

Recent advances in tumor-derived microvesicles reveal new insights into the cellular basis of cancer disease progression and the potential to translate this knowledge to innovative approaches for cancer diagnostics and personalized therapy. These heterogeneous membrane-bound sacs are shed from the surfaces of tumor cells into the extracellular environment. In addition to depositing paracrine information, these membrane vesicles can also fuse with cells in the microenvironment to alter recipient cell content and behavior. In this review by D'Souza-Schorey and Clancy, new and improved strategies for tumor-derived microvesicle identification, isolation, and capture and the implications in point-of-care diagnostics for cancer patients are discussed.
Keywords: oncosomes, shed microvesicles, tumor microenvironment
Abstract
Recent advances in the study of tumor-derived microvesicles reveal new insights into the cellular basis of disease progression and the potential to translate this knowledge into innovative approaches for cancer diagnostics and personalized therapy. Tumor-derived microvesicles are heterogeneous membrane-bound sacs that are shed from the surfaces of tumor cells into the extracellular environment. They have been thought to deposit paracrine information and create paths of least resistance, as well as be taken up by cells in the tumor microenvironment to modulate the molecular makeup and behavior of recipient cells. The complexity of their bioactive cargo—which includes proteins, RNA, microRNA, and DNA—suggests multipronged mechanisms by which microvesicles can condition the extracellular milieu to facilitate disease progression. The formation of these shed vesicles likely involves both a redistribution of surface lipids and the vertical trafficking of cargo to sites of microvesicle biogenesis at the cell surface. Current research also suggests that molecular profiling of these structures could unleash their potential as circulating biomarkers as well as platforms for personalized medicine. Thus, new and improved strategies for microvesicle identification, isolation, and capture will have marked implications in point-of-care diagnostics for cancer patients.
Intercellular communication is essential for embryonic development as well as the functional integrity of multicellular organisms. Although traditionally viewed as being mediated by soluble factors in the surrounding environment, the contribution of specialized membrane-enclosed sacs shed from the surface of cells to intercellular communication has recently become better appreciated (Ratajczak et al. 2006b; Mause and Weber 2010). Indeed, shed membrane vesicles serve to shuttle bioactive molecules between cells, and this cargo can modulate the extracellular environment (Ratajczak et al. 2006a; Cocucci et al. 2009). In addition to depositing paracrine information, these membrane vesicles can also fuse with cells in the microenvironment to alter recipient cell content and behavior. The latter may result from direct membrane fusion or the endocytosis of the extracellular vesicle into the target cell. The past few years have seen an explosion of research on these shed vesicles, largely because of their perceived roles in cancer, inflammation, coagulation, and stem cell renewal and expansion. Moreover, microvesicle presence in body fluids—including the blood—points to their promise as prospective biomarkers and prognostic indicators in the surveillance of various health conditions (van Doormaal et al. 2009; Fleissner et al. 2012).
Eukaryotic cells secrete a wide spectrum of membrane vesicles under physiological conditions, although aberrant release of microvesicles can arise in disease states, as has been well described in cancer (Al-Nedawi et al. 2009b; van Doormaal et al. 2009; Muralidharan-Chari et al. 2010; Lee et al. 2011). Microvesicles in cancer patients were first documented in the 1970s with their identification in cultures of spleen nodules and lymph nodes from a patient with Hodgkin's disease (Friend et al. 1978). The first key evidence for their role in disease progression came from the demonstration that vesicles shed spontaneously from highly metastatic B16 mouse melanoma cells (F10)—upon fusion with poorly metastatic B16 mouse melanoma cells (F1)—enabled F1 cells to metastasize to the lung (Poste and Nicolson 1980). These studies set the stage for further investigations into the importance of microvesicles in tumor progression. Since then, by virtue of their ability to harness select bioactive molecules and propagate the horizontal transfer of their cargo, shed microvesicles have been shown to have an enormous impact on tumor growth, survival, and spread, along with demonstrated effects on many stages of tumor progression, including angiogenesis, escape from immune surveillance, extracellular matrix (ECM) degradation, and metastasis (van Doormaal et al. 2009; Muralidharan-Chari et al. 2010). Here, we review recent advances on tumor-derived microvesicle research. We discuss and speculate on modes by which they are formed, mechanisms by which they modulate the tumor niche through horizontal transfer of bioactive molecules, and also isolation protocols that differentiate them from other shed vesicle populations. Their potential as cancer biomarkers and a personalized therapy platform is also considered.
Heterogeneity of tumor-derived extracellular vesicles
Tumor cells emit more than one type of membrane vesicle, each with unique morphological traits and functions. The best characterized of these are exosomes, 50- to 100-nm vesicles generated intracellularly in multivesicular bodies (MVBs) and released upon MVB fusion with the plasma membrane (Fig. 1). Exosomes are generated and shed from a wide spectrum of cell types, including tumor cells. They contain complex sets of cargo, depending on the physiological conditions in which they are generated and released, and once shed, they impact a variety of cellular responses, especially inflammatory responses, as has been described in several excellent reviews (Schorey and Bhatnagar 2008; Bobrie et al. 2009; Simons and Raposo 2009). Tumor cells undergoing apoptosis can also generate apoptotic bodies, which are relatively large membrane blebs (on the order of several micrometers in diameter) and formed by random blebbing of the plasma membrane (Saraste and Pulkki 2000). They contain fragmented nuclei as well as cytoplasmic organelles. Reports have indicated that the uptake of apoptotic bodies by cells in the tumor microenvironment can, in fact, influence the cellular response by transferring oncogenes to target cells (Bergsmedh et al. 2001) or facilitating immune suppression of CD8+ cytolytic T cells (Xie et al. 2009).
