in a change in the absorption spectra. J-aggregates induce a bathochromic shift (red shift) while H-aggregates induce a hypsochromic shift (blue shift). A third useful property of multiple chromophore systems is tunability: properties can readily be adjusted to suit application needs.53, 54 This can be done through geometric and directional control of the assembly, since the relative orientation and arrangement of chromophores play a crucial role in their photophysical properties. The examples below further illustrate some of these advantageous properties.
1.2. Building multichromophoric systems
The wide range of applications of multichromophoric systems has prompted the need to develop efficient methods to synthesize them. The assembly of multiple chromophores is typically accomplished using either covalent methods or self-assembly methods. Covalent assembly through synthetic methods produces well-defined structure, and is the most common approach used in the literature. However, this approach can be tedious in application.55
On the other hand, self-assembly allows large numbers of chromophores to be arrayed efficiently by relying on the intrinsic non-covalent interactions between monomers such as van der Waals forces, hydrophobic interactions, dipole interactions, and charge transfer, to drive the formation of supramolecular multichromophoric system. Directional and selective interactions such as hydrogen bonds have also been used to drive self-assembly,55-57 as has the use of templates to aid in the assembly of monomers.58 However, self-assembly is limited by a lack of methods for placing diverse dyes at precise locations.
In recent years, many research groups have explored the potential of the deoxyribose nucleic acid (DNA) as a template for arraying multichromophoric systems.59 This is the subject of this review, and it is another inspiration from nature, as the phosphodiester backbone of DNA acts in nature as a scaffold for the ordered arraying of nucleobases, which are in fact chromophoric but at wavelengths shorter than is usually desired. Building multichromophore systems from DNA offers distinct advantages, as synthesis can be done via automated synthesizers, and self-assembly strategies are well defined. Moreover, the DNA scaffold has been found to be functionally effective and highly advantageous in assembling chromophores relative to other molecular assembly strategies, and thus they are undergoing rapid growth in the literature.59
1.3. What is covered in this review
The aim of this review is to discuss the relatively new class of multichromophoric systems that harnesses the DNA phosphodiester backbone (and DNA analogue backbones) as a scaffold for arraying chromophores. The advantages that the DNA scaffold confers to the multichromophoric system are described in Section 2. The potential of functionalizing the DNA scaffold with series of chromophores has gained great interest in recent years. Many research labs have entered the field, and Section 3 provides an overview of the different DNA-scaffolded multichromophoric systems. DNA multichromophore systems have been used as fluorescent nucleic acid probes, in non-enzymatic fluorophore systems, in molecular beacons, in DNA microarrays, in DNA nanotechnology, in photonic wires, in DNA sensor devices, and a multitude of other applications. Their synthesis (Section 4), properties (Section 5) and uses (Section 6) are discussed below.
2. The Utility of DNA as a Scaffold for Multiple Chromophores
2.1. Organized structure and self-assembly
DNA duplex formation brings about a well-defined, regular right-handed helical structure defined by canonical Watson-Crick base-pairing and stacking interactions. The π-stacked base pairs are held at 3.4 Å apart by the deoxyribose-phosphodiester scaffold. Self-assembly of DNA oligonucleotides occurs in a predictable manner, which has made DNA a favored material for formation of nanostructures,60, 61 and thus has been used to build a variety of architectures for multiple applications.62, 63 In this light, Seeman and coworkers are pioneers in building DNA nanostructures.64 A variety of two-dimensional nanostructures has been constructed using complementary base pairing and B-DNA duplex formation.65, 66 In recent years, three-dimensional structures have been built using branched DNA motifs with sticky ends67 and more recently, the technology was brought further with the construction of curved 3-D DNA nanostructures.68
In addition, the selectivity, stability and directionality of DNA make it a useful and versatile template for the precise placement of functional auxiliaries such as nanoparticles69, 70, multivalent quantum dots71 and proteins such as streptavidin,72 fluorescent proteins73 and multi-enzyme complexes.74 DNA has also been employed in the directed assembly of conjugated polymers such as polyanilines.75 Interestingly, even in the absence of duplex structures, DNA confers useful structural organization. Oligomers and polymers of single-stranded DNA are helical and the bases are usually stacked,76-79 which allows for convenient organization of chromophores in molecules and assemblies that are smaller and more synthetically accessible than large double helical structures.
Excited-state interactions are encouraged in the DNA scaffold
In natural duplex DNA, adjacent base pairs are held at 3.4 Å apart in the common B-form. This is defined by the thickness of aromatic π-systems, and coincides with favorable distances at which excited-state interactions can occur. Notably, excimers and exciplexes are formed favorably when they are between 3.3 to 3.6 Å apart. Indeed, natural nucleobases interact with each other in the excited state (although they have very low efficiency as fluorophores), and excimers of adenine have been observed.80 With the advent of DNA modification technologies, new stacks of modified base pairs can also be engineered and incorporated into DNA to discover new capabilities. Introduction of multiple conjugates or aggregates of dyes induces emergent spectroscopic behavior, such as band narrowing and band shifts, that are not observed in single dyes.81 Using DNA as the organizing scaffold, chromophores can be arrayed at predictable positions and distances. Multiple examples of these interactions are given in Section 3.
2.2. Efficient enzymatic synthesis
Besides chemical synthesis methods (see 2.3), modified DNA oligonucleotides can also be built using enzymatic synthesis. Natural DNA is replicated by a large family of DNA and RNA polymerases. These enzymes play a crucial role in the high specificity of DNA replication. Base pair hydrogen bonding, base stacking and steric effects have been shown to play important roles in base recognition during replication and the relative contributions of these factors vary with different enzymes.82, 83
Fluorescently labeled nucleotides, with fluorophores located at the end of linker chains, are now routinely incorporated by polymerases into DNA, and this has important applications in DNA sequencing technologies. In contrast, enzymatic incorporation of nucleotides with chromophores directly attached to bases has achieved only moderate successes in recent reports. This could be attributed to steric bulk and lower flexibility between the chromophoric moiety and the nucleotide, hindering their incorporation at the polymerase active site. Examining possible rigid linkers, Burgess and coworkers found that TaqFS polymerase was only able to incorporate a fluoresceinated nucleotide with the longest linker comprising of one phenyl group and two ethynyl groups. Thus, the length of a rigid linker is an important factor in design.84
Rather than using linkers, fluorophores can be directly attached to deoxyribose in place of a nucleobase. An early study by Matray and Kool showed that a nucleotide built from the fluorescent hydrocarbon pyrene, a large flat aromatic molecule with high stacking abilities, was incorporated at a much higher efficiency opposite abasic sites than any of the natural nucleotides by the Klenow fragment of E. coli DNA polymerase I.85, 86 More recently, multiple incorporations of the same chromophore were also accomplished using the terminal deoxyribonucleotide transferase (TdT) enzyme;87 see Section 4 for more details. Finally, in addition to the use of native enzymes, the evolution of DNA polymerases through rational design and activity-based screening has produced mutant polymerases with improved capability for incorporating modified nucleotides such as the pyrene nucleotide88 and for more conventional nucleotides with fluorophores on linkers.89
2.3. Convenient automated synthesis
Automated solid-phase phosphoramidite chemistry for oligonucleotide synthesis has been well developed with high stepwise yields, allowing preparation of oligomers as large as 100 nt or longer.90-92 Facile synthesis of a large range of modified nucleotides to construct modified oligonucleotides has also been established.93, 94 Auxiliary agents such as chromophores and metal binding ligands can be derivatized to the common 5′-dimethoxytrityl-protected 3′-phosphoramidite form for direct incorporation during automated DNA synthesis. In addition, many chemical modifications can also be introduced post-synthetically.95
2.4. Water solubility
The phosphodiester backbone of DNA is negatively charged at every nucleotide unit in aqueous conditions. The resulting water solubility is extended to many modified oligonucleotides, conferring solubility to hydrophobic molecules conjugated to DNA.96 For example, foldamers of oligopyrene contain the hydrophobic pyrene that is usually insoluble in aqueous media. Using DNA as a backbone introduces charges to the system, rendering the oligopyrene foldamer water-soluble. Folding processes and self-organization of oligopyrene is also favored under aqueous conditions.97
2.5. Biological relevance
Multichromophoric systems that incorporate the ribose-phosphodiester backbone have an intrinsic advantage of ease of conjugation to natural nucleic acids, as they can be added via DNA synthesizer or through enzymatic methods. In addition, their structural similarity to the natural DNA duplex brings about minimal distortion, and in many cases, the helical structure can still be attained. In one study, new biological behavior was discovered through the use of DNA-multichromophoric systems. Li and coworkers found that HMGA proteins are able to unfold and refold DNA foldamers containing pyrene units, reversing the effect of pH and salt content.98 DNA-multichromophores have also been shown to be taken up into living cells and organisms,99 thus allowing them to be used as biological reporters. The availability of numerous cell uptake reagents for DNA suggests the future application of many DNA multichromophore systems in biological applications.
2.6. A limitation of DNA: Expense of large scales
Although automated oligonucleotide synthesis is well developed, it is still a multistep chemical synthesis. A typical cycle to incorporate one nucleotide involves four steps -coupling, capping, oxidation and detritylation. Oligonucleotides can be chemically synthesized efficiently up to as long as 150 bases but synthesis of longer oligomers faces low overall yields and occasional chemical damage. Thus better synthesis efficiency is still required.100 Moreover, the natural DNA components are relatively costly, and unnatural ones are yet more expensive. Solid-phase automated DNA synthesis is adequate for small laboratory scales in the micromole range, but large-scale production can incur a much higher cost.101 Thus although polyfluorophores in DNA have useful optical properties, the cost may be a barrier to large-scale industrial applications. As a result, most practical applications in the near term will be limited to small-scale applications and devices that use small quantities of these molecules and assemblies.
3. Varied Molecular Classes of DNA Multichromophores
A large preceding literature in DNA modification has enabled experimenters to assemble chromophores by multiple approaches. Chromophores can be introduced to the DNA through a non-nucleosidic approach, where the deoxyribose or ribose backbone is completely replaced and a linker is used to add the chromophore within a DNA duplex. Some research groups have also chosen to retain the nucleosidic sugar but replace the base with a chromophore of interest. This allows common sugar puckering and helical nature of the duplex to be retained, as long as the chromophores are relatively flat aromatic species, similar to the DNA natural bases. Besides base-replacement strategies, base modification has been a popular strategy for incorporating multiple chromophores. Chromophores are added as side chains to various positions of the base, designed to protrude into either the minor groove or the major groove of helical duplex DNA. Finally, non-covalent attachment of chromophores to duplex DNA has also generated a good deal of interest in the assembly of groove binding and intercalating dyes.
3.1. Chromophores replacing nucleosides
The five-membered ring of deoxyribose plays an important role in the three-dimensional structure of duplex DNA, conferring rigidity and chirality to the chain. On the other hand, it also adds structural complexity and multiple functional groups that must be addressed in the design and synthesis of modified DNAs. One advantage of introducing chromophores in the DNA duplex through a non-nucleosidic system is that alternative synthetic routes can be explored. Whereas typical nucleosidic chemistry can involve poorly selective, low-yielding glycosidation steps to form C-nucleosides or N-nucleosides, acyclic building blocks can potentially shorten and simplify synthetic routes by forming diastereomerically pure products.102 The use of sugar linkages also poses a concern for the stability of nucleosides in acidic conditions as depurination of natural nucleobases could occur. Thus, designed nonnucleoside linkers can in some cases improve the ease of synthesis and the stability of modified nucleotides.103 Flexible linkers may also allow aromatic chromophores to interact in the most favorable manner and optimize stacking interactions.
An important consideration when developing new acyclic scaffolds is to keep structural perturbations of the natural architecture to a minimum, in order to retain the helical and recognition properties of the duplex and its stability. Research groups using this method of attaching chromophores have carried out extensive studies on the helicity of the modified duplex. The lack of a continuous, pre-oriented sugar-phosphate backbone can in some cases be compensated for by other factors such as stacking interactions. Examples of non-nucleosidic chromophores are shown in Fig. 1.
Figure 1.

Examples of non-nucleosidic linked chromophores incorporated into DNAs. (A) Lewis’ carboxamide-linked pyrene; (B) Häner’s carboxamide-linked pyrene; (C) Asanuma’s threoninol linked pyrene; (D) Wagenknecht’s (S)-aminopropan-2,3-diol linked perylene bisimide; (E) Pederson’s glycerol linked pyrene; (F) Li’s tetraethylene glycol-linked perylene diimide.
3.1.1. The carboxamide linker
A simple carboxamide linker was first used by Letsinger and Wu to incorporate a non-nucleosidic stilbene-4,4′-dicarboxamide residue into an oligonucleotide strand.104 Upon hybridization of two complementary stilbene-labeled oligonucleotides, a characteristic excimer emission at 445 nm was observed and was reversible upon dissociation and association of the duplex. The stilbene dicarboxamide residue was found to promote base stacking and could be used as a stabilizing cap for double- and triple-stranded oligonucleotides.105 This stabilizing effect was harnessed by Lewis and coworkers to build hairpin structures for the study of DNA electron transfer.106 Using the same carboxamide linker, naphthalene-2,6-dicarboxamide,107 diphenylacetylene-4,4′-dicarboxamide,108, 109 phenanthrene-2,7-dicarboxamide,110 5,8-naphthalene diimide,110 pyrenedicarboxamide111 (Fig. 1A), perylenediimide112, 113 and perylenedicarboxamide114, 115 were also incorporated into hairpin oligonucleotides and used to study the distance-dependent photoinduced electron-transfer dynamics in duplex DNA.
Using a similar carboxamide linker, Häner and coworkers assembled a series of phenanthrenes,116, 117 phenanthrolines,118 pyrenes119 (Fig. 1B) and tetrathiafulvalene120 as non-nucleosidic base surrogates. When incorporated into DNA duplexes, phenanthroline derivatives were found to form more stable duplexes than phenanthrene analogs and propyl, butyl and pentyl linkers provided similar stabilizing effects.118 One to seven phenanthrenes were incorporated into a 21-mer oligonucleotide. Interestingly, only a small reduction in thermal stability (decrease of 8.3 °C) was experienced even when seven phenanthrenes replaced base pairs within the duplex. An interstrand stacking arrangement of the phenanthrene residues was predicted from the lengthening of the oligonucleotides as more phenanthrenes replacements were made (Fig. 2A).117 The stabilizing effect of the stacking interactions between phenanthrenes and pyrenes was also used in the formation of intramolecular triplex helices. Tm was increased by up to 15 °C, and the structure could be monitored by observing the excimer fluorescence signal of pyrene as well as exciton coupling in the CD spectrum.121
Figure 2.

Häner’s carboxamide-linked phenanthrenes (A) and pyrenes (B). (A) The molecular model of 14 phenanthrenes incorporated into a 21-mer oligonucleotide shows an extended structure, compared to the unmodified DNA, illustrating the interstrand stacking arrangement of the phenenthrenes. (B) A right-handed helical structure was observed when more than 12 pyrenes were stacked together. This chirality in the oligopyrene unit was induced by the neighboring DNA and is more pronounced when DNA is only on one end of the oligopyrene compared to being on both. Adapted with permission from refs 97 and 117. Copyright 2005 and 2009 Wiley-VCH Verlag GmbH & Co. KGaA.
In another study, two to seven pyrenes were incorporated into single oligonucleotide strands and hybridization of two such strands resulted in up to 14 pyrene residues stacked within a DNA duplex. Temperature-dependent UV/Vis spectroscopic studies showed two isosbestic points upon melting of the duplex, indicating the presence of both intra- and inter-strand stacking, which was corroborated further by observation of excimers and broadened absorption spectra.122 Interestingly, when more than ten pyrene units were present in the duplex, the oligopyrene stack adopted a well-defined right-handed helical conformation as confirmed by exciton-coupled CD experiments (Fig. 2B).123 This phenomenon was not observed in the single strands nor when less pyrene units were present. The intrinsic helical nature of the oligopyrene was exemplified when oligomers of pyrene carboxamides built with the achiral phosphodiester linkage and a single chiral 1,2-diaminocyclohexane unit formed stable helical hybrids in aqueous solution.124 Remarkably, a chiral pyrene oligomer containing a terminal 2′-deoxycytidine group was able to induce the formation of homochiral supramolecular helical polymers of achiral pyrene oligomers in random coils, displaying the organizational effects of intermolecular aromatic stacking and hydrophobic interactions.125
3.1.2. The threoninol linker
Using a chiral threoninol linker, Asanuma, Komiyama and coworkers incorporated a variety a chromophores into the DNA duplex, including azobenzene,126 methyl red,126 naphthyl red,126 pyrene (Fig. 1C), N,N-dimethylaniline,127 4′-methylthioazobenzene,128 4′-dimethylamino-2-nitroazobenzene,128 p-methylstilbazole129 and perylene.130 Melting temperatures remained similar to that of natural DNA and were elevated with multiple incorporations of methyl red chromophores.131 Sequential incorporation of methyl red chromophores without spacer groups between them resulted in H* aggregation as shown by a large hypsochromic shift (blue shift) – typical of H aggregates - and in addition, narrowing of the absorption band compared to that of the monomer (an observation that defines the highly ordered “H*” classification).132 Circular dichroism studies revealed an intense exciton coupled CD signal (in the absorption region of the methyl red aggregates), which increased with the number of chromophore residues. Interestingly, upon hybridization, the H* aggregate was transformed to a less highly ordered H-aggregate and this could be reversed upon dissociation when temperature was increased. Increased salt concentration promoted H* aggregation, demonstrating that hydrophobic stacking interaction between the dyes is the main driving force for aggregation. Stacking interactions between methyl red incorporations was further confirmed by NMR studies.131
Heteroaggregates where methyl red and naphthyl red units stacked alternately were prepared by the hybridization of a methyl red-DNA conjugate with a naphthyl red DNA complementary strand.126 Up to six alternating dyes were incorporated in a controlled and predetermined manner. When spacer groups were added between chromophores, both positive and negative CDs were strongly induced by hybridization, indicating the formation of a right-handed helix, closely resembling natural B-DNA (Fig. 3A). Conversely, when no spacer groups were used, no induced CD was observed, indicating that part of the helix was unwound in forming a strongly stacked structure (Fig. 3B). It was hypothesized that the flexibility of the threoninol scaffold allowed this structure to be formed without losing stability as shown by the higher melting temperature as the number of chromophore increased.133 Further studies on heterodimerization of the dyes were carried out with azobenzene derivatives such as 4′-methylthioazobenzene and 4′-dimethylamino-2-nitroazobenzene.128 When the difference in absorbance maxima of the dyes was smaller, the absorbance changes in the duplex form intensified.128 The incorporation of perylene and pyrene into fluorophore assemblies of dimers, trimers and hexamers on the D-threoninol scaffold formed multichromophoric dyes with a high emission intensity, large Stokes’ shifts and shifted emission maxima, mimicking properties of inorganic quantum dots.130
Figure 3.

Methyl red chromophores, M, incorporated into oligonucleotides using a threoninol linker showed a strong positive and negative CD when they were interlaced with spacer groups (A). This indicated that the chromophore stack was able to adopt a right-handed helical structure. Consecutive methyl red chromophores without spacer groups (B) were strongly stacked with one another, exhibiting strong excitonic interactions, as shown by the larger hypsochromic shift and narrower band in the absorption spectra. However, the helical structure was unwound and the CD spectra showed much weaker signals. Reproduced by permission of The Royal Society of Chemistry from ref 133.
3.1.3. The (S)-aminopropan-2,3-diol linker
The (S)-aminopropan-2,3-diol linker was adopted by Wagenknecht and coworkers to tether perylene-3,4:9,10-tetracarboxylic acid bisimides in DNA (Fig. 1D).134 This linker does not have the methyl group of the above threoninol linker, but has a similar stereochemistry to the 3′-position of 2′-deoxyribofuranosides. Up to six perylene bisimide dyes were incorporated in a DNA duplex, with three chromophores in each of the complementary strands, separated either by thymine or by an abasic spacer. Some thermal stability was lost when six perylene bisimides were incorporated, and Tm decreased by 4°C.135 Hydrophobic stacking and strong π-π excitonic interactions were observed by absorption and CD spectroscopy. Interestingly, CD spectroscopy showed that a left-handed helical organization of the dye was achieved when thymine was placed opposite to the dyes, while a right-handed helix was formed when they were abasic sites. Excimer emission at 660 nm was observed upon interstrand hybridization. Thymine residues between perylene bisimides were able to prevent self-quenching between the chromophores. Stacking of perylene bisimides tethered at the 5′ termini formed one-dimensional aggregates of DNA duplexes.136 Interestingly, when an intrastrand perylene bisimide dimer was used as a clamp around a base of interest, excimer formation between intrastrand perylene bisimides was only observed when mismatches occurred opposite this intervening base between the dyes. No excimer was formed in a completely matched duplex. Thus, this could be used for single nucleotide polymorphism detection.
The (S)-aminopropan-2,3-diol linker has also been used to site-selectively incorporate an intercalating phenanthridinium chromophore into a DNA duplex.137, 138 This ethidium residue has been used as a charge donor in the study of electron transfer in DNA139 and was also used to detect single base mismatches and abasic sites.140 Thiazole orange,141, 142 phenothiazine143 and indoles102 have also been incorporated using the same linker system.
3.1.4. The glycerol linker
Pedersen and coworkers used a glycerol linker to incorporate pyrene (Fig. 1E) into oligonucleotides, forming intercalating nucleic acids (INA).144, 145 The pyrene moiety was able to stack efficiently and increased the thermal stability of duplexes formed with complementary ssDNA. Interestingly, hybridization with ssRNA was destabilized. Additional incorporations of the acyclic pyrene modification further increased the difference in melting temperatures between INA/DNA and INA/RNA duplexes, up to 25.8°C higher for INA/DNA duplexes. In a further iteration of INA, twisted intercalating nucleic acids (TINA) were designed.146 These contain a phenylmethyl glycerol linker conjugated with an ethynylpyrene chromophore. para-TINA derivatives had greater stabilizing effects for Hoogsteen type triplexes and duplexes while ortho-TINA derivatives stabilized both Hoogsteen and Watson-Crick type complexes.
3.1.5. The tetraethylene glycol linker
Perylene tetracarboxylic diimides (PDI) were conjugated by Li and coworkers to oligodeoxynucleotides through a tetraethylene glycol linker (Fig. 1F).147 Dimers, trimers, tetramers and pentamers of perylene diimide were assembled into foldamers (Fig. 4). Hydrophobic effects governed the formation of loosely folded nanostructures, which upon heating became increasingly ordered in an aqueous medium. The addition of complementary DNA strands resulted in unfolding of the thermophilic foldamers.
Figure 4.

PDI pentamers linked to DNA using a tetraethylene glycol linker formed a π-stacked foldamer surrounded by DNA hairpins. The structure is thermophilic and can be unfolded upon DNA hybridization. Reprinted with permission from ref 147. Copyright 2003 American Chemical Society.
3.1.6 The triazolyl linker
To build a π-extended system with the chromophores, a bistriazolyl derivative linker was developed.148 Bistriazolyl linkers have been used previously as isosteres of the carboxamide linker described in Section 3.1.1, but they have a more rigid structure. Thus, a single pair of 1,6-bistriazolyl pyrenes adopt a well-defined helical arrangement as shown by exciton coupling in the CD spectra, which has not been observed in a single carboxamide linked pyrene pair.
3.1.7 The alkynyl linker
The alkynyl linker has been used to incorporate pyrenes149 and fluorenes150 into the DNA double helix. CD measurements provided evidence for the self-organization of dialkynylpyrenes into well-defined helical structure within the DNA duplex.149 The use of DNA to assemble fluorenes enabled the formation of fluorene excimers, as indicated by a broad structureless band around 400 nm in the emission spectra.
3.2. Chromophores replacing DNA bases
Many of the chromophores used in multichromophoric systems are flat aromatic molecules. In order to incorporate them into the DNA π-stack with the least amount of perturbation, some research groups have opted to replace nucleosidic bases with chromophores, retaining the natural deoxyribose scaffold. This topic has been independently reviewed recently.151 Chromophores are typically synthetically conjugated to the anomeric position of deoxyribose (Fig. 5), which is then derivatized for automated DNA synthesis. In many (but not all) cases, Watson-Crick hydrogen bonding is disrupted in these nucleosides. However, chromophores with extended π-systems generally increase hybrid stability by favoring stacking interactions.
Figure 5.

Examples of chromophores replacing nucleobases. (A) Kool’s β-pyrene nucleoside; (B) Leumann’s biphenyl nucleoside; (C) Inouye’s alkynyl pyrene nucleoside; (D) Seitz’s 1,1′-binaphthyl nucleoside; (E) Tor’s 2,2′-bipyridine nucleoside; (F) Asseline’s propyl-linked perylene nucleoside.
The first examples of simple aromatic hydrocarbon chromophores as nucleobase replacements involved naphthalene, phenanthrene and pyrene moieties substituted as deoxyribose glycosides.152 In some of the earliest work on multichromophore DNAs, Kool and coworkers studied a pyrene deoxyriboside (Fig. 5A) in duplex DNAs as a base replacement, paired opposite abasic residues. Such base pairs were found to be either slightly destabilizing or somewhat stabilizing to the DNA, depending on numbers of substitution. Duplexes composed of up to 20% of pyrene base pairs were described.85
Building further on this concept, Kool and coworkers have developed a system of base replacement chromophores, called oligodeoxyfluorosides (ODFs). Aromatic fluorophores such as pyrene (Fig. 5A), perylene, benzopyrene, terphenyl, terthiophene, dimethylaminostilbene, binaphthyl, ternaphthyl, quinacridone and others have been attached at anomeric position of deoxyribose, forming monomeric deoxyribosides (termed “fluorosides”).96, 152 These monomers were derivatized to their respective phosphoramidites and automated DNA synthesis was used to synthesize the oligomeric ODFs. Although they are single-stranded, absorption data suggested that they remain stacked, leading to ground state and excited state electronic interactions that vary with sequence.99, 153
Studies have shown that ODF dyes can exhibit properties not observed in common organic fluorophores. Large Stokes shifts of more than 200 nm have been observed (Fig. 6)153 and numerous excimers and exciplexes have been characterized. Multiple emission colors of ODF dyes, which can be excited at a single excitation wavelength, have been described. Combinatorial libraries of tetrameric ODFs have been synthesized to provide ease in the identification of sequences of ODFs with desired properties such as tuned emission or sensing of light exposure.96, 154 Highly efficient quenching and unusual sensing properties have also been reported in ODFs.155, 156 These multichromophores have also been substituted at the ends of duplex DNAs, where quenching and FRET donor properties have been observed.157, 158
Figure 6.

Fluorescence spectra of a series short single-stranded oligomers containing one to five pyrene nucleosides (Y). The formation of excimers when two or more Ys are present is apparent fomr the broad emission at 490 nm. Reprinted with permission from ref 153. Copyright 2005 American Chemical Society.
In a related approach, bipyridyl, biphenyl (Fig. 5B) and their derivatives159 were employed by Leumann as base substitutes for C-nucleosides as part of a zipper-like interstrand stacking recognition motif in duplex DNA.160, 161 Up to seven consecutive biphenyl and bipyridyl pairs were incorporated into the DNA duplex.162 The affinity of planar aromatic bipyridyl base pairs was found to be higher than that of an A-T base pair and comparable to that of a G-C base pair in natural DNA. Hetero-polymers of these C-nucleosides also exhibited stable interstrand stacking interactions. NMR and CD data provided evidence for a highly ordered, stacking arrangement of the chromophore pairs within the DNA double helix. Desolvation effects are likely to play an important role in the enhanced duplex stability as shown by the higher thermodynamic stability of pentafluorobiphenyl base pair incorporations compared to biphenyl base pairs.163 This thermodynamic stability was traced to entropic factors through isothermal titration calorimetry (ITC) experiments, and was compatible with a model that stacking of the hydrophobic base pairs is dominated by dehydration of the aromatic units upon duplex formation. Solution NMR structure studies revealed stacking interactions of biphenyl base pairs in a head to tail fashion in van der Waals contact with each other in a decamer duplex containing one biaryl pair 3”,5”-dinitrobiphenyl and 3”,4”-dimethoxybiphenyl C-nucleosides (Fig. 7).164 Phenanthrenyl deoxyribonucleosides were modified with a donor (NH2) and an acceptor (NO2) and were incorporated into oligodeoxynucleotides.165 The phenanthrenyl derivatives were found to form stable interstrand zipper-like stacking motifs within DNA. Thermal stability of duplexes varied depending on the type of substituent. The nitro-substituted derivative was an efficient quencher of the fluorescence of the others, indicating the presence of electron- or energy-transfer processes and electronic coupling between bases on opposite strands.
Figure 7.

Biphenyl rings stacked head to tail and with their neighboring base pairs as viewed perpendicular to the mean base pair plane (left) and in a representative low energy structure calculated from the NMR solution structure data of the B-DNA duplex. Reprinted with permission from ref 164.
Alkynyl C-nucleosides with pyrene (Fig. 5C), perylene and anthracene as base substitutes were synthesized by Inouye and coworkers.166 The interactions of these aromatic residues, fixed rigidly to the DNA scaffold via an alkyne bond, resulted in excimer and exciplex formation. Heinke and Seitz reported the use of a non-planar 1,1′-binaphthyl aromatic molecule as a base surrogate (Fig. 5D).167 The two naphthyl rings were nearly orthogonal but the structure was torsionally flexible due to rotation. Incorporation of one binaphthyl decreased duplex stability by 6-9°C. However, successive incorporations of binaphthyl nucleosides increased duplex stability. Modified duplexes also exhibited enhanced fluorescence properties upon multiple incorporations of binaphthyl residues. Weizmann and Tor replaced natural bases with metal-binding 2,2′-bipyridine (Fig. 5E), forming “ligandosides” built on the D-deoxyribose-phosphate backbone. Upon titration with various metal ions such as Cu+, Pd2+ and Ag+, new long wavelength bands were observed in the absorption spectra due to metal to ligand charge transfer (MLCT). Double stranded structures could be formed from oligomers with multiple incorporations of ligandosides.168, 169 Aubert and Asseline prepared perylene conjugates where perylene is covalently linked either directly to the anomeric position of 2′-deoxyribose or via a propyl linker. Perylene-modified nucleosides with the propyl linker (Fig. 5F) induced a greater stabilization effect. Having two incorporations of the perylene nucleoside was also more stabilizing for triplex and duplex structures.170
3.3. Chromophores as sidechains of DNA
Fluorophores have been commonly conjugated to DNA nucleosides as sidechains to natural bases. One important advantage of this mode of attachment is that classical Watson-Crick base pairing can be maintained. Chromophores can be attached generically to the 5-position of pyrimidines, the 7-position of 7-deazapurines or the 8-position of purines. Modifications at the first two of these positions generally do not affect the normal anti conformation of the corresponding nucleobase.171, 172 Purines modified at the 8-position are generally in the syn conformation, and thus Watson-Crick base pairing is adversely affected.173,174
Attachment of chromophores on the nucleobases is usually made through short phenylene or acetylene bridges or via a simple C-C bond (Fig. 8). This causes strong electronic coupling between chromophores and the nucleobases. Direct attachment of a chromophore and DNA base can cause steric hindrance, which an acetylene bridge can avoid. Sonogashira coupling is commonly used to attach ethynyl-substituted aromatics to iodo-modified nucleobases. Conjugation of multiple chromophores has also been carried out at the phosphodiester linkage of DNA. McLaughlin and coworkers explored multiple labeling of oligonucleotides with bimane fluorophores to enhance the detection limit for nucleic acids in the early 1990s (Fig. 8A).175
Figure 8.

Examples of chromophores linked as side chains of nucleosides. (A) Bimane conjugated to phosphorothioate ester of oligonucleotide; (B) 5-(1-pyrenyl)-2′-deoxyuridine; (C) 5-(1-ethylnylpyrenyl)-2′-deoxyadenosine; (D) 5-(tetraphenylporphyrin)-2′-deoxyuridine; (E) 3-[2-[(1-pyrenylacetyl)amino]ethyl]-2′-deoxythymidine; (F) 3′-deoxy-3′-[(trispyridylphenylporphyrin)benzoyl]amino-2′deoxythymidine.
In work by Wagenknecht and co-workers, clusters of aromatic chromophores have been covalently attached to the 5-position of uridine through a C-C bond, resulting in electronic coupling between uracil and the chromophore. Chromophores that have been used include pyrene (PydU; Fig. 8B), 1-ethynylpyrene, phenothiazine and Nile red.176 Exciplex formation was observed between uracil and pyrene, with spectra exhibiting an intense structureless red-shifted fluorescence emission band at 475 nm. Multiple incorporations of these dyes (up to five adjacent PydU moieties) formed a right-handed helical π -array along the major groove of the DNA duplex.177 The CD spectra of the duplex with five incorporations of PydU (shown in Figure 9) displayed a significant exciton-coupled CD signal in the absorption range of the PydU chromophore (305-425 nm) with a positive Cotton effect, characteristic of a right-handed helical conformation. Upon hybridization of such a modified strand to an unmodified complementary DNA, a 22-fold enhancement in fluorescence was observed.
Figure 9.

A significant exciton-coupled CD signal was observed in the pyrene chromophore region of the spectrum for DNA3a, where a sequence of five consecutive PydU were paired across adenines. This signal was not observed in DNA1, which is a single strand of five PydU incorporations. The introduction of one mismatched guanine in place of adenine in DNA3b was tolerated in the helix, but when the number of mismatches increased to two and five in DNA3c and DNA3d respectively, the CD signal was greatly decreased, indicating perturbation of the helical structure. Reprinted with permission from ref 177. Copyright 2006 Wiley-VCH Verlag GmbH & Co. KGaA.
Mismatches opposite the modified base sites resulted in a reduction of fluorescence intensity and a bathochromic shift in the emission maximum from 445 nm to 465 nm as the excitonic coupling between chromophores was disrupted. CD data also showed that at least 3 units of 1-ethynylpyrenes were required before a helical, π-stacked ordered structure was formed within the duplex.178 Alternating pyrene and phenothiazine chromophores linked to the 5-position of uracil also formed regular helical arrays along the major groove of the DNA duplex upon hybridization with an unmodified DNA strand.179
Employing a different base for attachment, Kim and coworkers conjugated ethynylpyrene to the C8-position of deoxyadenosine (APy; Fig. 8C). Excimers were formed upon duplex formation, indicating strong interstrand stacking interactions between the pyrenes.180 The change in fluorescence upon stacking of these modified bases was used in the design of quencher-free molecular beacons.181 Interestingly, when two APy residues were incorporated into an oligodeoxyadenylate strand, and placed 2 adenines apart from each other, a red-shifted fluorescence emission at 580 nm was observed (Fig. 10), with a long excited state lifetime of 42 ns.182 Experiments suggested that this band is a result of an intermolecular self-duplex formation, resulting in pyrenes interacting in the ground state through J-type exciton coupling.
Figure 10.

The incorporation of Kim’s ethynyl pyrene units at varying distances from each other in Strands A1 to A4 results in different fluorescence properties observed. When they are 2 nucleotides apart, a new emission band at 580 nm appeared. Adapted with permission from ref 182. Copyright 2007 American Chemical Society.
The Stulz group attached diphenylporphyrin (DPP)183 and tetraphenylporphyrin (TPP; Fig. 8D)184 to the 5-position of 2′-deoxyuridine through an acetylene linker. Up to 11 porphyrin-modified dU units were introduced into DNA, and molecular mechanics-based calculations suggested that single-stranded modified oligomers displayed helical stacking interactions between the chromophores as the minimum energy conformation (Fig. 11). When hybridized to an unmodified complementary DNA, or another modified strand to form a zipper-like heteropolymer,185 the porphyrin stack was assembled in the major groove of DNA, forming a stable right-handed helical array, as indicated by CD studies and further molecular mechanics simulations.
Figure 11.

Energy-minimized structures of eleven tetraphenylporphyrin substituted deoxyuridines (7D) incorporated into a duplex DNA (A) and single stranded DNA (B). Adapted with permission from ref 184. Copyright 2007 American Chemical Society.
One useful aspect of porphyrins as chromophores is that metal ions can be incorporated into the DNA duplex. Complementary strands of porphyrin-labeled DNA could be metalated separately to give mixed zinc(II)-metalated and free-base porphyrin arrays. Energy transfer between the zinc porphyrin and the free porphyrin resulted in quenching of the fluorescence of the zinc porphyrin.185
Ono and coworkers introduced pyrene at the 3-imino position of thymine through an amide bond (Fig. 8E). The pyrene-thymidine conjugate was then incorporated into oligonucleotides to form multi-pyrene arrays.186 Large increases in the pyrene excimer emission were observed as the number of pyrene incorporations increased from two to five in the single-stranded oligonucleotides. This excimeric emission was stable upon duplex formation, leaving the pyrene-thymidine units in the dangling end.
Proni and coworkers incorporated trispyridylphenyl porphyrin at the 3′-position of deoxyribose through a rigid amide bond (Fig. 8F).187 This porphyrin-modified residue was incorporated onto both ends of a double stranded DNA. Exciton coupling between two porphyrins was observed by exciton-coupled circular dichroism (EC-CD) and could be used as sensitive CD sensors for detecting conformational changes of the intervening helical DNA.
3.4. Use of other nucleic acid structures to array chromophores
Besides DNA, multi-labeling of other nucleic acid architectures, including RNA and locked nucleic acid (LNA) have also been carried out (Fig. 12).
Figure 12.