Figure 1.

Types of membrane vesicles released extracellularly by tumor cells. (1) Exosomes are formed and sequestered in preformed MVBs and released upon fusion of the MVB-limiting membrane with the plasma membrane. (2) Apoptotic bodies are formed by the random blebbing of the plasma membrane. Fragmented DNA and cytoplasmic organelles are packaged indiscriminately into these blebs. (3) TMVs contain selectively sorted cargo and form by the outward blebbing and fission of the plasma membrane. Nascent microvesicles at the cell surface may be a convergence point for multiple membrane trafficking pathways, including ARF6-regulated early endosome recycling, which directs specialized cargo to these sites of biogenesis.
Tumor cells also shed a heterogeneous population of vesicles that appear to be distinct from both exosomes and apoptotic bodies. These vesicles are larger than exosomes and can range from 200 nm to a few micrometers in diameter. Vesicles that fit these criteria—also referred to as shedding vesicles, oncosomes, microvesicles, microparticles, or ectosomes—are the focus of this review. Here, we refer to them as tumor-derived microvesicles (TMVs). Several reports suggest that they appear to be ubiquitously released by tumor cells, and the amounts shed increase with cell invasiveness or disease progression (Muralidharan-Chari et al. 2010). Importantly, TMVs are detected only upon acquisition of tumorigenic phenotypes and appear not to be shed in any detectable level by normal or “parental” cells (Ginestra et al. 1998). For example, while the normal mammary cell line MCF10A shows no detectable TMV release, invasive breast tumor lines such as MDA-MB 231 shed TMVs and do so to a much larger extent than MCF7 cells, a breast tumor cell line that is significantly less invasive. Consistent with this observation, ARF6, which is a component of TMVs (described further below), is not detected on extracellular vesicles shed from normal mammary cells, but is present on TMVs shed from breast tumor cell lines (A Sedgwick and C D'Souza-Schorey, unpubl.). Whether TMVs are similar in structure, composition, and/or function to extracellular vesicles of comparable size released from cells of the immune or nervous systems (e.g., platelets, neutrophils, and microglia) is still unclear and requires further investigation.
Of the utmost importance to the analysis of extracellular vesicles shed by tumor cells is the ability to isolate and capture individual shed vesicle populations. Given the different size profiles of exosomes, TMVs, and apoptotic bodies, researchers have used size-based separation procedures, such as centrifugation, as the primary means of vesicle isolation. For isolation of TMVs, overlap at the upper (with apoptotic bodies) and lower (with exosomes) ends of the TMV size distribution can often lead to inclusion of multiple vesicle types in subsequent analyses. Advances in sorting and capture capabilities may provide relief from these challenges and are explored in greater detail below. As also discussed further below, molecular profiling of individual shed vesicle populations will aid in designing more precise capture strategies.
How are TMVs formed?
TMVs are formed through unique cellular mechanisms. Unlike exosomes, which are released upon fusion of the limiting membrane of MVBs with the cell surface (Nickel 2005), or apoptotic bodies, which are formed by indiscriminate plasma membrane blebbing (Saraste and Pulkki 2000), the formation of TMVs likely involves distinct mechanisms. The result is outward budding and fission of vesicles from the tumor cell surface (Fig. 1). Protein profiling of shed microvesicles from human colon and prostate cell lines indicates that these membrane-bound structures harbor signaling proteins that affect cell metabolism, mRNA processing, angiogenesis, and cell growth and motility, in addition to molecules that are likely required for TMV biogenesis (Di Vizio et al. 2009; Choi et al. 2011). It is important to note, however, that due to the heterogeneity of both TMV populations and isolation techniques, one cannot exclude the possible contribution of several vesicle populations to the reported protein profiles. Nonetheless, given the complexity of protein cargo, likely both lateral redistribution at and vertical trafficking of cargo to the plasma membrane are involved. A recent study demonstrated that cellular proteins are selectively incorporated into TMVs via ARF6-regulated endosome recycling; some cell surface proteins were incorporated into TMVs with the exclusion of others (Muralidharan-Chari et al. 2009). ARF6 is a member of the ARF family of Ras-related small GTP-binding proteins, and its activation has been linked to the regulation of endosome recycling as well as actin remodeling at the cell periphery (Donaldson 2003; D'Souza-Schorey and Chavrier 2006). The expression and activation of ARF6 have been associated with the acquisition of tumor-invasive potential in cell and animal model systems (D'Souza-Schorey and Chavrier 2006). Thus, one mechanism by which ARF6 can promote cell invasion is by facilitating the formation of large microvesicles. Notably, activated ARF6 or ARF6-GTP appears to be enriched on the larger TMVs (sized at 200 nm to a few micrometers) but is barely detectable on the smaller vesicles that resemble exosomes (Muralidharan-Chari et al. 2009). This study also showed that MHC class I, β1-integrin receptors, and VAMP3—all of which trafficked to the plasma membrane via ARF6-regulated endosomal recycling—were present in nascent as well as shed TMVs. Intriguingly, however, transferrin receptors, which are abundant at the cell surface and also trafficked on ARF6 endosomes in some cell types, were not recruited to TMVs. In addition, cortactin and Tks5, components of invadopodia, which are membrane protrusions formed at the adherent and invasive front of the tumor cell, were also absent from shed TMVs. In addition to the cargo mentioned above, the ARF6-positive TMVs also contain MT1-MMP, which has been thought to traffic along the early and late endocytic recycling pathways (Jiang et al. 2001; Hakulinen et al. 2008). Collectively, these findings suggest that in tumor cells, specialized ARF6-postive recycling vesicles target protein cargo to the cell surface for incorporation into TMVs (Fig. 1). Receptor tyrosine kinases such as EGFRvIII, which are also present in TMVs (Al-Nedawi et al. 2008), may also be transported intracellularly to TMVs via early endocytic recycling, although this has yet to be demonstrated. Studies using prostate tumor cell lines show that Akt and caveolin-1 are present on TMVs (Di Vizio et al. 2009) and are also recruited to early endosomes (Scita and Di Fiore 2010). More recent studies have shown that caveolin-1 is targeted to MVBs and lysosomes (Hayer et al. 2010), leading one to speculate that membranes derived from these compartments may also direct cargo to TMVs. Thus, while the ARF6-regulated recycling pathway is one mechanism to direct cargo to sites of TMV biogenesis, nascent microvesicles at the cell surface may be a convergence point for multiple membrane trafficking pathways directing specialized cargo to these structures.