Examples of chromophores linked to non-DNA nucleic acid architectures. (A) 2-(1-ethynylpyrenyl)-adenosine; (B) 2′-O-(1-pyrenylmethyl)-uridine; (C) pyrenemethyl ara-uridine-2′-carbamate; (D) 2′-N-(1-pyrenyl)carbonyl-2′-amino-LNA monomer.
3.4.1. Multi-labeling of RNA
Engels and coworkers introduced 1-ethynylpyrene to the 2-position of adenine (Fig. 12A).188, 189 A broad unstructured fluorescence emission band at 437 nm, distinctly different from the 1-ethnylpyrene emission (λmax at 385, 405 and 425 nm) was observed, likely due to electronic coupling between pyrene and the nucleobase. This was previously observed by Wagenknecht as well.190 Upon hybridization, two pyrene fluorophores on opposite strands interacted with each other in the minor groove of RNA, forming excimers with a fluorescence maximum at 480 nm.
Yamana and coworkers covalently attached pyrene to the 2′-position of ribose (Fig. 12B) and arrayed the chromophore consecutively in a RNA strand (Fig. 13).191 Upon duplex formation, hypsochromicity of the pyrene absorption band was observed, together with a strong enhancement in pyrene excimer fluorescence. The studies provided evidence for ground state interaction of pyrenes resulting in the formation of H-aggregates, with pyrenes oriented outside the duplex. When pyrenes were incorporated with an alternating base to both strands of RNA and hybridized, a “zipper” array of chromophores was formed in the minor groove of the RNA duplex as demonstrated by the induced CD signal and a positive Cotton effect.192
Figure 13.

Schematic representation of two RNA sequences containing five 2’-pyrene labeled uridines hybridizing and forming a “zipper array” (left). Induced CD measurements showed that pyrenes are arranged in the minor groove of RNA as shown on right. Reprinted with permission from ref 192.
Korshun and coworkers used a rigid carbamate-based linker to attach pyrene at the 2′ position of ribose (Fig. 12C). In contrast to previous modifications, this modification is expected to direct pyrene to the major groove of duplexes. Destabilization of duplexes was more pronounced with RNA than with DNA complementary strands. Excimer emission was observed when two pyrene modifications were placed one base pair apart.193
3.4.2. Multi-labeling of LNA
LNAs (locked nucleic acids) have a methylene bridge connecting the 2′ and 4′ carbon atoms of ribose, thus locking the ribose in a C3′-endo structural conformation, which is found in duplex RNA and A-form duplex DNA. LNAs and 2′-amino-LNAs are known to increase thermal stability of duplexes substantially. These properties are further enhanced with the incorporation of aromatic chromophores at the 2′-position. For example, pyrene (Fig. 12D),194 (phenylethynyl)pyrene,195 perylene196 and coronene197 have been used to functionalize 2′-amino-LNAs and upon multiple incorporations of the chromophores into an oligonucleotide, lead to highly stabilizing effects with both DNA and RNA complements. Hybridization of an oligonucleotide containing four units of pyrene-functionalized 2′-amino LNA to either RNA or DNA complements generated a 69-fold increase in fluorescence intensity.198 The large enhancement was hypothesized to be due to the locked ribose fixing the orientation of pyrene in the minor groove, avoiding quenching interactions with nucleobases (Fig. 14). When multiple pyrene-2′-amino-LNA units were flanked with an abasic site on its 3′-side and paired against another abasic site, a large stabilization of the duplex resulted.199 This formation of this stable helical structure was hypothesized to be due to the intercalation of the pyrene moiety with the natural DNA base pair within the duplex, thus filling the void left by the pair of abasic sites. A similar pyrene/abasic stabilization effect was observed by Kool with the natural DNA backbone.200
Figure 14.

Lowest-energy calculated models of pyrene-labeled LNAs show the pyrenes protruding into the minor groove of the duplex, thus restricting its motion and aiding the increase in fluorescence. Reprinted with permission from ref 198. Copyright 2005 American Chemical Society.
3.5. Noncovalent DNA-associated dyes
Noncovalent interactions - such as hydrogen bonding, electrostatic forces, van der Waals forces and hydrophobic effects - drive the formation of higher-order structures in nucleic acids, which are critical for their biological functions. Noncovalent interactions generally avoid the chemoselectivity issues in covalent bonding but are also considerably weaker and can be transient. In recent studies, DNA has served as an attractive template for the assembly of dyes, inducing the formation of stacked, helical multichromophore aggregates.
Intercalating dyes have been studied with DNA for over four decades. Using the concept of multi-intercalation, Armitage and coworkers designed branched DNA templates to array intercalating dyes, forming fluorescent DNA “nanotags” (Fig. 15).201 Dye assemblies on three-dimensional DNA tetrahedrons have also been formed.201, 202 Oxazole yellow dyes such as YOYO and YO-PRO-1 can intercalate as frequently as every other base pair, keeping dyes ca. 7 Å apart. This allows relatively high densities of fluorescent dyes to be achieved, while separating them enough to prevent self-quenching. Dye assemblies exhibited favorable light harvesting properties and were able to carry out FRET to covalently bonded acceptor dyes.
Figure 15.

Schematic representation of fluorescent DNA nanotags where the intercalating dyes are held in place by duplex DNA. Both linear and branched templates can be formed as shown in the figure. Reprinted with permission from ref 201. Copyright 2007 American Chemical Society.
In another study by Armitage, symmetrical cationic benzothiazole cyanine dyes were shown to spontaneously assemble in the minor groove of a DNA duplex, which acted as a template to form helical J-aggregates.203-205 Co-facial dimeric cyanine dyes in the form of H-aggregate dimers aligned cooperatively in the end-to-end dimension, leading to the propagation of an aggregate up to hundreds of angstroms in length along a poly(dA-dT) template.204 Changes in temperature and ionic strength resulted in the conversion of the J-aggregate into a H-aggregate. These conclusions were supported by observations of the hypsochromic shift of λmax from 648 nm to 590 nm characteristic of a H-aggregate and a long wavelength band at 756 nm, characteristic of a J-aggregate, in the absorption spectra, together with a pronounced excitonic band in the induced CD spectra.203 The formation of this templated aggregate is dependent on dye structure and concentration and DNA sequence and length.206 Similar dye aggregates have also been built on PNA-related duplexes.207, 208
Single-stranded DNA has been used as an effective template for the self-assembly of an oligomeric strand of chromophores. Meijer and Schenning showed that a 40-mer oligodeoxythymidine strand was able to form organized helical structures with naphthalene and π-conjugated oligo(p-phenylene)vinylene (OPVT) through hydrogen bonding of the thymine with a complementary diaminotriazine moiety on the chromophores. π-π interactions between chromophores and hydrophobic interactions further stabilize the right-handed helical structure, as shown by the positive Cotton effect in the CD spectra (Fig. 16).209-211 The G-quadruplex structure has also been harnessed to array OPV chromophores. OPV chromophores were attached to the N-8 position of guanines and in the presence of salt, these guanosines exhibited a high propensity to form π-stacked quartets. π-conjugated OPV units were π-stacked in a J-type arrangement, resulting in an enhancement of fluorescence.212
Figure 16.

A single-stranded oligodeoxythymidine (dTn) acts as a template to array naphthalenes equipped with a diaminotriazine unit (NT) through hydrogen bonding, forming a right-handed helical structure. Reprinted with permission from ref 210. Copyright 2007 American Chemical Society.
Right-handed helical stacks of the chromophore are formed when two strands of oligoadenylic acid sandwich a bis-thymidine-appended oligophenylenevinylene strand (OPV-dT). Complementary A-T base pairing induced the binary self-assembly of this helical stack and the diameter of the stack could be controlled by varying the stoichiometry of the thymine and adenine components.213 A bis-thymidylic acid appended anthracene dye underwent similar binary self-assembly with two strands of dA-oligonucleotide, forming a triple helical J-aggregate of chromophores.214
Water-soluble porphyrins have also been shown by Pasternack and Gibbs to form long-range organized assemblies spontaneously using DNA as a template.215-217 These trans-bis(N-methylpyridinium-4-yl)diphenylporphyrins intercalated with DNA at low ionic strength (I < 5 mM) but aggregated on the nucleic acid backbone with increasing ionic strength. Large induced circular dichroism signals were produced and strong electronic coupling enabled these porphyrins to behave as an antenna system.217 It was also shown that the extent of the supramolecular porphyrin assemblies can be controlled through competitive binding with a nonaggregating porphyrin, tetrakis(N-methylpyridinium-4-yl)porphyrin gold (III), which binds to DNA only through intercalation.218
3.6. Tri- and tetra-FRET systems on DNA
Labeling of DNA with fluorescent dyes has become highly accessible to researchers. Many fluorophores are commercially available and can be appended to oligonucleotides site-specifically using well-developed post-synthetic strategies. Most applications of fluorescence detection involve the singular labeling of one fluorophore and perhaps a quencher to an oligonucleotide strand, such as in a molecular beacon. In this section, the utility of attaching more than one fluorophore at one time to either a single stranded DNA or DNA duplex is explored. DNA serves as the scaffold for site-selective attachment of multiple fluorophores through chemical synthesis. The number and position of fluorophores is controlled, and arrays can be built with predictable geometry and position.
Common fluorescent dyes such as 6-carboxyfluorescein, TAMRA and Cy5 have been arrayed in different combinations on a single-stranded oligonucleotide probe sequence to construct combinatorial fluorescence energy transfer tags (CFETs).219, 220 Eight different CFET tags were constructed from three fluorescent dyes. Using 6-carboxyfluorescein as the common donor, all the tags could be excited at a single wavelength (488 nm). Distance between acceptors and donors in each tag was varied using 1′,2′-dideoxyribose phosphate spacers. Since FRET is distance dependent, energy transfer efficiencies between dyes could be tuned, and each of the CFET tags exhibited a different fluorescent emission signature. Cassettes of FRET tags have been used in the multiplex detection of single nucleotide polymorphisms221 and as sequencing primers.222, 223 Combinatorial FRET tags have also been used in molecular beacons.87
The DNA double helix has been used in many laboratories to array chromophores for FRET. Thus, programmable arrangements of these conjugates can be constructed with regulated distances and geometry.224, 225 Multi-step FRET and energy migration can thus occur over relatively long distances along the DNA double helix. Energy transfer through five fluorophores, employing intermediary fluorophore units to reduce energy loss through inefficient FRET between donor and acceptor units, has been described (Fig. 17).226 In a related approach, the intercalating chromophore YO has been used as an energy mediator for homo-transfer of energy to reduce energy loss through the red-shifted energy cascade in hetero FRET.227
Figure 17.

Schematic representation of a multi-chromophore FRET array constructed through hybridization of multiple labeled fragments to a longer matrix oligo-DNA, thus allowing multi-step FRET to occur. Reprinted with permission from ref 226. Copyright 2003 American Chemical Society.
3.6.1. DNA photonic wires
Using a similar concept of hybridizing short multiply-labeled oligonucleotides, DNA photonic wires were constructed.11, 228 Up to five different fluorescent dyes (such as Rhodamine Green, tetramethylrhodamine, Atto 590, LightCycler Red, and Atto 680) were arrayed on the DNA duplex, and multistep energy transfer was observed using single molecule spectroscopy (Fig. 18). Unidirectional excited state energy transfer through this energy cascade with a spectral range of over 200 nm was achieved by careful selection of chromophores. The “photonic wire” spanned about 13.6 nm, and single-molecule studies showed that an estimated 10% of the DNA photonic wires constructed exhibited a FRET efficiency of up to 90%.10 However, multiple emission pathways were seen due to unfavorable conformational orientation of the dyes, incomplete hybridization and quenching by adjacent nucleotides.229 Besides traditional small-molecule fluorophores, fluorescent proteins have also been used in forming an energy transfer pair using sequence specific DNA hybridization. Efficient energy transfer between EYFP-DNA conjugate and Atto647-labeled DNA has been observed.73, 230 More recently, quantum dot-sensitized multivalent photonic wires have been constructed.231 This combines the multi-fluorophore assembly on DNA with quantum dots as ultraviolet energy harvesting donors, to form an energy transfer cascade over distances more than 15 nm.
Figure 18.

(Left) DNA photonic wire containing five different fluorophores, held in position through hybridization of the fluorophore-labeled oligonucleotides to a longer complementary strand, and its bulk excitation spectrum (λcoll = 703 nm; dotted line) and fluorescence spectrum (λex = 468 nm; solid line). (Right) Single molecule fluorescence image of the DNA photonic wire spin-coated onto a glass surface acquired using confocal scanning optical microscopy. Adapted with permission from ref 230. Copyright 2006 American Chemical Society.
3.7. Size-expanded DNA (xDNA)
Size-expanded DNAs have benzo-homologated bases and retain canonical Watson-Crick base pairing that is more stable than that of natural DNA.232-234 Structures of xDNA exhibit right-handed helices with base pairs and grooves widened by ca. 2.4Å.233 Increased π-conjugation of the bases (dxA, dxG, dxC, dxT) shifts the absorption and emission spectra to longer (visible) wavelengths, and the bases exhibit quantum yields of 0.3 to 0.6. Multiple xDNA monomers have been appended to a single stranded oligonucleotide and display interesting fluorescence properties upon hybridization with the complement. Multiple dxA incorporations produced a featureless long wavelength band at 520 nm, indicating that excimers of xA chromophores were formed (Fig. 19).235
Figure 19.

Emission spectra of oligonucleotide strands containing one to four benzo-expanded adenine (xA) nucleotides in a single strand (λex = 333 nm), showing excimer emission (~520 nm). Adapted with permission from ref 236. Copyright 2008 American Chemical Society.
4. Methods of Assembling DNA Multichromophores
The versatility of DNA as a scaffold is also exemplified by the different methods that can be used to synthesize the DNA multichromophore assembly. Here, we describe the chief methods that have been employed with success.
4.1. Automated synthesis and post-synthetic labeling
Automated chemical synthesis of oligonucleotides on solid support with phosphoramidite monomers is one of the most common methods of making DNA multichromophoric systems.236 Modified nucleoside analogs, some containing deoxyribose and some not, are typically chemically synthesized and derivatized to the 5′-DMT-protected, 3′-phosphoramidite intermediate for automated DNA synthesis of oligonucleotides. Oligonucleotide synthesis with phosphoramidite coupling has been extensively studied and proceeds with high-yielding steps,100, 237 which allows a wide range of lengths to be accessible.
For multichromophore systems in which bases are replaced with the chromophores of interest, C-nucleoside monomers are common. C-analogs are more stable than the natural N-nucleosides towards enzymatic and acid catalyzed hydrolysis. C-nucleosides have been synthesized for various applications such as drug discovery, tools for chemical biology and fluorescent labeling of DNA238 and the various synthetic methods have been reviewed.239, 240 The chromophores studied are often aromatic hydrocarbon derivatives and have been synthesized using coupling chemistries such as organocadmium and organozinc chemistry,241 Friedel-Crafts alkylation,242 Heck coupling243 and cuprate glycosylation.244
For chromophores introduced as sidechains of DNA nucleosides, several positions have been shown to have favorable effects. In pyrimidines, position C5 has been shown to be optimal for incorporation of functional groups as it minimizes the perturbation of the double helical structure. In purines, both positions N7 (C7) and C8 are synthetically amenable for modification, although (as mentioned above) 8-position substitutions can alter base conformation.
Palladium-catalyzed cross-coupling chemistry such as Sonogashira, Heck and Suzuki-Miyaura coupling is typically used to introduce chromophore labels to 5-halo or 5-ethynyl pyridmidines and C8-halogenatived purines. Cross-coupling can be performed before solid phase synthesis, and then the modified nucleotide is used for oligonucleotide synthesis. Alternatively, they can also be carried out after synthesis of the oligonucleotide. In one example, ethynylpyrene-modified uracils were synthesized on the solid phase using Sonogashira-type coupling.245
Post-synthetic modification strategies are also valuable in labeling oligonucleotides, especially in cases where chemical incompatibility with DNA synthesis conditions precludes the use of the synthesizer for direct chromophore addition. Recent work in bioorthogonal methodologies such as Diel-Alder reaction, click chemistry and Staudinger ligation have been used to functionalize oligonucleotides.246-249 Typically, small chemical functional groups are introduced to the natural nucleotides, to be incorporated to oligonucleotides through solid-phase synthesis. Thereafter, chemoselective labeling with the desired chromophore is then carried out post-oligonucleotide synthesis. For example, using TMS and TIPS-protected alkynes, Carell and coworkers were able to post-synthetically modify DNA with up to three different labels.250
4.2. In situ enzymatic RNA synthesis
T7 RNA polymerase has been used by Tor and coworkers for the incorporation of fluorescent ribonucleotides into RNA oligonucleotides through in vitro transcription.251 A furan appended to the 5-position of pyrimidine was the basis for a fluorescent nucleoside analogue. Starting with commercially available 5-iodouridine, cross coupling with 2-(tributylstannyl)furan yielded the furan-conjugated uridine. Next, derivatization to the 5′ triphosphate yielded the substrate for transcription. Transcription proceeded efficiently, yielding a 224-fold amplification relative to the amount of DNA template used.252 The use of T7 RNA polymerase has also been extended to transcribe multiple incorporations of 5′-thiophene-uridine ribonucleotides.253
A fluorescent unnatural base pair system that is capable of undergoing both replication and transcription has also been designed by Hirao and coworkers. A strongly fluorescent base analog, 7-(2,2′-bithien-5-yl)-imidazo[4,5-b]pyridine was shown to be selectively incorporated into both DNA and RNA templates opposite pyrrole-2-carbaldehyde using Klenow fragment and T7 RNA polymerase respectively, allowing for site selective analysis of local structure of nucleic acids.254 Enzymatic incorporation of the modified ribonucleotide precludes the potentially complicated protecting group chemistry required for solid phase RNA synthesis and postlabeling.
4.3. DNA polymerase synthesis
As mentioned briefly in Section 2.2, Matray and Kool showed that the pyrene nucleotide was incorporated selectively opposite an abasic site in a template strand by the Klenow fragment of E. coli DNA polymerase I (Fig. 20). The efficiency of this incorporation was comparable to that of a natural base pair and selectivity for the abasic site was between 102 to104-fold.86 This study showed that neither hydrogen bonding nor canonical purine-pyrimidine shapes is necessary for enzymatic synthesis of a base pair by this enzyme, but that steric complementarity was an important factor.85 The work established that fluorescent base replacements (even those that do not have H-bonded pairing) can be substrates for polymerases.
Figure 20.
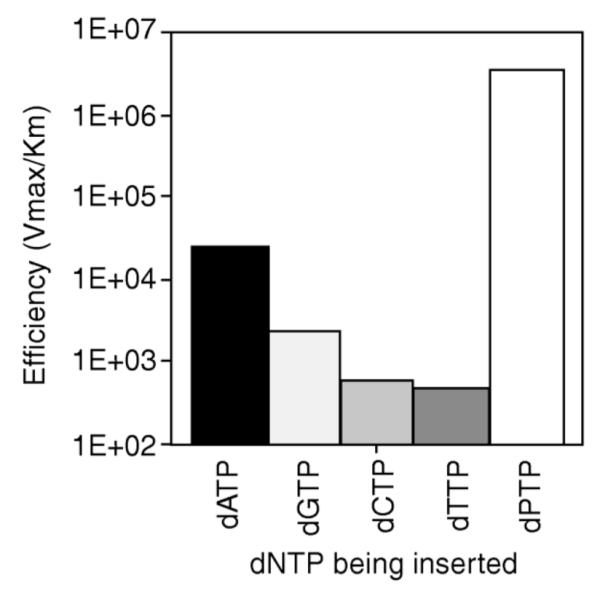
Fidelity experiments with β-pyrene nucleoside triphosphate (dPTP) showed that the Klenow fragment of E. coli DNA polymerase I preferentially inserted dPTP opposite an abasic site on the template strand, compared to the natural nucleotides. Reproduced with permission from Ref. 86. Copyright 1999 Nature Publishing Group.
Using chromophores directly labeled on the 5-position of uridines and 8-position of guanosines, Marx, Wagenknecht and coworkers tested their accommodation by DNA polymerases in primer extension studies. Klenow fragment, human DNA polymerase β (Pol β) and Dpo4 of the Y-family were able to insert the correct complementary A opposite a template containing pyrene and BODIPY-modified uridines. Pyrene-modified guanosine posed the most problems as Pol β was not able to insert any nucleotide opposite it and none of the polymerases tested can bypass PydG. All three polymerases were able to bypass modified uridines in the primer extension experiments, although enzymatic activities were lower.255
Large base pairs containing fluorescent benzo-expanded bases (xDNA and yDNA) were studied as potential polymerase substrates by Kool and coworkers. xDNA bases were found to be tolerated by Dpo4 polymerase and Klenow fragment polymerase I in primer extension studies. Extension of these large base pairs were much more efficient with Dpo4, which is known to be more flexible, compared to the more rigid Kf enzyme.256 yDNA, similar in size but with a different geometry than xDNA, was tested in PCR amplification and in cloning, albeit with limited success due to problems in mismatching.257 Interestingly, xDNA performed more efficiently, and up to four successive (and fluorescent) xDNA pairs could be formed by the Dpo4 enzyme.258 Moreover, in vivo experiments with E. coli also demonstrated that two of the size-expanded bases, xA and xC, can encode natural sequences correctly and efficiently.221
Since many of the chromophores in these multichromophoric systems are hydrophobic and sterically diverse, the evolution of a mutant polymerase that can carry out enzymatic synthesis may be helpful. Holliger and coworkers carried out directed evolution of a polymerase using compartmentalized self-replication (CSR) and were able to isolate a polymerase (5D4) that can extent 5-nitroindole and 5-nitroindole-3-carboxamide, with efficiencies similar or even exceeding those of Watson-Crick base pairs. 5D4 was also able to extend fluorescent pyrene-abasic pairs and isocarbostyril-azaindole pairs. PCR amplification was carried out with high fidelity.88
Highly modified DNA can be achieved through the combination of PCR with modified triphosphates, followed by further derivatization through a bioorthogonal reaction. Alkyne-modified nucleotide triphosphates were incorporated by family B polymerases Pwo, Deep Vent exo- and KOD XL. After incorporating the anchor alkyne groups, click chemistry could be performed on the amplicons at a high efficiency of 95%.259-261 In addition to this approach, it has recently become possible to incorporate a nucleoside triphosphate modified with an aldehyde functionality enzymatically into DNA through PEX and PCR. Pwo polymerase and KOD XL polymerase were shown to exhibit excellent tolerance to aldehyde-modified triphosphates and multiple incorporations were observed. The PCR product can then be treated with arylhydrazines for bioconjugation and staining purposes.262
4.4. Synthesis by terminal transferase and methyltransferase
Template-independent polymerases offer the possibility of accommodating sterically large fluorophores at the terminus of DNA. Cho and Kool demonstrated multiple incorporations of pyrene nucleotide using the terminal deoxynucleotide transferase (TdT) enzyme and the 5′-triphosphate derivative of the nucleoside. Both α- and β-anomers of pyrene nucleotide were shown to function as substrates for TdT enzyme and up to four incorporations were observed for the β-anomer and up to three incorporations were made of the α-anomer (Fig. 21). Strong pyrene excimer fluorescence was observed in the mulifluorophore oligomeric products.87 More recently, the TdT enzyme has also been used to incorporate benzo-expanded deoxynucleoside analogs, dxATP and dxCTP, forming fluorescent homopolymers.263
Figure 21.
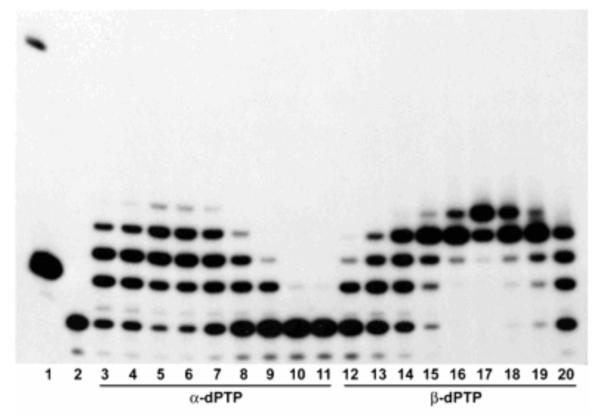
Polyacrylamide gel data showing multiple additions of pyrene nucleoside triphosphate at the end of a DNA primer. Lane 1: 10 bp size marker, lane 2: primer only, lanes 3-11: TdT reaction with increasing concentrations (2.5–640 μm) of α -dPTP, lanes 12-20: TdT reaction with increasing concentrations (2.5-640 μm) of β-dPTP. TdT was capable of incorporating up to three α-dPTPs or four β-dPTPs. Reprinted with permission from ref 87. Copyright 2006 Wiley-VCH Verlag GmbH & Co. KGaA.
The AdoMet method is another promising method for enzymatic labeling of DNA with various small molecules.264 DNA methyltransferases catalyze the transfer of an activated methyl group from S-adenosyl-L-methionine (SAM or AdoMet) to adenine or cytosine residues in a sequence specific manner. By modifying AdoMet with various functionalities, Weinhold and coworkers have been able to deliver fluorophores265 and functional groups such as aziridines,266 amines,267 allyls and alkynes268 into DNA, and functional groups can then be further derivatized with fluorophores and affinity probes.266
4.5. Synthesis of combinatorial libraries
In most cases, incorporation of multiple chromophores into DNA has been done via design, and have involved only one or two types of dyes. However, when several monomer components become available and different numbers and sequences are contemplated, the complexity increases geometrically. Moreover, it is often not possible to predict the electronic interactions between dyes given multiple forms of energy and excitation transfer that are possible. This complexity and unpredictability can be addressed by constructing large libraries of multichromophores and selecting for desired properties. This has been an approach taken by Kool with the aforementioned ODF polyfluorophores. Observing the ODF multichromophores on polystyrene beads under a fluorescence microscope provided a convenient method for screening. In this light, Gao and Kool reported the construction of a library of oligodeoxyfluorosides (ODFs) in which fluorescent base replacements were assembled in combinations on polyethylene glycol-polystyrene beads. A single-stranded tetrameric library of 256 members was built from 4 monomers.96 In a more complex example, 11 different monomeric deoxyfluorosides were used to build a tetrameric library, containing 14,641 sequence variants.154 These libraries were synthesized using the split-and-pool method, so that each bead contained only one type of tetrameric chromophore, and the sequences were encoded by molecular tags developed by Still.269, 270 More recently, 23 multispectral dye candidates were selected from a combinatorial library built from 8 monomers, yielding 4096 different tetramer ODFs (Fig. 22).99 These multicolor combinatorial library beads harbor significant potential in the development of multichromophore sensors for a wide range of species.
Figure 22.

(A) A representative image from a combinatorial library of tetrameric oligofluorosides (ODFs) synthesized from eight monomeric fluorosides on PEG-polystyrene beads. Image was acquired with an excitation filter at 340-380 nm, with long-pass emission filter >400 nm. (B) Structure of a typical tetrameric ODF. Reprinted with permission from ref 99. Copyright 2009 American Chemical Society.
As mentioned above (Section 3.6), combinatorial FRET tags can be synthesized from a small number of fluorescent dyes. Distances between dyes can be tuned to vary FRET efficiencies and thus the fluorescent signature.219, 220
5. Properties of DNA Multichromophores
5.1. New emission properties
Commercially available small-molecule fluorophores are widely used in fluorescence labeling experiments in the biological context. However, DNA multichromophoric systems have been able to address some of the limitations of these traditional organic labels. Such limitations include low photostability and limited light harvesting properties and brightness. In addition, there are difficulties in using traditional dyes in multiplexed experiments, since there is a requirement for a different excitation wavelength for each dye in the experiment.
Some of these limitations have been explored by the development of oligodeoxyfluorosides (ODFs) by Kool and coworkers. As described above, these are multichromophores built on a single-stranded DNA backbone, and prepared on a DNA synthesizer. The use of combinatorial library synthesis greatly increases the number of different fluorophores that can be synthesized and screened for a desired property, and libraries of >14000 members have been prepared to date.
Using this approach, a set of 23 different ODFs spanning the visible spectrum of emission, but using a single wavelength of excitation, was identified. The set of ODFs ranging from blue to green, yellow and red were constructed with just four different fluoroside monomers (Fig. 23).99 Such diverse emission properties were possible due to the interactions of monomers at close proximity on the DNA deoxyribose backbone. Multiple examples were given in which the tetramers exhibited emission properties not present in the monomers. Interestingly, examples of sequence-specific emission were noted, including anagrams, where different ordering of the same components yielded distinct emissions. Several of these multispectral ODFs were used to label antibodies for application in multispectral labeling of cellular antigens.271
Figure 23.

A set of 23 different ODFs spanning the visible spectrum using a single wavelength of excitation at 354 nm. Reprinted with permission from ref 99. Copyright 2009 American Chemical Society.
A recent interest in developing fluorophores with white light emission has stemmed from potential applications in white light emitting diodes (LEDs) and in liquid crystalline displays.31 Varghese and Wagenknecht used the DNA scaffold to array fluorophores ethynyl pyrene, a blue-green fluorophore, and ethynyl Nile Red, a red fluorophore, conjugated as side chains to the 5-position of 2′-deoxyuridine and incorporated into a duplex oligonucleotide. When both fluorophores were placed adjacent to each other, highly efficient excitation energy transfer occurred and white light emission was observed. Duplex formation was found to be critical in the interaction between the fluorophores as white light emission was only observed in the duplex state (Fig. 24).272
Figure 24.

White light emission observed from a duplex DNA doubly labeled with both ethynyl pyrene and Nile Red (dsDNA3; right vial image; λex=380 nm). Single stranded DNA3 (ssDNA3) which was also doubly-labeled yielded a red fluorescence as seen in middle vial image. DNA1 and DNA 2 are respectively labeled with either pyrene or Nile Red only. Reprinted with permission from ref 273. Copyright 2009 Wiley-VCH Verlag GmbH & Co. KGaA.
Other studies have shown that DNA-multichromophores can interact with small-molecule dyes via FRET interactions. FRET donor properties have been observed in ODF oligomers of Kool. ODFs were synthesized on the 3′ end of a DNA oligomer and hybridized to a complementary strand labeled with a commercially available acceptor fluorophore. Förster energy transfer was observed through steady-state emission measurements and time-resolved measurements, indicating energy transfer from excimers and exciplexes of ODFs to fluorophores such as TAMRA and Cy5.157 This suggests the use of DNA multichromophores as biophysical tools and as reporters of DNA hybridization.
5.2. Sensors
The unique fluorescence emission properties of multichromophoric systems have been put to good use through the development of sensing systems. The Häner and Nakatani groups developed a photoresponsive optical switch by using a photoswitchable ligand in conjunction with non-nucleosidic dipropynyl pyrene probes.273 Upon irradiation with light at 360 nm, the cis structure of the ligand was induced, resulting in the hybridization of two pyrene-labeled strands. Interstrand stacking of the two pyrenes resulted in excimer formation and a green fluorescence at 520 nm was observed. With irradiation at the UV wavelength, 430 nm, the trans isomer of the ligand prevented hybridization of the pyrene probes. Thus, only the monomer emission of pyrene at 430 nm was observed.
The efficient quenching observed in some DNA-multichromophore structures can be used to develop sensitive reporters of enzyme activity. In a proof-of-principle study, dabcyl quencher was conjugated to oligodeoxyfluorosides through an ester linker to form a quenched esterase sensor. Large fluorescence turn-on signals of more than 60-fold were observed in the presence of esterase activities both in vitro and in live human cells.274 A related ODF approach was used to develop three differently colored DNA-polychromophoric sensors of protease activity. These were initially dark due to an appended quencher, and yielded fluorogenic signals with different colors denoting specific caspase enzymes.275
Besides biomolecular detection, Kool and coworkers have also shown that multichromophores built on the DNA backbone can act as sensors of small molecules in the vapor phase, yielding varied responses to different analytes. The ease of building combinatorial libraries of ODFs provided a rapid way to screen for effective sensor compounds. When applied to vapor analytes such as acrolein, nitrobenzene or methyl iodide, a variety of responses, such as quenching, red shifts and blue shifts in fluorescence, unique to each analyte, could be detected.276, 277 Recently this strategy was applied to sensing of vapor-phase metabolites above bacteria cultures, allowing the discrimination of three types of pathogenic bacteria.278
By incorporating aromatic metal ligands as DNA base replacements into combinatorial libraries of oligodeoxyfluorosides, Kool and coworkers have also discovered fluorescent sensors for different metal ions. A wide variety of fluorescence responses were afforded by the combinatorial libraries of oligodeoxyfluorosides and the use of solid-phase combinatorial library screening allowed for responses toward various metal ions to be tested efficiently. In one example, a light-up sensor that responds selectively to Ag+ was found.279 In a separate study, a set of eight fluorescence-quenching metal ions (Co2+, Ni2+, Cu2+, Hg2+, Pb2+, Ag+, Cr3+ and Fe3+) could be distinguished based on a the responses of as few as two fluorescent ODFs.280
5.3. Fluorescence changes upon hybridization
One of the most useful aspects of fluorescence is its sensitivity to environment. Fluorophores that respond to environmental differences have been very useful for detecting and imaging many biological processes such as DNA hybridization, conformation changes in nucleic acids, nucleic acid-protein interactions and single nucleotide polymorphisms (SNPs). SNPs, in particular, have biomedical importance due to their connection with many genetic diseases.281-283 The ability to detect SNPs can be important in early disease detection, but requires probes that are very sensitive to a single mismatched nucleotide.
DNA multichromophoric systems have been used in solution-based fluorescence DNA detection assays. The use of color changes upon interaction of multiple fluorophores is particularly attractive in the development of probes. In particular, the fluorescence emission change upon the formation of pyrene excimers have been used by many different groups for reporting on DNA hybridization,284, 285 SNPs286, 287 and G-quadruplex formation.288 Indeed, the color change upon excimer formation of pyrene has been used widely as a tool for detecting biomolecular changes.284, 289-291 Pyrene nucleosides in which the chromophores replaced the nucleobases were used in a dual probe system by Kool and coworkers. The adjacent hybridization of singly labeled pyrene probes to a complementary oligonucleotide brings two pyrene fluorophores to close proximity and favorable geometry, resulting in excimer formation. In the experiments, monomer emission at 398 nm was suppressed while the excimer emission at 489 nm was enhanced by three-fold. This was used to detect a C → A point mutation found in the H-ras oncogene.292
Saito and coworkers used a doubly labeled pyrene probe to detect an insertion polymorphism (Fig. 25). Excimer fluorescence was 13 times higher than hybridization with the wild-type sequence. This fluorescence change was used to detect a G insertion after the 256th nucleotide of exon 13 in the βEnaC gene encoding for he epithelial sodium channel β-subunit associated with Liddle’s syndrome, an autosomal dominant form of hypertension with variable clinical expression.293
Figure 25.

Schematic diagram of Saito’s doubly-labeled pyrene probe, capable of detecting an insertion polymorphism. Reprinted with permission from ref 294. Copyright 2004 American Chemical Society.
A deletion polymorphism was detected using pyrene attached to a threoninol backbone synthesized by Kashida et. al. (Fig. 26). Two of these pyrene moieties were tethered on both sides of the nucleotide of interest in an oligonucleotide. Upon hybridization with wild-type DNA, excimer formation between the two pyrenes was hindered by the fully complementary base pair. However, in the presence of a deletion, a three base bulge occurred, bringing two pyrene moieties close together, yielding excimer formation. A two-base deletion was also identified using the same principle.286 Perylene tethered via threoninol was also used for the detection of single-base deletion mutations.294
Figure 26.

Schematic diagram of using threoninol-linked pyrene probes to detect deletion mutations. Reproduced by permission of The Royal Society of Chemistry from ref 287.
Pyrene excimers were also used to identify mismatch SNPs. Pyrene-functionalized 2′-amino-LNA monomers were incorporated into a dual probe system. Upon hybridization with a fully complementary DNA, excimer fluorescence was observed. Single mismatches resulted in low excimer emission due to decreased thermal stability and helix geometry changes.287
Pyrene chromophores linked to oligonucleotides via a flexible alkyl linker were used in mRNA detection in a pyrene binary probe design. Sensorin mRNA was detected through the excimer formation of two pyrene labeled probes. The use of time-resolved measurements increased the signal to background ratio of detecting mRNA in a cell extract.295
The incorporation of multichromophoric systems in molecular beacons296-299 has increased the versatility of these DNA hybridization probes. Classical beacons require both a fluorophore and a quencher to be labeled on DNA. In quencher-free molecular beacons using pyrene excimers, pyrene-labeled nucleosides are incorporated into the hairpin stem so that in the hairpin conformation, green fluorescence is observed. Upon duplex formation with the complementary target strand, the pyrenes are further apart and a blue fluorescence results.181 In another example of a wavelength shifting molecular beacon, non-nucleosidic thiazole orange and thiazole red chromophores were incorporated into the hairpin stem. Upon hybridization of the target strand, a large 140 mm change in emission was observed; thus both states were readily distinguishable.300 In another format of quencher free molecular beacon, ethynylpyrene-labeled 2′-deoxyadenosine and 2′deoxyuridine can be quenched through photoelectron transfer to neighboring C, T and G bases. This has the advantage of simplifying synthesis by omitting the conjugation of separate quencher chromophores.301, 302
Pyrene has also been observed to form exciplexes with guanosine upon stacking interactions. The resultant color change from blue to green has been used in the detection of G-quadruplex formation.303 Besides pyrenes, pairs of perylene diimides and thiazole orange142 have been incorporated into molecular beacons. Strong excitonic interactions between the chromophores result in a red-shifted excimer emission. In contrast, the H-dimer formation between dicyanomethylenedihydrofuran (DCDHF) dyes result in self-quenching. When incorporated into a molecular beacon, a self-quenched intramolecular dimer (SquID) molecular beacon was formed. Quenching efficiency of 97.2% was reported.304 Asanuma’s threoninol linked chromophores were used in the synthesis of an in-stem molecular beacon where the fluorophore perylene is stacked in the stem region of a molecular beacon with the quencher. The quenchers dabcyl, anthraquinone or azobenzene were tested in separate experiments.305
Non-nucleosidic chromophores 1,8-dialkynylpyrene and perylene diimide were incorporated by Häner into the stem of a molecular beacon. In the hairpin structure, interstrand stacking of the electron-rich pyrenes and electron-poor perylene diimides resulted in the formation of a donor-acceptor complex and suppression of fluorescence emission (Fig. 27). Upon hybridization with the complement, a large excimer signal from the two pyrenes was generated and target DNAs could be detected at low nanomolar concentrations.306
Figure 27.