As indicated above, genetic material—including mRNAs, microRNAs (miRNAs), genomic DNA, and retrotransposons—is also found in microvesicles derived from a wide spectrum of tumor cell lines, including glioblastoma, colon, and gastric cancers (Lee et al. 2011). Here again, due to the heterogeneity of both TMV populations and isolation techniques, one cannot exclude the possible contribution of several vesicle populations to the studies described. It is unclear how nucleic acids are targeted to the tumor cell surface or specific intracellular compartments. That there is enrichment of certain nucleic acids in microvesicles suggests that similar to protein cargo, nucleic acids may also be selectively packaged into microvesicles. While it is possible that cytoplasmic nucleic acids might traffic to sites of microvesicle biogenesis as complexes along with lipids or proteins on cytoskeletal tracks (Ondrej et al. 2007; Holt and Bullock 2009), how genomic DNA might be incorporated is less clear. One possibility is that amplified genomic sequences escape into the cytoplasm during mitosis following nuclear envelope breakdown and are then trafficked as complexes to specific sites in cells for packaging into TMVs.
TMV biogenesis and shedding occur at regions on the cell membrane that appear to be enriched in specific lipids. One of these required lipids is cholesterol; pharmacological depletion of cellular cholesterol inhibits microvesicle release (Del Conde et al. 2005; Pilzer et al. 2005). Similarly, phosphatidylserine (PS) is exposed on the extracellular leaflet of TMVs (Lima et al. 2009; Muralidharan-Chari et al. 2009). This topological reversal may facilitate microvesicle formation by promoting shape changes in the plasma membrane that are conducive to membrane curvature and detachment from the underlying cytoskeleton to enable microvesicle formation.
Fission of TMVs from the cell surface to allow “shedding” is facilitated by actin–myosin-based contraction via a process that also appears to involve the ARF6 protein. Studies have presented a regulatory role for ARF6-mediated ERK activation in facilitating localized activation of myosin light chain kinase and, subsequently, the phosphorylation of myosin light chain to promote the release of TMVs from invasive melanoma cells (Muralidharan-Chari et al. 2009). This ERK induction required ARF6-induced activation of PLD. A study examining PS externalization in platelets showed that phosphatidylinositol 4,5-bisphosphate (PIP2) is necessary for efficient PS externalization (Bucki et al. 2001), providing a potential link to ARF6-regulated PIP2 generation (Honda et al. 1999) and PS externalization. Of note, blocking ARF6 activation in a melanoma cell line drastically reduced TMV release, and ARF6 is detected on TMVs released from a spectrum of tumor cell lines (Muralidharan-Chari et al. 2009), suggesting that common pathways dictate TMV biogenesis. The demonstration that TMV shedding is linked to suppression of Diaphanous-related formin-3 (DRF3) expression provides further evidence for the involvement of actin cytoskeleton-based fission (Di Vizio et al. 2009). DRF3 is a signaling protein that binds small Rho family GTPases and has been implicated in actin nucleation (Peng et al. 2003). Loss of DRF3 promotes cellular changes conducive to the formation of microvesicles and the acquisition of the amoeboid phenotype (Di Vizio et al. 2009). Intriguingly, in this regard, more recent work has implicated RhoA signaling in the generation of TMVs with downstream activation of ROCK (Rho-associated coiled-coiled-containing protein kinase) and LIM kinase downstream from RhoA activation (Li et al. 2012). Cross-talk between the ARF6 and Rho signaling pathways is essential to the coordination of TMV release. Also interesting to note are the commonalities between mechanisms and signaling pathways that govern TMV formation and membrane blebbing at the cell surface (Charras et al. 2006; Gadea et al. 2007). TMVs differ from membrane blebs in that they are in fact shed from the cell surface, rather than retracted. One hypothesis worth considering is that mechanisms that favor retraction are compromised during disease pathogenesis. This reduction in bleb retraction could facilitate TMV shedding, which becomes even more pronounced as tumors become more aggressive. Given emerging literature documenting the unexpected ways TMVs can influence disease progression, understanding the mechanisms involved in TMV biogenesis and shedding at the tumor cell surface is important, as it could provide novel and key cancer therapeutic strategies.