A donor-acceptor complex formed between perylene diimide (E) and pyrene (Y) in the molecular beacon form. Upon hybridization with the target strand, E and Y units are separated and fluorescence from the pyrene excimer can be observed. Reproduced with permission from ref 307. Copyright 2010 Wiley-VCH Verlag GmbH & Co. KGaA.
5.4. Exciton transfer
The exciton theory proposed by Kasha in 1965 describes the formation of H-aggregates and J-aggregates when excited states of chromophores interact with one another, forming a delocalized excited state lower in energy (in J-aggregates) upon end-to-end interaction since the π-conjugation area is larger.307-309 A higher energy excited state is formed in H-aggregates, which result from face-to-face orientation. The molecular exciton theory has been mainly applied to the H- and J-aggregates consisting of homo dyes. Recently, a detailed experimental study was carried out by Asanuma and coworkers to test the molecular exciton theory for heterodimeric π-stacked chromophores.128 Using azobenzene-derivatized chromophores attached to DNA through the threoninol linker, they were able to systematically tune the difference in absorption maxima of two chromophores in the heterodimer through substitutions on the azobenzene chromophore. Bathochromism and hypochromism were observed, verifying the coherent coupling between in heterodimers as predicted by the molecular exciton theory.
Excitonic coupling between excited states of chromophores can be useful for probing biomolecular interactions. Using non-nucleosidic carboxamide linkers, stilbene chromophores were attached by Lewis and coworkers to both ends of a DNA hairpin.310 Exciton-coupled circular dichroism (EC-CD) was observed between these stilbene chromophores separated by up to 11 A-T base pairs. Both the sign and intensity of the EC-CD spectra were dependent on the number of base pairs between them.311 In another report, Burin and coworkers proposed the use of sum rules derived from UV absorption spectra and EC-CD spectra to determine the exciton coupling between monomeric excited states.312 Thus, excitonic coupling studied with these optical methods is sensitive to molecular structure, geometry and conformation, and it could be used as a “biomolecular ruler”, as an alternative to Förster resonance energy transfer (FRET) methods.
5.5. Charge transfer
The ability to incorporate different chromophores and multiple chromophores at desired positions of the oligonucleotide strand provided researchers with a tool to study electron transfer in DNA. For instance, Wagenknecht’s phenothiazine- and pyrene-modified uridines were used to selectively photoreduce pyrimidine bases C and T in order to initiate an electron hopping process in DNA.143, 313 On the other hand, ethidium and thiazole orange have lower oxidation potentials than all the natural nucleobases and thus were used as charge donors to 5-nitroindole and 7-deazaguanine.138, 314, 315
Lewis’ non-nucleosidic carboxamide-linked chromophores were used in hairpin DNA to study photoinduced electron transfer. Chromophores such as pyrene,316 perylene diimide317, 318 and anthraquininone319 were incorporated at various positions of DNA hairpin conjugates and served as good electron donors to study the excited state and charge transfer dynamics.
5.6. Superquenching
Highly efficient “superquenching” effects have been observed recently in DNA multichromophoric systems. Superquenching was initially observed in conjugated polymers, and results from mobile excited states that move along the polymer chain.43-45 The ability of a fluorescent assembly to be quenched with high efficiency leads to highly sensitive on/off detection in sensor constructs.3, 4, 46 Similar high efficiency quenching has been demonstrated in single-stranded oligodeoxyfluorosides of Kool. Methyl viologen was an effective quencher of ODFs containing several adjacent pyrene monomers, with quenching efficiencies characterized by Stern-Volmer constants of up to 108 M−1.156 Quenching of ODFs was not affected upon conjugation to oligonucleotides. The efficient quenching was used effectively in sensors for esterases,274 described in Section 5.2. Efficient quenching of ODFs was also demonstrated using dabcyl, another commonly used quencher in DNA.158
The interstrand stacking interaction between pyrene and perylenediimide nucleosides (PDI) also promoted the highly efficient quenching of pyrene excimer fluorescence by PDI units.320 Both the pyrene and perylene diimide non-nucleosidic base surrogates were incorporated in the middle of respective DNA strands. Upon DNA hybridization, a change in the exciton chirality in the CD spectra was observed, together with a reduction in the amplitude of the CD signal. This indicated that a larger separation between PDI units and an alternating, zipper-like arrangement of pyrene and PDI. Thus, quenching was brought about by the formation of a non-fluorescent pyrene-PDI complex and enhanced energy transfer through favorable interstrand stacking interactions.
5.7. Applications in cells
The oligodeoxyfluorosides of Kool have been shown to be cell-permeable despite their negative charge. In preliminary studies they were used to label cells and zebrafish embryos (Fig. 28). Multicolor labels could be imaged with a single excitation wavelength for multispectral imaging.99 This is of potential utility in multiplexed imaging of multiple biological species.
Figure 28.

Multicolor labeling of zebrafish embryos with four different ODF fluorophores at (A) 24 h post fertilization and (B) 48 h post fertilization. Real color images taken with an epifluorescence microscope using a long pass emission filter (λex=354 nm). Reprinted with permission from ref 99. Copyright 2009 American Chemical Society.
Esterase sensors developed using dabcyl-quenched oligodeoxyfluorosides have also been shown to be permeable to mammalian cells and are functional within live cells, displaying a fluorescence turn-on signal in response to esterase activity.274 A set of three different colored oligodeoxyfluoroside (ODF) probes have been used together with the dabcyl quencher to synthesize caspase chemosensor.275 The quencher and ODFs were linked via a short caspase recognition peptide sequence. By using a unique ODF for each caspase substrate, these caspase chemosensors were able to target capases 2, 9 and 9, yielding a fluorescent signal upon detecting caspase activity. The biggest advantage of these probes is that they can be used to detect caspase activity in live cells. In this case, intracellular delivery was achieved by a commercial DNA transfection reagent. No wash step was necessary as these probes are dark in their unreacted state and were only rendered fluorescent with caspase activity. Enzyme sensors developed based on fluorescent DNA have been recently reviewed.321
Doubly-labeled thiazole orange probes have been observed to interact through H-aggregation in the excited state and display a red-shifted emission wavelength. When labeled on 2′-O-methyl-RNA, the probes demonstrate resistance to nucleases and thus can be used for long term monitoring of intracellular RNAs.322 A 13-mer perylene-LNA probe complementary to a target mRNA was transfected into HEK 293 cells and its fluorescence was shown to colocalize with its target without being masked by autofluorescence of the cells.196
6. Conclusions and Future Prospects
6.1. Unusual properties of DNA multichromophores
Although there are several other classes of molecular scaffolds used to assemble chromophores, the use of DNA to arrange multiple chromophores offers a set of properties that are difficult to attain otherwise. The rigidity and predictability of the DNA structure, the water solubility, and the ease of synthesis and assembly all lend real utility to this approach. The use of DNA as a scaffold also enables the assembly of structures with a wide range of sizes, from a few Angstroms to several nanometers and larger.323 Recently, the hydrophobic self-assembly of non-nucleosidic perylenediimides (PDI) conjugated to both ends of 8-mer DNA duplexes formed supramolecular polymers with linear regions more than 1 μm.324 These linear end-to-end assemblies of between 10 to 30 DNA-PDI dumbbells monomers are formed in the presence of 100mM of NaCl, driven by hydrophobic effects.
The various systems described throughout this review share a common feature of having unusual photophysical properties that are not observed in the monomeric states of the chromophores. For example, the interaction of multiple excited states at close proximity results in the formation of excimers and charge transfer exciplexes, yielding new fluorescence properties. Ground state interactions result in the formation of H-dimers and J-aggregates, giving changes in absorption. These differences vary with the number of monomers and orientation of monomers with respect to one another, thus creating a new spectrum of fluorescence properties. Interactions between monomers have also resulted in phenomena such as efficient quenchin and resonance energy transfer. These have been useful in the construction of sensors and reporters for multiple applications. This wide variety of properties and applications is unusual among multichromophore scaffolds.
6.2. Remaining challenges
Beyond a few relatively simple examples, it is often difficult to predict how multiple chromophores will interact, even in the relatively rigid and predictable DNA context. Thus, continued progress in understanding the molecular photophysics of aromatic molecules, individually and in concert, and of how charges and excited states move between chromophores, will be helpful in the design of better-performing molecular systems for applications in biology and materials sciences.325, 326 Advances both in experimental mechanistic studies and in theoretical methods will be useful in this regard.
Because interchromophore interactions depend strongly on distance, orientation and local structure, we expect that a greater understanding of self-assembly using π-stacking and molecular designs that combine those features with hydrogen bonding and metal-ligand bonding could simplify the structure of the building blocks for artificial photosynthetic complexes, while retaining their ability to assemble complex, photofunctional structures. Some examples of using DNA to direct the assembly energy transfer complexes for light harvesting have been reported recently.327-329 In the coming years we expect to see many new classes of hybrid DNA structures that arrange chromophores in new ways.
Finally, it is likely that DNA multichromophores will see increasing use in biology, medicine and materials sciences. The need for new and better materials to meet the demands of photonics, solar energy conversion, and sensing devices will continue to drive the development of rigid, well-characterized materials such as DNA multichromophoric systems.330 Their unusual fluorescence properties also present opportunities for the development of new devices for use in biomedical detection and diagnosis of diseases. Future progress in all these areas will no doubt continue at a rapid pace.
ACKNOWLEDGEMENTS
We acknowledge the National Institutes of Health (GM063587 and GM067201) for support. Y.N.T acknowledges Agency for Science, Technology and Research (A*STAR), Singapore, for a graduate fellowship.
Biography

Eric Kool received his Ph.D. degree from Columbia University in 1988 and was a postdoctoral fellow at the California Institute of Technology before taking a position as Assistant Professor at the University of Rochester in 1990. In 1999 he moved to Stanford University, where he is Professor of Chemistry. His research interests include the study of DNA replication and repair, development of templated chemistry for imaging cellular RNAs, design of new genetic systems, and discovery of new DNA-based fluorophores and sensors. His work has been recognized by the ACS Pfizer Award in Enzyme Chemistry and the ACS Cope Scholar Award.

Yin Nah Teo received her B.Sc. degree in Chemistry from the University of Bristol. She then moved to Stanford University, where performed her Ph.D. thesis research under the guidance of Prof. Eric Kool. Her thesis involved the synthesis and study of a novel set of fluorophores appended on the DNA deoxyribose scaffold. She was a recipient of the Public Service Commission Scholarship and the National Science Scholarship from the Agency of Science, Technology and Research (A*STAR) in Singapore. Currently she is a scientist at the Molecular Engineering Lab in A*STAR headed by Prof. Sydney Brenner, and she continues to pursue her research interest in bioconjugation, modified nucleic acids and development of imaging probes.
REFERENCES
- (1).Schwartz E, Le Gac S, Cornelissen JJ, Nolte RJ, Rowan AE. Chem. Soc. Rev. 2010;39:1576. doi: 10.1039/b922160c. [DOI] [PubMed] [Google Scholar]
- (2).Hide F, Díaz-García MA, Schwartz BJ, Heeger AJ. Acc. Chem. Res. 1997;30:430. [Google Scholar]
- (3).McQuade DT, Pullen AE, Swager TM. Chem. Rev. 2000;100:2537. doi: 10.1021/cr9801014. [DOI] [PubMed] [Google Scholar]
- (4).Thomas SW, Joly GD, Swager TM. Chem. Rev. 2007;107:1339. doi: 10.1021/cr0501339. [DOI] [PubMed] [Google Scholar]
- (5).Ho HA, Najari A, Leclerc M. Acc. Chem. Res. 2008;41:168. doi: 10.1021/ar700115t. [DOI] [PubMed] [Google Scholar]
- (6).De Schryver FC, Vosch T, Cotlet M, Van der Auweraer M, Mullen K, Hofkens J. Acc. Chem. Res. 2005;38:514. doi: 10.1021/ar040126r. [DOI] [PubMed] [Google Scholar]
- (7).Adronov A, Fréchet J. Chem. Commun. 2000;18:1701. [Google Scholar]
- (8).Astruc D, Boisselier E, Ornelas C. Chem. Rev. 2010;110:1857. doi: 10.1021/cr900327d. [DOI] [PubMed] [Google Scholar]
- (9).Garcia-Parajo MF, Hernando J, Sanchez Mosteiro G, Hoogenboom JP, van Dijk EM, van Hulst NF. ChemPhysChem. 2005;6:819. doi: 10.1002/cphc.200400630. [DOI] [PubMed] [Google Scholar]
- (10).Tinnefeld P, Heilemann M, Sauer M. ChemPhysChem. 2005;6:217. doi: 10.1002/cphc.200400513. [DOI] [PubMed] [Google Scholar]
- (11).Heilemann M, Kasper R, Tinnefeld P, Sauer M. J. Am. Chem. Soc. 2006;128:16864. doi: 10.1021/ja065585x. [DOI] [PubMed] [Google Scholar]
- (12).Lewis FD. Photochem. Photobiol. 2005;81:65. doi: 10.1562/2004-09-01-ir-299.1. [DOI] [PubMed] [Google Scholar]
- (13).Hossain MA, Mihara H, Ueno A. J. Am. Chem. Soc. 2003;125:11178. doi: 10.1021/ja036427y. [DOI] [PubMed] [Google Scholar]
- (14).SolladieA N, Hamel A, Gross M. Tet. Lett. 2000;41:6075. [Google Scholar]
- (15).Heinze K, Hempel K. Chem.-Eur. J. 2009;15:1346. doi: 10.1002/chem.200801864. [DOI] [PubMed] [Google Scholar]
- (16).Huang Y-S, Yang X, Schwartz E, Lu LP, Albert-Seifried S, Finlayson CE, Koepf M, Kitto HJ, Ulgut B, Otten MBJ, Cornelissen JJLM, Nolte RJM, Rowan AE, Friend RH. J. Phys. Chem. B. 2011;115:1590. doi: 10.1021/jp1071605. [DOI] [PubMed] [Google Scholar]
- (17).Wolffs M, Delsuc N, Veldman D, Nguyên VA, Williams RM, Meskers SCJ, Janssen RAJ, Huc I, Schenning APHJ. J. Am. Chem. Soc. 2009;131:4819. doi: 10.1021/ja809367u. [DOI] [PubMed] [Google Scholar]
- (18).Soto C, Blum A, Vora G, Lebedev N, Meador C, Won A, Chatterji A, Johnson J, Ratna B. J. Am. Chem. Soc. 2006;128:5184. doi: 10.1021/ja058574x. [DOI] [PubMed] [Google Scholar]
- (19).Stewart G, Fox M. J. Am. Chem. Soc. 1996;118:4354. [Google Scholar]
- (20).Bhosale R, Mísek J, Sakai N, Matile S. Chem Soc Rev. 2010;39:138. doi: 10.1039/b906115k. [DOI] [PubMed] [Google Scholar]
- (21).Peng KY, Chen SA, Fann WS. J. Am. Chem. Soc. 2001;123:11388. doi: 10.1021/ja011493q. [DOI] [PubMed] [Google Scholar]
- (22).Chen C-H, Liu K-Y, Sudhakar S, Lim T-S, Fann W, Hsu C-P, Luh T-Y. J. Phys. Chem. B. 2005;109:17887. doi: 10.1021/jp051909c. [DOI] [PubMed] [Google Scholar]
- (23).Talukdar P, Bollot G, Mareda J, Sakai N, Matile S. J. Am. Chem. Soc. 2005;127:6528. doi: 10.1021/ja051260p. [DOI] [PubMed] [Google Scholar]
- (24).Bhosale S, Sisson AL, Talukdar P, Fürstenberg A, Banerji N, Vauthey E, Bollot G, Mareda J, Röger C, Würthner F, Sakai N, Matile S. Science. 2006;313:84. doi: 10.1126/science.1126524. [DOI] [PubMed] [Google Scholar]
- (25).Matile S, Vargas Jentzsch A, Montenegro J, Fin A. Chem Soc Rev. 2011;40:2453. doi: 10.1039/c0cs00209g. [DOI] [PubMed] [Google Scholar]
- (26).Greyson EC, Stepp BR, Chen X, Schwerin AF, Paci I, Smith MB, Akdag A, Johnson JC, Nozik AJ, Michl J, Ratner MA. J. Phys. Chem. B. 2010;114:14223. doi: 10.1021/jp909002d. [DOI] [PubMed] [Google Scholar]
- (27).Günes S, Neugebauer H, Sariciftci NS. Chem. Rev. 2007;107:1324. doi: 10.1021/cr050149z. [DOI] [PubMed] [Google Scholar]
- (28).Hardin B, Hoke E, Armstrong P, Yum J, Comte P, Torres T, Fréchet J, Nazeeruddin M, Grätzel M, McGehee M. Nat. Photonics. 2009;3:406. [Google Scholar]
- (29).Coakley K, McGehee M. Chem. Mater. 2004;16:4533. [Google Scholar]
- (30).Zang L, Che Y, Moore JS. Acc. Chem. Res. 2008;41:1596. doi: 10.1021/ar800030w. [DOI] [PubMed] [Google Scholar]
- (31).Abbel R, Grenier C, Pouderoijen MJ, Stouwdam JW, Leclère PELG, Sijbesma RP, Meijer EW, Schenning APHJ. J. Am. Chem. Soc. 2009;131:833. doi: 10.1021/ja807996y. [DOI] [PubMed] [Google Scholar]
- (32).Burrell A, Officer D, Plieger P, Reid D. Chem. Rev. 2001;101:2751. doi: 10.1021/cr0000426. [DOI] [PubMed] [Google Scholar]
- (33).Choi M-S, Yamazaki T, Yamazaki I, Aida T. Angew. Chem., Int. Ed. 2004;43:150. doi: 10.1002/anie.200301665. [DOI] [PubMed] [Google Scholar]
- (34).Scholes G, Fleming G. J. Phys. Chem. B. 2000;104:1854. [Google Scholar]
- (35).Cheng Y-C, Fleming GR. Annu. Rev. Phys. Chem. 2009;60:241. doi: 10.1146/annurev.physchem.040808.090259. [DOI] [PubMed] [Google Scholar]
- (36).Jang S, Newton M, Silbey R. Phys. Rev. Lett. 2004;92:218301. doi: 10.1103/PhysRevLett.92.218301. [DOI] [PubMed] [Google Scholar]
- (37).Jang S, Newton MD, Silbey RJ. J. Phys. Chem. B. 2007;111:6807. doi: 10.1021/jp070111l. [DOI] [PubMed] [Google Scholar]
- (38).Scholes GD, Rumbles G. Nat. Mater. 2006;5:683. doi: 10.1038/nmat1710. [DOI] [PubMed] [Google Scholar]
- (39).Scholes GD. ACS Nano. 2008;2:523. doi: 10.1021/nn700179k. [DOI] [PubMed] [Google Scholar]
- (40).Goodson TG. Acc. Chem. Res. 2005;38:99. doi: 10.1021/ar020247w. [DOI] [PubMed] [Google Scholar]
- (41).Hansen MR, Graf R, Sekharan S, Sebastiani D. J. Am. Chem. Soc. 2009;131:5251. doi: 10.1021/ja8095703. [DOI] [PubMed] [Google Scholar]
- (42).Wang Z, Enkelmann V, Negri F, Mullen K. Angew. Chem., Int. Ed. 2004;43:1972. doi: 10.1002/anie.200353245. [DOI] [PubMed] [Google Scholar]
- (43).Swager TM. Acc. Chem. Res. 1998;31:201. [Google Scholar]
- (44).Zhou Q, Swager TM. J. Am. Chem. Soc. 1995;117:12593. [Google Scholar]
- (45).Zhou Q, Swager TM. J. Am. Chem. Soc. 1995;117:7017. [Google Scholar]
- (46).Chen L, McBranch D, Wang H, Helgeson R, Wudl F, Whitten D. Proc. Natl. Acad. Sci. U.S.A. 1999;96:12287. doi: 10.1073/pnas.96.22.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Duan X, Liu L, Feng F, Wang S. Acc. Chem. Res. 2010;43:260. doi: 10.1021/ar9001813. [DOI] [PubMed] [Google Scholar]
- (48).Gaylord B, Heeger A, Bazan G. J. Am. Chem. Soc. 2003;125:896. doi: 10.1021/ja027152+. [DOI] [PubMed] [Google Scholar]
- (49).Kumaraswamy S, Bergstedt T, Shi X, Rininsland F, Kushon S, Xia W, Ley K, Achyuthan K, Mcbranch D, Whitten D. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7511. doi: 10.1073/pnas.0402367101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wosnick JH, Mello CM, Swager TM. J. Am. Chem. Soc. 2005;127:3400. doi: 10.1021/ja043134b. [DOI] [PubMed] [Google Scholar]
- (51).Andrews DL. J. Nanophotonics. 2008;2:022502. [Google Scholar]
- (52).Hoeben FJ, Jonkheijm P, Meijer EW, Schenning AP. Chem. Rev. 2005;105:1491. doi: 10.1021/cr030070z. [DOI] [PubMed] [Google Scholar]
- (53).Datta A, Pati SK. Chem. Soc. Rev. 2006;35:1305. doi: 10.1039/b605478a. [DOI] [PubMed] [Google Scholar]
- (54).Bazan GC. J. Org. Chem. 2007;72:8615. doi: 10.1021/jo071176n. [DOI] [PubMed] [Google Scholar]
- (55).Aratani N, Kim D, Osuka A. Acc. Chem. Res. 2009;42:1922. doi: 10.1021/ar9001697. [DOI] [PubMed] [Google Scholar]
- (56).Wasielewski MR. Acc. Chem. Res. 2009;42:1910. doi: 10.1021/ar9001735. [DOI] [PubMed] [Google Scholar]
- (57).Kim D, Osuka A. Acc. Chem. Res. 2004;37:735. doi: 10.1021/ar030242e. [DOI] [PubMed] [Google Scholar]
- (58).Anderson S, Anderson HL, Sanders JKM. Acc. Chem. Res. 1993;26:469. [Google Scholar]
- (59).Wengel J. Org. Biomol. Chem. 2004;2:277. doi: 10.1039/b313986g. [DOI] [PubMed] [Google Scholar]
- (60).Endo M, Sugiyama H. ChemBioChem. 2009;10:2420. doi: 10.1002/cbic.200900286. [DOI] [PubMed] [Google Scholar]
- (61).Feldkamp U, Niemeyer CM. Angew. Chem., Int. Ed. 2006;45:1856. doi: 10.1002/anie.200502358. [DOI] [PubMed] [Google Scholar]
- (62).Seeman NC. Nature. 2003;421:427. doi: 10.1038/nature01406. [DOI] [PubMed] [Google Scholar]
- (63).Lu Y, Liu J. Acc. Chem. Res. 2007;40:315. doi: 10.1021/ar600053g. [DOI] [PubMed] [Google Scholar]
- (64).Seeman NC. Mol. Biotechnol. 2007;37:246. doi: 10.1007/s12033-007-0059-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Dietz H, Douglas SM, Shih WM. Science. 2009;325:725. doi: 10.1126/science.1174251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Douglas SM, Dietz H, Liedl T, Högberg B, Graf F, Shih WM. Nature. 2009;459:414. doi: 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Zheng J, Birktoft JJ, Chen Y, Wang T, Sha R, Constantinou PE, Ginell SL, Mao C, Seeman NC. Nature. 2009;461:74. doi: 10.1038/nature08274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Han D, Pal S, Nangreave J, Deng Z, Liu Y, Yan H. Science. 2011;332:342. doi: 10.1126/science.1202998. [DOI] [PubMed] [Google Scholar]
- (69).Macfarlane RJ, Lee B, Hill HD, Senesi AJ, Seifert S, Mirkin CA. Proc. Natl. Acad. Sci. U.S.A. 2009;106:10493. doi: 10.1073/pnas.0900630106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Rosi NL, Mirkin CA. Chem. Rev. 2005;105:1547. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]
- (71).Tikhomirov G, Hoogland S, Lee PE, Fischer A, Sargent EH, Kelley SO. Nat. Nanotechnol. 2011;6:485. doi: 10.1038/nnano.2011.100. [DOI] [PubMed] [Google Scholar]
- (72).Yan H, Park SH, Finkelstein G, Reif JH, LaBean TH. Science. 2003;301:1882. doi: 10.1126/science.1089389. [DOI] [PubMed] [Google Scholar]
- (73).Kukolka F, Schoeps O, Woggon U, Niemeyer CM. Bioconjug. Chem. 2007;18:621. doi: 10.1021/bc060143w. [DOI] [PubMed] [Google Scholar]
- (74).Niemeyer CM, Koehler J, Wuerdemann C. ChemBioChem. 2002;3:242. doi: 10.1002/1439-7633(20020301)3:2/3<242::AID-CBIC242>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- (75).Datta B, Schuster GB, McCook A, Harvey SC, Zakrzewska K. J. Am. Chem. Soc. 2006;128:14428. doi: 10.1021/ja0648413. [DOI] [PubMed] [Google Scholar]
- (76).Chen RL, Fong P. J. Theor. Biol. 1969;22:180. doi: 10.1016/0022-5193(69)90086-1. [DOI] [PubMed] [Google Scholar]
- (77).Goddard NL, Bonnet G, Krichevsky O, Libchaber A. Phys. Rev. Lett. 2000;85:2400. doi: 10.1103/PhysRevLett.85.2400. [DOI] [PubMed] [Google Scholar]
- (78).Chen P, Li CM. Small. 2007;3:1204. doi: 10.1002/smll.200700049. [DOI] [PubMed] [Google Scholar]
- (79).Ramprakash J, Lang B, Schwarz FP. Biopolymers. 2008;89:969. doi: 10.1002/bip.21044. [DOI] [PubMed] [Google Scholar]
- (80).Olaso-González G, Merchán M, Serrano-Andrés L. J. Am. Chem. Soc. 2009;131:4368. doi: 10.1021/ja808280j. [DOI] [PubMed] [Google Scholar]
- (81).Varghese R, Wagenknecht H-A. Chem. Commun. 2009;19:2615. doi: 10.1039/b821728a. [DOI] [PubMed] [Google Scholar]
- (82).Kool ET. Annu. Rev. Biochem. 2002;71:191. doi: 10.1146/annurev.biochem.71.110601.135453. [DOI] [PubMed] [Google Scholar]
- (83).Kool ET. Annu. Rev. Biophys. Biomol. Struct. 2001;30:1. doi: 10.1146/annurev.biophys.30.1.1. [DOI] [PubMed] [Google Scholar]
- (84).Thoresen LH, Jiao G-S, Haaland WC, Metzker ML, Burgess K. Chem.-Eur. J. 2003;9:4603. doi: 10.1002/chem.200304944. [DOI] [PubMed] [Google Scholar]
- (85).Matray T, Kool E. J. Am. Chem. Soc. 1998;120:6191. doi: 10.1021/ja9803310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Matray TJ, Kool ET. Nature. 1999;399:704. doi: 10.1038/21453. [DOI] [PubMed] [Google Scholar]
- (87).Li X, Li Z, Mart AA, Jockusch S, Stevens N, Akins DL, Turro NJ, Ju J. Photochem. Photobio. Sci. 2006;5:896. doi: 10.1039/b605936h. [DOI] [PubMed] [Google Scholar]
- (88).Loakes D, Gallego J, Pinheiro VB, Kool ET, Holliger P. J. Am. Chem. Soc. 2009;131:14827. doi: 10.1021/ja9039696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Ramsay N, Jemth AS, Brown A, Crampton N, Dear P, Holliger P. J. Am. Chem. Soc. 2010;132:5096. doi: 10.1021/ja909180c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Caruthers MH. Science. 1985;230:281. doi: 10.1126/science.3863253. [DOI] [PubMed] [Google Scholar]
- (91).Caruthers MH, Barone AD, Beaucage SL, Dodds DR, Fisher EF, McBride LJ, Matteucci M, Stabinsky Z, Tang JY. Method Enzymol. 1987;154:287. doi: 10.1016/0076-6879(87)54081-2. [DOI] [PubMed] [Google Scholar]
- (92).LeProust EM, Peck BJ, Spirin K, McCuen HB, Moore B, Namsaraev E, Caruthers MH. Nucleic Acids Res. 2010;38:2522. doi: 10.1093/nar/gkq163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Verma S, Eckstein F. Annu. Rev. Biochem. 1998;67:99. doi: 10.1146/annurev.biochem.67.1.99. [DOI] [PubMed] [Google Scholar]
- (94).Cobb AJA. Org. Biomol. Chem. 2007;5:3260. doi: 10.1039/b709797m. [DOI] [PubMed] [Google Scholar]
- (95).Benner SA. Acc. Chem. Res. 2004;37:784. doi: 10.1021/ar040004z. [DOI] [PubMed] [Google Scholar]
- (96).Gao J, Strässler C, Tahmassebi D, Kool ET. J. Am. Chem. Soc. 2002;124:11590. doi: 10.1021/ja027197a. [DOI] [PubMed] [Google Scholar]
- (97).Häner R, Samain F, Malinovskii VL. Chem.-Eur. J. 2009;15:5701. doi: 10.1002/chem.200900369. [DOI] [PubMed] [Google Scholar]
- (98).Wan W, Wang W, Li ADQ. ChemBioChem. 2008;9:304. doi: 10.1002/cbic.200700389. [DOI] [PubMed] [Google Scholar]
- (99).Teo YN, Wilson JN, Kool ET. J. Am. Chem. Soc. 2009;131:3923. doi: 10.1021/ja805502k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Beaucage S, Iyer R. Tetrahedron. 1992;48:2223. [Google Scholar]
- (101).Sinha ND. Method. Mol. Biol. 1993;20:437. doi: 10.1385/0-89603-281-7:437. [DOI] [PubMed] [Google Scholar]
- (102).Barbaric J, Wanninger-Weiß C, Wagenknecht H-A. Eur. J. Org. Chem. 2009;3:364. [Google Scholar]
- (103).Kashida H, Liang X, Asanuma H. Curr. Org. Chem. 2009;13:1065. [Google Scholar]
- (104).Letsinger RL, Wu T. J. Am. Chem. Soc. 1994;116:811. [Google Scholar]
- (105).Letsinger R, Wu T. J. Am. Chem. Soc. 1995;117:7323. [Google Scholar]
- (106).Lewis FD, Wu T, Zhang Y, Letsinger RL, Greenfield SR, Wasielewski MR. Science. 1997;277:673. doi: 10.1126/science.277.5326.673. [DOI] [PubMed] [Google Scholar]
- (107).Lewis FD, Zhang Y, Liu X, Xu N, Letsinger RL. J. Phys. Chem. B. 1999;103:2570. [Google Scholar]
- (108).Letsinger RL, Wu T, Yang J-S, Lewis FD. Photochem. Photobio. Sci. 2008;7:854. doi: 10.1039/b805452e. [DOI] [PubMed] [Google Scholar]
- (109).Lewis FD, Liu X, Miller SE, Wasielewski MR. J. Am. Chem. Soc. 1999;121:9746. [Google Scholar]
- (110).Lewis F, Kalgutkar R, Wu Y, Liu X, Liu J. J. Am. Chem. Soc. 2000;122:12346. [Google Scholar]
- (111).Daublain P, Siegmund K, Hariharan M, Vura-Weis J, Wasielewski MR, Lewis FD, Shafirovich V, Wang Q, Raytchev M, Fiebig T. Photochem. Photobio. Sci. 2008;7:1501. doi: 10.1039/b813995d. [DOI] [PubMed] [Google Scholar]