TMVs and the horizontal transfer of bioactive molecules
Once shed, TMVs enable the horizontal transfer of bioactive content and can thus have a profound influence on the tumor microenvironment. In addition to depositing paracrine signals, microvesicles have been shown to interact with cells in the tumor microenvironment, promoting signaling responses in the target cells (Fig. 2). Microvesicle–target cell interaction results in either the fusion of the microvesicle with the target cell or endocytosis of the microvesicle (Gasser et al. 2003; Losche et al. 2004; Eken et al. 2008; Pluskota et al. 2008). Here we discuss recent advances and speculations as to how individual sets of bioactive effectors—namely, proteins and nucleic acids—contained in TMVs can affect cancer progression.
Figure 2.
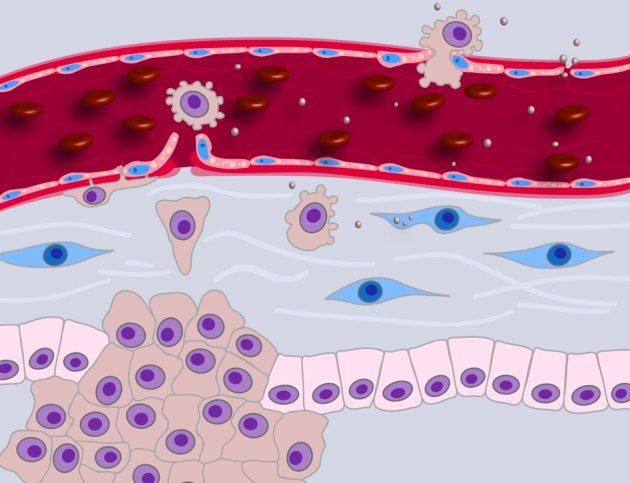
TMV-mediated modulation of the tumor microenvironment. Accruing literature suggests that shed TMVs can condition the stromal microenvironment to promote angiogenesis, evasion of the immune response, tumor invasion, and, potentially, metastasis. TMVs released from tumor cells (brown) can be taken up by cells in the tumor microenvironment, such as carcinoma-associated fibroblasts (blue), with consequences for target cell behavior. They can also interact with the extracellular matrix by depositing paracrine information or facilitating matrix degradation, thereby creating paths of least resistance.
Proteins in TMVs
Oncogenic receptors form an important group of bioactive cargoes packaged in TMVs that can significantly modulate the microenvironment. For example, when a nonaggressive population of tumor cells was exposed to EGFRvIII protein obtained from aggressive glioma TMVs, mitogen-activated protein kinase (MAPK) and Akt were induced, leading to morphological transformation of these recipient cells and an increase in anchorage-independent growth (Al-Nedawi et al. 2008). Notably, treatment of the TMVs with EGFRvIII kinase inhibitor or masking their exposed PS residues with annexin V diminished the aforementioned signaling responses in recipient cells, further suggesting a link between the downstream effects in the recipient cells and the acquisition of EGFRvIII from TMVs (Al-Nedawi et al. 2008, 2009b). This raises the question of whether PS recognition signals might be present on target cells, analogous to those present on mononuclear phagocytes that recognize apoptotic membranes (Zhou 2007). Another example is the AXL receptor in TMVs shed from chronic lymphoblastic leukemia (CLL). AXL-positive TMVs conditioned the bone marrow to enhance CLL disease progression (Ghosh et al. 2010).
TMVs are also loaded with proteases, which provide an additional means of matrix degradation, and likely promote focal proteolysis at more distant sites to create a path of least resistance for metastatic cells. Accordingly, MMP-2, MMP-9, MT1-MMP, and their zymogens urokinase-type plasminogen activator (uPA) and EMMPRIN have been discovered within TMVs (Ginestra et al. 1998; Angelucci et al. 2000; Taraboletti et al. 2002; Hakulinen et al. 2008; Muralidharan-Chari et al. 2009). It has been proposed that β1 integrins facilitate the interaction of shed vesicles with the ECM and that the characteristically acidic pH of the tumor environment promotes microvesicle burst and content release (Giusti et al. 2008). This mode of matrix degradation might be especially important, as tumor cells traverse long distances and assume the amoeboid phenotype that is characterized by high levels of nonapoptotic blebbing, reduced protease activities, and a deformable plasma membrane (Paluch et al. 2006; Pinner and Sahai 2008; Sanz-Moreno et al. 2008; Friedl and Wolf 2010). The amoeboid phenotype is distinct from the “mesenchymal” phenotype, in which the cells are elongated and fibroblast-like (Friedl and Wolf 2003; Sahai and Marshall 2003). Amoeboid movement through tissue spaces can be rapid and only minimally dependent on repetitive cycles of membrane attachment to ECM and retraction. TMV shedding from amoeboid tumor cells could condition the extracellular milieu through deposition of proteases and other paracrine signals to allow the cell to traverse and invade at long distances (Fig. 2). This hypothesis would also be consistent with findings that support an absolute dependency on protease-mediated degradation of tumor invasion through ECM (Sabeh et al. 2009).