- (112).Zheng Y, Long H, Schatz GC, Lewis FD. Chem. Commun. 2005;38:4795. doi: 10.1039/b509754a. [DOI] [PubMed] [Google Scholar]
- (113).Hariharan M, Zheng Y, Long H, Zeidan TA, Schatz GC, Vura-Weis J, Wasielewski MR, Zuo X, Tiede DM, Lewis FD. J. Am. Chem. Soc. 2009;131:5920. doi: 10.1021/ja900347t. [DOI] [PubMed] [Google Scholar]
- (114).Lewis FD, Zhang L, Zuo X. J. Am. Chem. Soc. 2005;127:10002. doi: 10.1021/ja0524402. [DOI] [PubMed] [Google Scholar]
- (115).Lewis F, Zhang L, Kelley R, McCamant D, Wasielewski M. Tetrahedron. 2007;63:3457. [Google Scholar]
- (116).Langenegger S, Häner R. Helv. Chim. Acta. 2002;85:3414. [Google Scholar]
- (117).Langenegger SM, Häner R. ChemBioChem. 2005;6:2149. doi: 10.1002/cbic.200500366. [DOI] [PubMed] [Google Scholar]
- (118).Langenegger S, Häner R. Tetrahedron Lett. 2004;45:9273. [Google Scholar]
- (119).Langenegger S, Häner R. Chem. Commun. 2004;24:2792. doi: 10.1039/b412831a. [DOI] [PubMed] [Google Scholar]
- (120).Bouquin N, Malinovskii VL, Guegano X, Liu S-X, Decurtins S, Häner R. Chem.-Eur. J. 2008;14:5732. doi: 10.1002/chem.200800505. [DOI] [PubMed] [Google Scholar]
- (121).Trkulja I, Häner R. J. Am. Chem. Soc. 2007;129:7982. doi: 10.1021/ja0715501. [DOI] [PubMed] [Google Scholar]
- (122).Samain F, Malinovskii VL, Langenegger SM, Häner R. Bioorgan. Med. Chem. 2008;16:27. doi: 10.1016/j.bmc.2007.04.052. [DOI] [PubMed] [Google Scholar]
- (123).Malinovskii VL, Samain F, Häner R. Angew. Chem., Int. Ed. 2007;46:4464. doi: 10.1002/anie.200700891. [DOI] [PubMed] [Google Scholar]
- (124).Häner R, Garo F, Wenger D, Malinovskii VL. J. Am. Chem. Soc. 2010;132:7466. doi: 10.1021/ja102042p. [DOI] [PubMed] [Google Scholar]
- (125).Nussbaumer AL, Studer D, Malinovskii VL, Häner R. Angew. Chem., Int. Ed. 2011;50:5490. doi: 10.1002/anie.201100677. [DOI] [PubMed] [Google Scholar]
- (126).Kashida H, Asanuma H, Komiyama M. Angew. Chem., Int. Ed. 2004;43:6522. doi: 10.1002/anie.200460870. [DOI] [PubMed] [Google Scholar]
- (127).Kashida H, Komiyama M, Asanuma H. Chem. Lett. 2006;35:934. [Google Scholar]
- (128).Fujii T, Kashida H, Asanuma H. Chem.-Eur. J. 2009;15:10092. doi: 10.1002/chem.200900962. [DOI] [PubMed] [Google Scholar]
- (129).Kashida H, Ito H, Fujii T, Hayashi T, Asanuma H. J. Am. Chem. Soc. 2009;131:9928. doi: 10.1021/ja9013002. [DOI] [PubMed] [Google Scholar]
- (130).Kashida H, Sekiguchi K, Liang X, Asanuma H. J. Am. Chem. Soc. 2010;132:6223. doi: 10.1021/ja101007d. [DOI] [PubMed] [Google Scholar]
- (131).Kashida H, Tanaka M, Baba S, Sakamoto T, Kawai G, Asanuma H, Komiyama M. Chem.-Eur. J. 2006;12:777. doi: 10.1002/chem.200500111. [DOI] [PubMed] [Google Scholar]
- (132).Asanuma H, Shirasuka K, Takarada T, Kashida H, Komiyama M. J. Am. Chem. Soc. 2003;125:2217. doi: 10.1021/ja021153k. [DOI] [PubMed] [Google Scholar]
- (133).Kashida H, Fujii T, Asanuma H. Org. Biomol. Chem. 2008;6:2892. doi: 10.1039/b806406g. [DOI] [PubMed] [Google Scholar]
- (134).Wagner C, Wagenknecht H-A. Org. Lett. 2006;8:4191. doi: 10.1021/ol061246x. [DOI] [PubMed] [Google Scholar]
- (135).Baumstark D, Wagenknecht H-A. Chem.-Eur. J. 2008;14:6640. doi: 10.1002/chem.200800514. [DOI] [PubMed] [Google Scholar]
- (136).Baumstark D, Wagenknecht H-A. Angew. Chem., Int. Ed. 2008;47:2612. doi: 10.1002/anie.200705237. [DOI] [PubMed] [Google Scholar]
- (137).Huber R, Amann N, Wagenknecht H. J. Org. Chem. 2004;69:744. doi: 10.1021/jo0355404. [DOI] [PubMed] [Google Scholar]
- (138).Valis L, Wang Q, Raytchev M, Buchvarov I, Wagenknecht H-A, Fiebig T. Proc. Natl. Acad. Sci. U.S.A. 2006;103:10192. doi: 10.1073/pnas.0600957103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Amann N, Huber R, Wagenknecht H. Angew. Chem., Int. Ed. 2004;43:1845. doi: 10.1002/anie.200353153. [DOI] [PubMed] [Google Scholar]
- (140).Valis L, Amann N, Wagenknecht H-A. Org. Biomol. Chem. 2005;3:36. doi: 10.1039/b414672g. [DOI] [PubMed] [Google Scholar]
- (141).Menacher F, Rubner M, Berndl S, Wagenknecht H-A. J. Org. Chem. 2008;73:4263. doi: 10.1021/jo8004793. [DOI] [PubMed] [Google Scholar]
- (142).Berndl S, Wagenknecht H-A. Angew. Chem., Int. Ed. 2009;48:2418. doi: 10.1002/anie.200805981. [DOI] [PubMed] [Google Scholar]
- (143).Wagner C, Wagenknecht H-A. Org. Biomol. Chem. 2008;6:48. doi: 10.1039/b708904j. [DOI] [PubMed] [Google Scholar]
- (144).Christensen UB, Pedersen EB. Nucleic Acids Res. 2002;30:4918. doi: 10.1093/nar/gkf624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Nielsen CB, Petersen M, Pedersen EB, Hansen PE, Christensen UB. Bioconjug. Chem. 2004;15:260. doi: 10.1021/bc0341932. [DOI] [PubMed] [Google Scholar]
- (146).Filichev VV, Astakhova IV, Malakhov AD, Korshun VA, Pedersen EB. Chem.-Eur. J. 2008;14:9968. doi: 10.1002/chem.200800380. [DOI] [PubMed] [Google Scholar]
- (147).Wang W, Wan W, Zhou H-H, Niu S, Li ADQ. J. Am. Chem. Soc. 2003;125:5248. doi: 10.1021/ja0341900. [DOI] [PubMed] [Google Scholar]
- (148).Werder S, Malinovskii VL, Häner R. Org. Lett. 2008;10:2011. doi: 10.1021/ol8006474. [DOI] [PubMed] [Google Scholar]
- (149).Bittermann H, Siegemund D, Malinovskii VL, Häner R. J. Am. Chem. Soc. 2008;130:15285. doi: 10.1021/ja806747h. [DOI] [PubMed] [Google Scholar]
- (150).Wenger D, Malinovskii VL, Häner R. Chem. Commun. 2011;47:3168. doi: 10.1039/c0cc05125j. [DOI] [PubMed] [Google Scholar]
- (151).Malinovskii VL, Wenger D, Häner R. Chem. Soc. Rev. 2010;39:410. doi: 10.1039/b910030j. [DOI] [PubMed] [Google Scholar]
- (152).Ren RXF, Chaudhuri NC, Paris PL, Rumney S, Kool ET. J. Am. Chem. Soc. 1996;118:7671. doi: 10.1021/ja9612763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Cuppoletti A, Cho Y, Park JS, Strassler C, Kool ET. Bioconjug. Chem. 2005;16:528. doi: 10.1021/bc0497766. [DOI] [PubMed] [Google Scholar]
- (154).Gao J, Watanabe S, Kool ET. J. Am. Chem. Soc. 2004;126:12748. doi: 10.1021/ja046910o. [DOI] [PubMed] [Google Scholar]
- (155).Kim SJ, Kool ET. J. Am. Chem. Soc. 2006;128:6164. doi: 10.1021/ja0581806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Wilson JN, Teo YN, Kool ET. J. Am. Chem. Soc. 2007;129:15426. doi: 10.1021/ja075968a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Teo YN, Kool ET. Bioconjug. Chem. 2009;20:2371. doi: 10.1021/bc9003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Teo YN, Wilson JN, Kool ET. Chem.-Eur. J. 2009;15:11551. doi: 10.1002/chem.200901607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Zahn A, Leumann CJ. Chem.-Eur. J. 2008;14:1087. doi: 10.1002/chem.200701345. [DOI] [PubMed] [Google Scholar]
- (160).Brotschi C, Häberli A, Leumann CJ. Angew. Chem., Int. Ed. 2001;40:3012. doi: 10.1002/1521-3773(20010817)40:16<3012::AID-ANIE3012>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- (161).Brotschi C, Leumann CJ. Angew. Chem., Int. Ed. 2003;42:1655. doi: 10.1002/anie.200250516. [DOI] [PubMed] [Google Scholar]
- (162).Brotschi C, Mathis G, Leumann C. Chem.-Eur. J. 2005;11:1911. doi: 10.1002/chem.200400858. [DOI] [PubMed] [Google Scholar]
- (163).Zahn A, Brotschi C, Leumann CJ. Chem.-Eur. J. 2005;11:2125. doi: 10.1002/chem.200401128. [DOI] [PubMed] [Google Scholar]
- (164).Johar Z, Zahn A, Leumann CJ, Jaun B. Chem.-Eur. J. 2008;14:1080. doi: 10.1002/chem.200701304. [DOI] [PubMed] [Google Scholar]
- (165).Grigorenko NA, Leumann CJ. Chem.-Eur. J. 2009;15:639. doi: 10.1002/chem.200801135. [DOI] [PubMed] [Google Scholar]
- (166).Chiba J, Takeshima S, Mishima K, Maeda H, Nanai Y, Mizuno K, Inouye M. Chem.-Eur. J. 2007;13:8124. doi: 10.1002/chem.200700559. [DOI] [PubMed] [Google Scholar]
- (167).Hainke S, Seitz O. Angew. Chem., Int. Ed. 2009;48:8250. doi: 10.1002/anie.200903194. [DOI] [PubMed] [Google Scholar]
- (168).Weizman H, Tor Y. J. Am. Chem. Soc. 2001;123:3375. doi: 10.1021/ja005785n. [DOI] [PubMed] [Google Scholar]
- (169).Weizman H, Tor Y. Chem. Commun. 2001;5:453. [Google Scholar]
- (170).Aubert Y, Asseline U. Org. Biomol. Chem. 2004;2:3496. doi: 10.1039/b410695d. [DOI] [PubMed] [Google Scholar]
- (171).Seela F, Zulauf M. Helv. Chim. Acta. 1999;82:1878. [Google Scholar]
- (172).Wagenknecht H-A. Angew. Chem., Int. Ed. 2009;48:2838. doi: 10.1002/anie.200900327. [DOI] [PubMed] [Google Scholar]
- (173).Taniguchi Y, Kool ET. J. Am. Chem. Soc. 2007;129:8836. doi: 10.1021/ja071970q. [DOI] [PubMed] [Google Scholar]
- (174).Valis L, Mayer-Enthart E, Wagenknecht H-A. Bioorgan. Med. Chem. 2006;16:3184. doi: 10.1016/j.bmcl.2006.03.063. [DOI] [PubMed] [Google Scholar]
- (175).Conway N, McLaughlin L. Bioconjug. Chem. 1991;2:452. doi: 10.1021/bc00012a013. [DOI] [PubMed] [Google Scholar]
- (176).Varghese R, Wagenknecht HA. Chem.-Eur. J. 2010;16:9040. doi: 10.1002/chem.201001136. [DOI] [PubMed] [Google Scholar]
- (177).Mayer-Enthart E, Wagenknecht H-A. Angew. Chem., Int. Ed. 2006;45:3372. doi: 10.1002/anie.200504210. [DOI] [PubMed] [Google Scholar]
- (178).Barbaric J, Wagenknecht H-A. Org. Biomol. Chem. 2006;4:2088. doi: 10.1039/b605251g. [DOI] [PubMed] [Google Scholar]
- (179).Mayer-Enthart E, Wagner C, Barbaric J, Wagenknecht H. Tetrahedron. 2007;63:3434. [Google Scholar]
- (180).Hwang G, Seo Y, Kim B. Tetrahedron Lett. 2005;46:1475. [Google Scholar]
- (181).Seo Y, Hwang G, Kim B. Tetrahedron Lett. 2006;47:4037. [Google Scholar]
- (182).Seo YJ, Rhee H, Joo T, Kim BH. J. Am. Chem. Soc. 2007;129:5244. doi: 10.1021/ja069069i. [DOI] [PubMed] [Google Scholar]
- (183).Bouamaied I, Nguyen T, Rühl T, Stulz E. Org. Biomol. Chem. 2008;6:3888. doi: 10.1039/b813584c. [DOI] [PubMed] [Google Scholar]
- (184).Fendt L-A, Bouamaied I, Thöni S, Amiot N, Stulz E. J. Am. Chem. Soc. 2007;129:15319. doi: 10.1021/ja075711c. [DOI] [PubMed] [Google Scholar]
- (185).Nguyen T, Brewer A, Stulz E. Angew. Chem., Int. Ed. 2009;48:1974. doi: 10.1002/anie.200805657. [DOI] [PubMed] [Google Scholar]
- (186).Kosuge M, Kubota M, Ono A. Tetrahedron Lett. 2004;45:3945. [Google Scholar]
- (187).Balaz M, Holmes AE, Benedetti M, Rodriguez PC, Berova N, Nakanishi K, Proni G. J. Am. Chem. Soc. 2005;127:4172. doi: 10.1021/ja043373z. [DOI] [PubMed] [Google Scholar]
- (188).Kwon T, Piton N, Grünewald C, Engels JW. Nucleos. Nucleot. Nucl. 2007;26:1381. doi: 10.1080/15257770701534048. [DOI] [PubMed] [Google Scholar]
- (189).Grünewald C, Kwon T, Piton N, Förster U, Wachtveitl J, Engels JW. Bioorgan. Med. Chem. 2008;16:19. doi: 10.1016/j.bmc.2007.04.058. [DOI] [PubMed] [Google Scholar]
- (190).Trifonov A, Raytchev M, Buchvarov I, Rist M, Barbaric J, Wagenknecht H, Fiebig T. J. Phys. Chem. B. 2005;109:19490. doi: 10.1021/jp052108c. [DOI] [PubMed] [Google Scholar]
- (191).Nakamura M, Ohtoshi Y, Yamana K. Chem. Commun. 2005;41:5163. doi: 10.1039/b507808c. [DOI] [PubMed] [Google Scholar]
- (192).Nakamura M, Murakami Y, Sasa K, Hayashi H, Yamana K. J. Am. Chem. Soc. 2008;130:6904. doi: 10.1021/ja801054t. [DOI] [PubMed] [Google Scholar]
- (193).Dioubankova NN, Malakhov AD, Stetsenko DA, Gait MJ, Volynsky PE, Efremov RG, Korshun VA. ChemBioChem. 2003;4:841. doi: 10.1002/cbic.200300678. [DOI] [PubMed] [Google Scholar]
- (194).Hrdlicka PJ, Babu BR, Sørensen MD, Wengel J. Chem. Commun. 2004;13:1478. doi: 10.1039/b404446k. [DOI] [PubMed] [Google Scholar]
- (195).Astakhova IV, Korshun VA, Wengel J. Chem.-Eur. J. 2008;14:11010. doi: 10.1002/chem.200801077. [DOI] [PubMed] [Google Scholar]
- (196).Astakhova IV, Korshun VA, Jahn K, Kjems J, Wengel J. Bioconjug. Chem. 2008;19:1995. doi: 10.1021/bc800202v. [DOI] [PubMed] [Google Scholar]
- (197).Gupta P, Langkjær N, Wengel J. Bioconjug. Chem. 2010;21:513. doi: 10.1021/bc900421r. [DOI] [PubMed] [Google Scholar]
- (198).Hrdlicka PJ, Babu BR, Sørensen MD, Harrit N, Wengel J. J. Am. Chem. Soc. 2005;127:13293. doi: 10.1021/ja052887a. [DOI] [PubMed] [Google Scholar]
- (199).Kumar TS, Madsen AS, Østergaard ME, Wengel J, Hrdlicka PJ. J. Org. Chem. 2008;73:7060. doi: 10.1021/jo800551j. [DOI] [PubMed] [Google Scholar]
- (200).Guckian KM, Schweitzer BA, Ren RXF, Sheils CJ, Tahmassebi DC, Kool ET. J. Am. Chem. Soc. 2000;122:2213. doi: 10.1021/ja9934854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Benvin AL, Creeger Y, Fisher GW, Ballou B, Waggoner AS, Armitage BA. J. Am. Chem. Soc. 2007;129:2025. doi: 10.1021/ja066354t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Oezhalici-Uenal H, Armitage BA. ACS Nano. 2009;3:425. doi: 10.1021/nn800727x. [DOI] [PubMed] [Google Scholar]
- (203).Seifert J, Connor R, Kushon S, Wang M, Armitage B. J. Am. Chem. Soc. 1999;121:2987. [Google Scholar]
- (204).Wang M, Silva G, Armitage B. J. Am. Chem. Soc. 2000;122:9977. [Google Scholar]
- (205).Hannah KC, Gil RR, Armitage BA. Biochemistry. 2005;44:15924. doi: 10.1021/bi051298e. [DOI] [PubMed] [Google Scholar]
- (206).Hannah KC, Armitage BA. Acc. Chem. Res. 2004;37:845. doi: 10.1021/ar030257c. [DOI] [PubMed] [Google Scholar]
- (207).Dilek I, Madrid M, Singh R, Urrea CP, Armitage BA. J. Am. Chem. Soc. 2005;127:3339. doi: 10.1021/ja045145a. [DOI] [PubMed] [Google Scholar]
- (208).Smith J, Olson D, Armitage B. J. Am. Chem. Soc. 1999;121:2686. [Google Scholar]
- (209).Janssen PGA, Jabbari-Farouji S, Surin M, Vila X, Gielen JC, de Greef TFA, Vos MRJ, Bomans PHH, Sommerdijk NAJM, Christianen PCM, Leclere P, Lazzaroni R, van der Schoot P, Meijer EW, Schenning APHJ. J. Am. Chem. Soc. 2009;131:1222. doi: 10.1021/ja808075h. [DOI] [PubMed] [Google Scholar]
- (210).Janssen PGA, Vandenbergh J, van Dongen JLJ, Meijer EW, Schenning APHJ. J. Am. Chem. Soc. 2007;129:6078. doi: 10.1021/ja0711967. [DOI] [PubMed] [Google Scholar]
- (211).Janssen PGA, Ruiz-Carretero A, González-Rodríguez D, Meijer EW, Schenning APHJ. Angew. Chem., Int. Ed. 2009;48:8103. doi: 10.1002/anie.200903507. [DOI] [PubMed] [Google Scholar]
- (212).Gonzalez-Rodriguez D, Janssen PGA, Martin-Rapun R, Cat ID, Feyter SD, Schenning APHJ, Meijer EW. J. Am. Chem. Soc. 2010;132:4710. doi: 10.1021/ja908537k. [DOI] [PubMed] [Google Scholar]
- (213).Iwaura R, Hoeben FJM, Masuda M, Schenning APHJ, Meijer EW, Shimizu T. J. Am. Chem. Soc. 2006;128:13298. doi: 10.1021/ja064560v. [DOI] [PubMed] [Google Scholar]
- (214).Iwaura R, Ohnishi-Kameyama M, Iizawa T. Chem.-Eur. J. 2009;15:3729. doi: 10.1002/chem.200802537. [DOI] [PubMed] [Google Scholar]
- (215).Pasternack RF, Giannetto A, Pagano P, Gibbs EJ. J. Am. Chem. Soc. 1991;113:7799. [Google Scholar]
- (216).Pasternack R, Bustamante C, Collings P. J. Am. Chem. Soc. 1993;115:5393. [Google Scholar]
- (217).Pasternack RF, Goldsmith JI, Szép S, Gibbs EJ. Biophys. J. 1998;75:1024. doi: 10.1016/S0006-3495(98)77591-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Scolaro LM, Romeo A, Pasternack RF. J. Am. Chem. Soc. 2004;126:7178. doi: 10.1021/ja049669m. [DOI] [PubMed] [Google Scholar]
- (219).Tong A, Li Z, Jones G, Russo J, Ju J. Nat. Biotechnol. 2001;19:756. doi: 10.1038/90810. [DOI] [PubMed] [Google Scholar]
- (220).Tong AK, Jockusch S, Li Z, Zhu HR, Akins DL, Turro NJ, Ju J. J. Am. Chem. Soc. 2001;123:12923. doi: 10.1021/ja016904h. [DOI] [PubMed] [Google Scholar]
- (221).Tong AK, Ju J. Nucleic Acids Res. 2002;30:e19. doi: 10.1093/nar/30.5.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Ju J, Ruan C, Fuller C, Glazer A, Mathies R. Proc. Natl. Acad. Sci. U.S.A. 1995;92:4347. doi: 10.1073/pnas.92.10.4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Ju J, Glazer A, Mathies R. Nucleic Acids Res. 1996;24:1144. doi: 10.1093/nar/24.6.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Ohya Y, Yabuki K, Komatsu M, Ouchi T. Polym. Adv. Technol. 2000;11:845. [Google Scholar]
- (225).Ohya Y, Yabuki K, Hashimoto M, Nakajima A, Ouchi T. Bioconjug. Chem. 2003;14:1057. doi: 10.1021/bc034028m. [DOI] [PubMed] [Google Scholar]
- (226).Vyawahare S, Eyal S, Mathews KD, Quake SR. Nano Lett. 2004;4:1035. [Google Scholar]
- (227).Hannestad JK, Sandin P, Albinsson B. J. Am. Chem. Soc. 2008;130:15889. doi: 10.1021/ja803407t. [DOI] [PubMed] [Google Scholar]
- (228).Heilemann M, Tinnefeld P, Sanchez Mosteiro G, Garcia Parajo M, Van Hulst NF, Sauer M. J. Am. Chem. Soc. 2004;126:6514. doi: 10.1021/ja049351u. [DOI] [PubMed] [Google Scholar]
- (229).Sánchez-Mosteiro G, van Dijk EMHP, Hernando J, Heilemann M, Tinnefeld P, Sauer M, Koberlin F, Patting M, Wahl M, Erdmann R, van Hulst NF, García-Parajó MF. J. Phys. Chem. B. 2006;110:26349. doi: 10.1021/jp064701f. [DOI] [PubMed] [Google Scholar]
- (230).Kukolka F, Mueller BK, Paternoster S, Arndt A, Niemeyer CM, Braeuchle C, Lamb DC. Small. 2006;2:1083. doi: 10.1002/smll.200600202. [DOI] [PubMed] [Google Scholar]
- (231).Boeneman K, Prasuhn DE, Blanco-Canosa JB, Dawson PE, Melinger JS, Ancona M, Stewart MH, Susumu K, Huston A, Medintz IL. J. Am. Chem. Soc. 2010;132:18177. doi: 10.1021/ja106465x. [DOI] [PubMed] [Google Scholar]
- (232).Scopes DI, Barrio JR, Leonard NJ. Science. 1977;195:296. doi: 10.1126/science.188137. [DOI] [PubMed] [Google Scholar]
- (233).Liu H, Gao J, Lynch SR, Saito YD, Maynard L, Kool ET. Science. 2003;302:868. doi: 10.1126/science.1088334. [DOI] [PubMed] [Google Scholar]
- (234).Krueger AT, Lu H, Lee AHF, Kool ET. Acc. Chem. Res. 2007;40:141. doi: 10.1021/ar068200o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Krueger AT, Kool ET. J. Am. Chem. Soc. 2008;130:3989. doi: 10.1021/ja0782347. [DOI] [PubMed] [Google Scholar]
- (236).Virta P, Katajisto J, Niittymaeki T, Lönnberg H. Tetrahedron. 2003;59:5137. [Google Scholar]
- (237).Goodchild J. Bioconjug. Chem. 1990;1:165. doi: 10.1021/bc00003a001. [DOI] [PubMed] [Google Scholar]
- (238).Asseline U. Curr. Org. Chem. 2006;10:491. [Google Scholar]
- (239).Wu Q, Simons C. Synthesis. 2004;2004:1533. [Google Scholar]
- (240).Hocek M. Chem. Rev. 2009;109:6729. doi: 10.1021/cr9002165. [DOI] [PubMed] [Google Scholar]
- (241).Chaudhuri NC, Kool ET. Tetrahedron Lett. 1995;36:1795. [Google Scholar]
- (242).Hainke S, Arndt S, Seitz O. Org. Biomol. Chem. 2005;3:4233. doi: 10.1039/b509846g. [DOI] [PubMed] [Google Scholar]
- (243).Wellington KW, Benner SA. Nucleos. Nucleot. Nucl. 2006;25:1309. doi: 10.1080/15257770600917013. [DOI] [PubMed] [Google Scholar]
- (244).Hainke S, Singh I, Hemmings J, Seitz O. J. Org. Chem. 2007;72:8811. doi: 10.1021/jo7016185. [DOI] [PubMed] [Google Scholar]
- (245).Rist M, Amann N, Wagenknecht H. Eur. J. Org. Chem. 2003;13:2498. [Google Scholar]
- (246).Gierlich J, Burley GA, Gramlich PME, Hammond DM, Carell T. Org. Lett. 2006;8:3639. doi: 10.1021/ol0610946. [DOI] [PubMed] [Google Scholar]
- (247).Weisbrod SH, Marx A. Chem. Commun. 2008;44:5675. doi: 10.1039/b809528k. [DOI] [PubMed] [Google Scholar]
- (248).Gutsmiedl K, Wirges CT, Ehmke V, Carell T. Org. Lett. 2009;11:2405. doi: 10.1021/ol9005322. [DOI] [PubMed] [Google Scholar]
- (249).Gramlich PME, Wirges CT, Manetto A, Carell T. Angew. Chem., Int. Ed. 2008;47:8350. doi: 10.1002/anie.200802077. [DOI] [PubMed] [Google Scholar]
- (250).Gramlich PME, Warncke S, Gierlich J, Carell T. Angew. Chem., Int. Ed. 2008;47:3442. doi: 10.1002/anie.200705664. [DOI] [PubMed] [Google Scholar]
- (251).Srivatsan SG, Tor Y. J. Am. Chem. Soc. 2007;129:2044. doi: 10.1021/ja066455r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Srivatsan SG, Tor Y. Nat. Protoc. 2007;2:1547. doi: 10.1038/nprot.2007.222. [DOI] [PubMed] [Google Scholar]
- (253).Srivatsan SG, Tor Y. Chem.-Asian. J. 2009;4:419. doi: 10.1002/asia.200800370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Kimoto M, Mitsui T, Yokoyama S, Hirao I. J. Am. Chem. Soc. 2010;132:4988. doi: 10.1021/ja100806c. [DOI] [PubMed] [Google Scholar]
- (255).Wanninger-Weiß C, Di Pasquale F, Ehrenschwender T, Marx A, Wagenknecht H-A. Chem. Commun. 2008;12:1443. doi: 10.1039/b718002k. [DOI] [PubMed] [Google Scholar]
- (256).Lu H, Lynch SR, Lee AHF, Kool ET. ChemBioChem. 2009;10:2530. doi: 10.1002/cbic.200900434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Chelliserrykattil J, Lu H, Lee AHF, Kool ET. ChemBioChem. 2008;9:2976. doi: 10.1002/cbic.200800339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Delaney JC, Gao J, Liu H, Shrivastav N, Essigmann JM, Kool ET. Angew. Chem., Int. Ed. 2009;48:4524. doi: 10.1002/anie.200805683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Gierlich J, Gutsmiedl K, Gramlich PME, Schmidt A, Burley GA, Carell T. Chem.-Eur. J. 2007;13:9486. doi: 10.1002/chem.200700502. [DOI] [PubMed] [Google Scholar]
- (260).Gramlich PME, Wirges CT, Gierlich J, Carell T. Org. Lett. 2008;10:249. doi: 10.1021/ol7026015. [DOI] [PubMed] [Google Scholar]
- (261).Gutsmiedl K, Fazio D, Carell T. Chem.-Eur. J. 2010;16:6877. doi: 10.1002/chem.201000363. [DOI] [PubMed] [Google Scholar]
- (262).Raindlová V, Pohl R, Sanda M, Hocek M. Angew. Chem., Int. Ed. 2010;49:1064. doi: 10.1002/anie.200905556. [DOI] [PubMed] [Google Scholar]
- (263).Jarchow-Choy SK, Krueger AT, Liu H, Gao J, Kool ET. Nucleic Acids Res. 2011;39:1586. doi: 10.1093/nar/gkq853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Klimasauskas S, Weinhold E. Trends Biotechnol. 2007;25:99. doi: 10.1016/j.tibtech.2007.01.006. [DOI] [PubMed] [Google Scholar]
- (265).Pljevaljcic G, Pignot M, Weinhold E. J. Am. Chem. Soc. 2003;125:3486. doi: 10.1021/ja021106s. [DOI] [PubMed] [Google Scholar]
- (266).Pljevaljcí G, Schmidt F, Scheidig AJ, Lurz R, Weinhold E. ChemBioChem. 2007;8:1516. doi: 10.1002/cbic.200700294. [DOI] [PubMed] [Google Scholar]
- (267).Lukinavicius G, Lapiene V, Stasevskij Z, Dalhoff C, Weinhold E, Klimasauskas S. J. Am. Chem. Soc. 2007;129:2758. doi: 10.1021/ja0691876. [DOI] [PubMed] [Google Scholar]
- (268).Dalhoff C, Lukinavicius G, Klimasǎuskas S, Weinhold E. Nat. Chem. Biol. 2006;2:31. doi: 10.1038/nchembio754. [DOI] [PubMed] [Google Scholar]
- (269).Ohlmeyer MH, Swanson RN, Dillard LW, Reader JC, Asouline G, Kobayashi R, Wigler M, Still WC. Proc. Natl. Acad. Sci. U.S.A. 1993;90:10922. doi: 10.1073/pnas.90.23.10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Nestler H, Bartlett P, Still W. J. Org. Chem. 1994;59:4723. [Google Scholar]
- (271).Guo J, Wang S, Dai N, Teo YN, Kool ET. Proc. Natl. Acad. Sci. U.S.A. 2011;108:3493. doi: 10.1073/pnas.1017349108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Varghese R, Wagenknecht H-A. Chem.-Eur. J. 2009;15:9307. doi: 10.1002/chem.200901147. [DOI] [PubMed] [Google Scholar]
- (273).Uno S.-n., Dohno C, Bittermann H, Malinovskii VL, Häner R, Nakatani K. Angew. Chem., Int. Ed. 2009;48:7362. doi: 10.1002/anie.200903251. [DOI] [PubMed] [Google Scholar]
- (274).Dai N, Teo YN, Kool ET. Chem. Commun. 2010;46:1221. doi: 10.1039/b926338a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Dai N, Guo J, Teo YN, Kool ET. Angew. Chem., Int. Ed. 2011;50:5105. doi: 10.1002/anie.201007805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Samain F, Ghosh S, Teo YN, Kool ET. Angew. Chem., Int. Ed. 2010;49:7025. doi: 10.1002/anie.201002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Samain F, Dai N, Kool ET. Chem.-Eur. J. 2011;17:174. doi: 10.1002/chem.201002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Koo C-K, Wang S, Gaur RL, Samain F, Banaei N, Kool ET. Chem. Commun. 2011;47:11435. doi: 10.1039/c1cc14871k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Tan SS, Teo YN, Kool ET. Org Lett. 2010;12:4820. doi: 10.1021/ol1019794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Tan SS, Kim SJ, Kool ET. J. Am. Chem. Soc. 2011;133:2664. doi: 10.1021/ja109561e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Wang DG, Fan JB, Siao CJ, Berno A, Young P, Sapolsky R, Ghandour G, Perkins N, Winchester E, Spencer J, Kruglyak L, Stein L, Hsie L, Topaloglou T, Hubbell E, Robinson E, Mittmann M, Morris MS, Shen N, Kilburn D, Rioux J, Nusbaum C, Rozen S, Hudson TJ, Lipshutz R, Chee M, Lander ES. Science. 1998;280:1077. doi: 10.1126/science.280.5366.1077. [DOI] [PubMed] [Google Scholar]
- (282).McCarthy JJ, Hilfiker R. Nat. Biotechnol. 2000;18:505. doi: 10.1038/75360. [DOI] [PubMed] [Google Scholar]
- (283).Altshuler D, Daly MJ, Lander ES. Science. 2008;322:881. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Ebata K, Masuko M, Ohtani H, Kashiwasake-Jibu M. Photochem. Photobiol. 1995;62:836. doi: 10.1111/j.1751-1097.1995.tb09144.x. [DOI] [PubMed] [Google Scholar]
- (285).Yamana K, Iwai T, Ohtani Y, Sato S, Nakamura M, Nakano H. Bioconjug. Chem. 2002;13:1266. doi: 10.1021/bc025530u. [DOI] [PubMed] [Google Scholar]
- (286).Kashida H, Asanuma H, Komiyama M. Chem. Commun. 2006;26:2768. doi: 10.1039/b604776a. [DOI] [PubMed] [Google Scholar]
- (287).Umemoto T, Hrdlicka PJ, Babu BR, Wengel J. ChemBioChem. 2007;8:2240. doi: 10.1002/cbic.200700408. [DOI] [PubMed] [Google Scholar]
- (288).Zhu H, Lewis FD. Bioconjug. Chem. 2007;18:1213. doi: 10.1021/bc060279u. [DOI] [PubMed] [Google Scholar]
- (289).Lewis FD, Zhang Y, Letsinger RL. J. Am. Chem. Soc. 1997;119:5451. [Google Scholar]
- (290).Mahara A, Iwase R, Sakamoto T, Yamana K, Yamaoka T, Murakami A. Angew. Chem., Int. Ed. 2002;41:3648. doi: 10.1002/1521-3773(20021004)41:19<3648::AID-ANIE3648>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- (291).Maeda H, Maeda T, Mizuno K, Fujimoto K, Shimizu H, Inouye M. Chem.-Eur. J. 2006;12:824. doi: 10.1002/chem.200500638. [DOI] [PubMed] [Google Scholar]
- (292).Paris PL, Langenhan JM, Kool ET. Nucleic Acids Res. 1998;26:3789. doi: 10.1093/nar/26.16.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Okamoto A, Ichiba T, Saito I. J. Am. Chem. Soc. 2004;126:8364. doi: 10.1021/ja049061d. [DOI] [PubMed] [Google Scholar]
- (294).Kashida H, Takatsu T, Asanuma H. Tetrahedron Lett. 2007;48:6759. [Google Scholar]
- (295).Marti AA, Li X, Jockusch S, Li Z, Raveendra B, Kalachikov S, Russo JJ, Morozova I, Puthanveettil SV, Ju J, Turro NJ. Nucleic Acids Res. 2006;34:3161. doi: 10.1093/nar/gkl406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Fujimoto K, Shimizu H, Inouye M. J. Org. Chem. 2004;69:3271. doi: 10.1021/jo049824f. [DOI] [PubMed] [Google Scholar]
- (297).Yamana K, Fukunaga Y, Ohtani Y, Sato S, Nakamura M, Kim WJ, Akaike T, Maruyama A. Chem. Commun. 2005;19:2509. doi: 10.1039/b502033f. [DOI] [PubMed] [Google Scholar]
- (298).Conlon P, Yang CJ, Wu Y, Chen Y, Martinez K, Kim Y, Stevens N, Marti AA, Jockusch S, Turro NJ, Tan W. J. Am. Chem. Soc. 2008;130:336. doi: 10.1021/ja076411y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Matsumoto K, Shinohara Y, Bag SS, Takeuchi Y, Morii T, Saito Y, Saito I. Bioorgan. Med. Chem. 2009;19:6392. doi: 10.1016/j.bmcl.2009.09.060. [DOI] [PubMed] [Google Scholar]
- (300).Holzhauser C, Wagenknecht H-A. Angew. Chem., Int. Ed. 2011;50:7268. doi: 10.1002/anie.201101968. [DOI] [PubMed] [Google Scholar]
- (301).Seo YJ, Ryu JH, Kim BH. Org. Lett. 2005;7:4931. doi: 10.1021/ol0518582. [DOI] [PubMed] [Google Scholar]
- (302).Venkatesan N, Seo YJ, Kim BH. Chem. Soc. Rev. 2008;37:648. doi: 10.1039/b705468h. [DOI] [PubMed] [Google Scholar]
- (303).Seo YJ, Lee IJ, Yi JW, Kim BH. Chem. Commun. 2007;27:2817. doi: 10.1039/b707278c. [DOI] [PubMed] [Google Scholar]
- (304).Conley NR, Pomerantz AK, Wang H, Twieg RJ, Moerner WE. J. Phys. Chem. B. 2007;111:7929. doi: 10.1021/jp073310d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (305).Kashida H, Takatsu T, Fujii T, Sekiguchi K, Liang X, Niwa K, Takase T, Yoshida Y, Asanuma H. Angew. Chem., Int. Ed. 2009;48:7044. doi: 10.1002/anie.200902367. [DOI] [PubMed] [Google Scholar]
- (306).Häner R, Biner SM, Langenegger SM, Meng T, Malinovskii VL. Angew. Chem., Int. Ed. 2010;49:1227. doi: 10.1002/anie.200905829. [DOI] [PubMed] [Google Scholar]
- (307).Kasha M. Rev. Mod. Phys. 1959;31:162. [Google Scholar]
- (308).Kasha M. Radiat. Res. 1963;20:55. [PubMed] [Google Scholar]
- (309).Kasha M, Rawls H, El-Bayoumi M. Pure Appl. Chem. 1965;11:371. [Google Scholar]
- (310).Lewis FD, Liu X, Wu Y, Zuo X. J. Am. Chem. Soc. 2003;125:12729. doi: 10.1021/ja036449k. [DOI] [PubMed] [Google Scholar]
- (311).Lewis FD, Zhang L, Liu X, Zuo X, Tiede D, Long H, Schatz GC. J. Am. Chem. Soc. 2005;127:14445. doi: 10.1021/ja0539387. [DOI] [PubMed] [Google Scholar]
- (312).Burin AL, Armbruster ME, Hariharan M, Lewis FD. Proc. Natl. Acad. Sci. U.S.A. 2009;106:989. doi: 10.1073/pnas.0808513106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (313).Wanninger-Weiß C, Wagenknecht H-A. Eur. J. Org. Chem. 2008;1:64. [Google Scholar]
- (314).Prunkl C, Berndl S, Wanninger-Weiß C, Barbaric J, Wagenknecht H-A. Phys. Chem. Chem. Phys. 2010;12:32. doi: 10.1039/b914487k. [DOI] [PubMed] [Google Scholar]
- (315).Kaden P, Mayer-Enthart E, Trifonov A, Fiebig T, Wagenknecht H-A. Angew. Chem., Int. Ed. 2005;44:1636. doi: 10.1002/anie.200462592. [DOI] [PubMed] [Google Scholar]
- (316).Siegmund K, Daublain P, Wang Q, Trifonov A, Fiebig T, Lewis FD. J. Phys. Chem. B. 2009;113:16276. doi: 10.1021/jp907323d. [DOI] [PubMed] [Google Scholar]
- (317).Zeidan TA, Carmieli R, Kelley RF, Wilson TM, Lewis FD, Wasielewski MR. J. Am. Chem. Soc. 2008;130:13945. doi: 10.1021/ja803765r. [DOI] [PubMed] [Google Scholar]
- (318).Carmieli R, Zeidan TA, Kelley RF, Mi Q, Lewis FD, Wasielewski MR. J. Phys. Chem. A. 2009;113:4691. doi: 10.1021/jp900230q. [DOI] [PubMed] [Google Scholar]
- (319).Lewis FD, Thazhathveetil AK, Zeidan TA, Vura-Weis J, Wasielewski MR. J. Am. Chem. Soc. 2010;132:444. doi: 10.1021/ja908470d. [DOI] [PubMed] [Google Scholar]
- (320).Bouquin N, Malinovskii VL, Häner R. Chem. Commun. 2008;17:1974. doi: 10.1039/b802193g. [DOI] [PubMed] [Google Scholar]
- (321).Dai N, Kool ET. Chem. Soc. Rev. 2011;40:5756. doi: 10.1039/c0cs00162g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Kubota T, Ikeda S, Yanagisawa H, Yuki M, Okamoto A. Bioconjug. Chem. 2009;20:1256. doi: 10.1021/bc900120a. [DOI] [PubMed] [Google Scholar]
- (323).Khakshoor O, Kool ET. Chem. Commun. 2011;47:7018. doi: 10.1039/c1cc11021g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Neelakandan PP, Pan Z, Hariharan M, Zheng Y, Weissman H, Rybtchinski B, Lewis FD. J. Am. Chem. Soc. 2010;132:15808. doi: 10.1021/ja1076525. [DOI] [PubMed] [Google Scholar]
- (325).Hsu C-P. Acc. Chem. Res. 2009;42:509. doi: 10.1021/ar800153f. [DOI] [PubMed] [Google Scholar]
- (326).Zgierski MZ, Fujiwara T, Lim EC. Acc. Chem. Res. 2010;43:506. doi: 10.1021/ar9002043. [DOI] [PubMed] [Google Scholar]
- (327).Kumar CV, Duff MR., Jr. J. Am. Chem. Soc. 2009;131:16024. doi: 10.1021/ja904551n. [DOI] [PubMed] [Google Scholar]
- (328).Dutta PK, Varghese R, Nangreave J, Lin S, Yan H, Liu Y. J. Am. Chem. Soc. 2011;133:11985. doi: 10.1021/ja1115138. [DOI] [PubMed] [Google Scholar]
- (329).Stein IH, Steinhauer C, Tinnefeld P. J. Am. Chem. Soc. 2011;133:4193. doi: 10.1021/ja1105464. [DOI] [PubMed] [Google Scholar]
- (330).Pinheiro AV, Han D, Shih WM, Yan H. Nat Nanotechnol. 2011;6:763. doi: 10.1038/nnano.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]