TMVs are also thought to carry cargo to modulate the immune response of the tumor microenvironment. This aspect of TMV function has been extensively reviewed and is an important feature of disease progression (Giebel and Wodarz 2006; Valenti et al. 2007; Thery et al. 2009). Examples of how TMVs aid in evasion of the immune response are as follows: Direct fusion of microvesicles produced by human melanoma or colorectal carcinoma cells with monocytes inhibits monocyte differentiation to antigen-presenting cells but promotes release of immunosuppressive cytokines, inhibited cytolytic T-cell activation, and function (Valenti et al. 2006). Other reports have shown that TMVs with exposed FasL, a ligand of the death receptor Fas (CD95), will induce apoptosis in activated T cells and thereby abrogate the potential of these effectors to kill tumor cells (Andreola et al. 2002; Huber et al. 2005). Furthermore, TMVs shed by lymphoblastoma cells contain latent membrane protein (LMP-I), another immune-suppressing transmembrane protein, which inhibits leukocyte proliferation (Flanagan et al. 2003).
TMVs are also thought to harbor proangiogenic regulators, including VEGF and bFGF (Brill et al. 2005; Taraboletti et al. 2006). Angiogenesis is a hallmark feature of tumor growth and survival and is characterized by endothelial cell proliferation to form blood vessels that infiltrate into the tumor (Carmeliet 2005). Al-Nedawi et al. (2009a) showed that the transfer of EGFR from TMVs shed by human cancer cell lines harboring the activated EGFR mutation results in the onset of VEGF and VEGF receptor expression in endothelial cells. TMVs shed from ovarian cancer cells have also been shown to promote the angiogenic capabilities of endothelial cells via a mechanism that requires CD147 (also known as extracellular matrix metalloproteinase inducer) found on shed TMVs (Millimaggi et al. 2007). In addition, reports indicate that proteins present on TMVs stimulate secretion of several proangiogenic factors by stromal fibroblasts to facilitate angiogenesis via the proliferation of endothelial cells (Tang and Conti 2004). TMVs have also been shown to support coagulation. One hypothesis put forth to describe the increased blood coagulation observed in cancer patients focuses on the presence of procoagulation factors in TMVs to promote a clotting cascade in the extracellular environment (Zwicker et al. 2009). Microvesicles shed from endothelial cells, monocytes, and platelets harboring similar cargo would only help to amplify such a coagulation response.
Nucleic acids in TMVs
Nucleic acids packaged in TMVs include mRNAs, miRNAs, noncoding RNAs (ncRNAs), genomic DNA, and cDNAs. The presence of nucleic acids, specifically mRNA, was first reported in shed vesicles released from stem cells in 2006 (Ratajczak et al. 2006a) and then 1 year later from mast cells (Valadi et al. 2007). These compelling and unexpected discoveries led to the fascinating idea that perhaps shed microvesicles, like cell-extruded viruses, can transfer genetic information between cells. As described here, and previously for protein cargo, this type of intercellular transfer of genetic information was shown to have a marked effect on the tumor microenvironment. In a classic example, TMVs shed from glioblastomas contain mRNAs that promote tumor growth, invasion, and immune repression (Skog et al. 2008). These TMV mRNAs are taken up by brain endothelial cells in culture to promote angiogenesis. TMVs containing mRNAs have also been reported for colon and gastric cancers (Baran et al. 2009; Hong et al. 2009). Interestingly, there appears to be overlap in transcripts contained in TMVs from these different cell types, suggesting that TMVs, independent of tissue source, likely have common influences on the surrounding microenvironment.
miRNA molecules have also been described in TMVs shed from several tumor cell lines, including lung, glioblastomas, and gastric cancers (Skog et al. 2008; Rabinowits et al. 2009; Ohshima et al. 2010). Through the negative regulation of multiple mRNA targets in recipient cells (Croce 2009), miRNAs have the potential to markedly affect the tumor microenvironment. However, the mechanisms by which miRNAs in TMVs might facilitate changes to the transcriptome of target cells are far from understood. More recently, TMVs released from CD105-positive human renal cancer stem cells were shown to stimulate angiogenesis and the formation of a lung premetastatic niche. Molecular characterization of these CD105-positive TMVs included a set of proangiogenic mRNAs and miRNAs implicated in tumor progression and metastases (Grange et al. 2011).
Retrotransposons, cDNAs, and ncRNAs have also been reported to be present in TMVs (Balaj et al. 2011). It is intriguing that a fairly large component of nucleic acids packaged into TMVs is ncRNAs, although their role in disease progression awaits further research. Some ncRNA cargoes have been implicated in cell function and gene regulation at both the transcriptional and post-transcriptional levels (Gibb et al. 2011). Retrotransposon RNAs such as LINE-1, HERV-K, and Alu have also been detected in TMVs (Balaj et al. 2011). The same study showed that when HERV-K-rich TMVs derived from human medulloblastoma tumor cells were exposed to human endothelial cells, the HERV-K sequences in the recipient endothelial cells were significantly elevated. By inserting themselves into the genome, these retrotransposon elements have the capacity to modulate and manipulate genomic content. Although normally silent, during cancer, they may be activated and result in an increased number of these repetitive elements, which enhances genome plasticity (Cordaux and Batzer 2009). Genomic DNA as well as cDNAs have been found in TMVs released from glioblastoma tumors. The source of these DNA elements is not fully understood, but the decrease in DNA content in TMVs when tumor cells are treated with inhibitors of DNA replication (such as L-mimosine) suggests that amplified genomic DNA sequences generated from DNA replication (which may enter the cytoplasm during mitosis) could be the source of DNA in TMVs (Balaj et al. 2011). All of the above point toward the idea that TMVs are important emerging transmitters of genetic information in the tumor microenvironment.
Thus, the most direct and potentially significant consequence of TMV release is their ability to modulate the behaviors of stromal cell populations. Once integrated, the consequences to target cell function are now recognized, but the exact fate of the cargo in recipient cells requires further investigation. As alluded to above, TMVs might fuse with the plasma membrane of target cells or be taken up by endocytosis, but the detailed mechanisms involved require further exploration. Do TMVs use specialized pathways for endocytosis? Do they interact with target cells by random contact, or are there “homing” signals that direct them toward specific cell populations? Do they fuse with specific intracellular organelles upon internalization, or are contents released into the cytoplasm? Does the molecular cargo dictate the fate of TMVs in recipient cells, or are common oncogenic-specific mechanisms involved? These are just a few of the plethora of questions that remain to be addressed in order to better understand mechanisms involved in TMV-mediated intercellular communication. The answers, in turn, could provide novel strategies for therapeutic intervention that specifically target modulation of the tumor microenvironment.
TMVs as cancer biomarkers and platforms for personalized therapy
While it is important to recognize the potential heterogeneity of TMV populations, the release of TMVs into body fluids such as blood and urine calls attention to their promise as circulating biomarkers in the surveillance of disease progression. TMVs have been detected in the serum of glioblastoma patients, and removal of the tumor correlates with depletion of circulating TMVs (Skog et al. 2008). Similarly, TMVs have also been detected in the circulation of patients with other cancers, including breast, lung, ovary, prostate, colorectal, and gastric cancers (van Doormaal et al. 2009). Since TMV cargo, both proteins and nucleic acids, appears to be selectively sorted to TMVs, the TMV proteome and nucleic acid profile is likely indicative of molecular changes in the tumor. Furthermore, given that common mechanisms are involved in TMV biogenesis and the overlap in protein and nucleic acid cargo, it is possible that one may identify common markers irrespective of tissue origin. However, TMVs could also present markers that are tissue-specific. A combination of these general and tissue-specific markers would constitute a unique, specific, and identifiable biosignature for individual cancers. This approach is distinct from analysis of whole tissue or unfractionated body fluid and in fact may also be highly advantageous, particularly if TMVs indeed concentrate molecular changes that occur in the tumor, as it would increase the sensitivity of detecting critical markers of cancer progression. Furthermore, as both the genomic and proteomic profiles of tumors change with disease progression and/or in response to treatment, TMVs might also be useful in disease staging and assessing therapeutic responsiveness.
If unique biosignatures on TMVs are amenable to capture, it would make their sampling over time a preferred method to monitor changes to the tumor in response to treatment, especially for tissues such as the ovary or pancreas, where repeated biopsies of these organs is unrealistic. Protein profiling of TMVs from colorectal and oral cancers suggest that these markers were not consistently detected in early stage cancers (Ghiran and Kuo 2010). However, this may be due to the isolation of the tumor from the circulation. The development of sensitive capture platforms from reasonably accessible body fluids (besides blood) that are exposed to the primary tumor—for example, saliva, ascites, or pleural effusions—could afford efficient early stage detection. One complicating factor, however, is the presence of shed vesicles from other nontumor cell types also in direct contact with these body fluids. Thus, equally significant is the development of strategies to selectively capture TMVs separate from other shed vesicle populations. The enrichment of specific nucleic acids, proteins, and lipids in TMVs has the potential to yield an isolation platform selective to TMVs. Current methods used to discriminate between various shed vesicle populations found in peripheral fluids are summarized further below.
With the hypothesis that TMVs are representative of molecular changes in the tumor, profiling of TMVs could form the basis of personalized, targeted cancer therapies, especially as more reliable and rapid profiling technologies become available. It is well accepted that delineating the molecular makeup of a tumor could provide invaluable insight into treatment options. For example, the oncogenic receptor HER-2/neu, in addition to being up-regulated in breast cancer, is also elevated in a relatively smaller subset of other cancers, such as gastric and ovarian cancers (Hung and Lau 1999). Indeed, HER-2 has been detected in serum microvesicles derived from gastric tumors (Baran et al. 2009). This latter group of patients would benefit from existing treatment strategies that target the HER-2 receptor (Arteaga et al. 2011). Along similar lines, somatic mutations in coding genes as well as cancer-specific miRNAs or RNAs can be profiled in serologic TMVs. This information may be vital, as therapeutic treatments are coupled to “oncogenic” mutations and cellular changes. Ultimately, the detection of such targets in TMVs could pave the way for potential cancer diagnostic and therapeutic strategies that could preclude the need for major surgical intervention and tumor biopsies.
Isolation and separation formats for shed vesicle populations
As described above, the clinical promise of TMVs prompts the development of platforms to enable the isolation of low-abundance microvesicles in body fluids. TMVs comprise a relatively small fraction of the bulk fluids from which they can be isolated. Hence, a growing number of enrichment and isolation techniques have been aimed at efforts to effectively concentrate and capture these structures in a minimally invasive fashion for use as diagnostic or prognostic indicators. Many of these techniques, developed for the isolation of shed vesicle populations from a myriad of sources, show renewed promise for use in isolating and characterizing TMVs (Table 1). Isolation and capture of microvesicles can be broadly separated into two primary categories: techniques that are based on the physical properties (size, density, and diffusivity) of the microvesicles and those that are based on biochemical affinity (Radisic et al. 2006).
Table 1.
Techniques for characterization, separation, and isolation of shed vesicle populations

Isolation protocols that separate based on physical characteristics of shed vesicle populations have predominantly relied on processing large fluid volumes with macroscale instrumentation. Because these sorting methodologies rely on intrinsic properties of the microvesicles themselves, they have the distinct advantage of requiring no prior knowledge of cargo content. Serial centrifugation, for example, has been routinely used in a manner that first separates cells and cell debris before pelleting a larger vesicle fraction and, finally, an exosome fraction. Larger particles have often been removed by first subjecting the samples to brief centrifugation at 300–2000g. Given the size distribution of TMVs, these particles can then be isolated from the resulting supernatant through additional centrifugation at 10,000–20,000g (Abrahams et al. 2003; Muralidharan-Chari et al. 2009; Maguire et al. 2012). Exosomes, with their size an order of magnitude smaller than TMVs, can be extracted from the final supernatant by prolonged centrifuging at 100,000g or more (Thery et al. 2006). In addition to pelleting by sedimentation at varied levels of applied force, researchers have also attempted to separate these populations by flotation into either a linear sucrose gradient or a sucrose cushion as a means to further purify the respective populations. Centrifugation can also be used in conjunction with other size-based mechanisms for isolation of extracellular vesicles (Choi et al. 2011). Rood et al. (2010) demonstrated that they were able to separate vesicles from highly abundant proteins by subjecting the pellets obtained by ultracentrifugation to size exclusion chromatography. These methods do generate a relatively homogeneous population of either larger vesicles or exosomes, although the process can result in contamination with other cellular or media components.
Size-based analysis can also be accomplished by examining the light-scattering properties of particles within a fluid medium. All particles in a fluid—including shed vesicles—experience Brownian motion. When illuminated by a laser, suspended particles appear as small bright spots under a microscope. NanoSight has automated the process of nanoparticle tracking analysis (NTA), which tracks the Brownian movement of these bright spots through time-lapse video frames. Since the velocity of Brownian motion depends on the solvent viscosity, particle diameter (the smaller the particle, the higher the velocity), and temperature, NanoSight is able to approximate a hydrodynamic diameter using a variation of the Stokes-Einstein equation. NTA works well on particles ranging in size from 50 nm up to 1–2 μm in diameter and can readily measure particle diameters in a polydispersed sample (Dragovic et al. 2011). Dynamic light scattering (also known as quasielastic light scattering or photon correlation spectroscopy) also correlates Brownian motion with particle size. Unlike NTA, however, dynamic light scattering measures fluctuations in the intensity of scattered light that can then be used to mathematically determine size distributions (Lawrie et al. 2009). This method is capable of measuring particles ranging in size from 1 nm to 6 μm in diameter but is best suited to monodisperse samples (Starostina et al. 2008; Dieckmann et al. 2009). Raman spectroscopy may also be used to determine the diameter of particles that fit within the probe volume (on the order of 0.3 μm3), including microvesicles. Since Raman spectroscopy is a quantitative technique, the signal strength is proportional to microvesicle volume, thereby allowing for relative size estimation (Buehler et al. 1991; Pully et al. 2011). Atomic force microscopy (AFM) provides topographical imaging at subnanometer resolution by scanning a sample surface (without physical contact) with a tipped cantilever and translating tip deflection into a three-dimensional image of the surface (Binnig et al. 1986). Given the lateral and vertical resolution, AFM can be used to measure the relative size distribution of microvesicles once bound to an extremely flat surface. The high resolution results in better size detection of polydisperse samples than dynamic light scattering; however, surface binding of microvesicles may adversely affect the morphology and accurate determination of real diameter (Siedlecki et al. 1999; Hoo et al. 2008; Yuana et al. 2010).
Flow cytometers are capable of sorting and capturing microvesicles based on both physical characteristics (size) and affinity (using fluorescently conjugated antibodies). To determine distributions of vesicles, cytometers make use of a combination of both forward and side-scatter measurements. In this technique, researchers often used a series of polystyrene beads with defined sizes to calibrate the equipment and then gate the experimental population at or just below 1-μm-diameter beads (Robert et al. 2009; Wysoczynski and Ratajczak 2009; Herrera et al. 2010). However, this analysis is limited to vesicle populations that are >200–300 nm in diameter due to constraints imposed by the wavelength of the laser (Gelderman and Simak 2008; Doeuvre et al. 2009). As a result, only a small fraction of the smaller vesicles can be detected using only forward and side-scatter technology. A further drawback to this technique applied to extracellular vesicles is that quantitative sizing is determined by a comparison of the scattering intensity of calibration beads with that of microvesicles. Scattering intensity, however, is not merely defined by the object size, but rather by a combination of absorption, shape, refractive index, and size (van der Pol et al. 2010). To further refine populations, microvesicles can be fixed and incubated with fluorophore-conjugated antibodies to known surface markers. Subpopulations of microvesicles are then investigated based on the presence or absence of fluorescent emission (Cherian et al. 2003; Combes et al. 2004; Pawelczyk et al. 2009).
Exosomes are typically too small to be analyzed on their own by flow cytometry. Thus, when attempting to characterize exosome populations by fluorescent-activated cytometry, they are first bound to beads prior to incubation with fluorescently labeled antibodies. The desire to generate isolation systems that are more readily amenable to diagnostic use has led to a growth in the design of microfluidic, lab-on-a-chip type systems capable of separating and capturing particles of different sizes. Many techniques have been developed using microfluidics to separate microparticles, using methods such as a combination of centrifugal force and graduated mechanical gap (Maruyama et al. 2010), flow splitting and recombining (so-called biomimetic devices that rely on size-based variations in particle behavior when in laminar flow) (Takagi et al. 2005; Yamada and Seki 2006; Andersen et al. 2009); optical fractionation (MacDonald et al. 2003; Ladavac et al. 2004; Milne et al. 2007; Smith et al. 2007), or deterministic lateral displacement arrays, which function much like a particle sieve (Huang et al. 2004; Mohamed et al. 2004; Loutherback et al. 2010). Research has also demonstrated that it is possible to isolate shed microvesicles from the cells of origin based on size discrimination using dielectrophoretic sorting. This method differentiates microvesicles from cells on the basis of their total membrane capacitance, which is in turn proportional to the total surface area of the cell/vesicle membrane (Shim et al. 2011). In addition to being a surface marker-free mechanism for sorting microvesicles, dielectrophoresis can be coupled to impedance measures, providing added benefit during the examination of biological samples (Zhiwei et al. 2008).
Microfluidic systems have also been developed that can sort microparticles based on affinity to specific ligands. Magnetophoretic sorting, for example, introduces ferromagnetic beads that have been functionalized by the addition of ligands, which can bind to and be used to isolate and capture specific microvesicle populations (Yao et al. 2010; Plouffe et al. 2012). Similarly, it is possible to functionalize the flow chamber itself, allowing for the capture and isolation of the target microvesicles containing the specific surface markers (Zheng et al. 2007). These microfluidic affinity chromatography devices often consist of glass and polymer chambers where the interior surfaces have been coated with antibodies, yielding a chamber that provides high surface area/volume ratios and short residence times (given the small total volume), with very high specificity in research isolating circulating tumor cells.
The development of multiplex platforms to enable the detection and capture of low-abundance microvesicles in body fluids has the potential to shift the paradigm of cancer care. The molecular stratification of tumors is often necessary in phase I and phase II clinical trials for potential therapeutics. Furthermore, frequent cataloging of readouts of the tumor's response or resistance to these agents is also required. While tumor imaging remains a useful tool, new and improved methods—especially ones that allow for the rapid and sequential evaluations of serologic biomarkers or those present in other body fluids, such as urine, ascites, spinal fluid, or saliva—could be transformative. Whether TMVs represent such a biomarker still requires intense investigation, but emerging evidence suggests that strategies for TMV identification, isolation, and capture will have marked implications in cancer diagnostics and therapeutics.
Concluding remarks
TMVs are packaged with abundant genetic and cellular information that can markedly impact the tumor microenvironment and, as a result, disease progression. Further investigation into the mechanisms by which cargo is selectively packaged into TMVs and those by which TMVs mediate communication between the tumor and the extracellular milieu will bolster current understanding of how tumor cells have adopted a rather specialized and sophisticated mode to spread and metastasize. Enhanced knowledge of TMV biogenesis could also determine their efficacy as therapeutic targets. The presence of TMVs in body fluids has also heightened general interest in their potential as circulating biomarkers. Molecular profiling of these structures would not only provide potential biomarkers for diagnostic purposes, but the information could also develop into critical and highly effective platforms for personalized medicine. Of vital importance to these endeavors is the development of protocols to identify and isolate TMVs and distinguish them from other shed populations. While this review highlights recent developments on large microvesicles shed by tumor cells, further investigation along these lines will help to better define and characterize these vesicles and also appreciate their roles in disease progression.
Acknowledgments
We thank Dr. Jeffrey Schorey for critical reading of the manuscript. We gratefully acknowledge support on related research in the D'Souza-Schorey laboratory from the National Cancer Institute, the Indiana Clinical and Translational Sciences Institute, and the Advanced Diagnostics and Therapeutics Initiative at the University of Notre Dame.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.192351.112.
References
- Abrahams VM, Straszewski SL, Kamsteeg M, Hanczaruk B, Schwartz PE, Rutherford TJ, Mor G 2003. Epithelial ovarian cancer cells secrete functional Fas ligand. Cancer Res 63: 5573–5581 [PubMed] [Google Scholar]
- Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J 2008. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol 10: 619–624 [DOI] [PubMed] [Google Scholar]
- Al-Nedawi K, Meehan B, Kerbel RS, Allison AC, Rak J 2009a. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc Natl Acad Sci 106: 3794–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Nedawi K, Meehan B, Rak J 2009b. Microvesicles: Messengers and mediators of tumor progression. Cell Cycle 8: 2014–2018 [DOI] [PubMed] [Google Scholar]
- Andersen KB, Levinsen S, Svendsen WE, Okkels F 2009. A generalized theoretical model for ‘continuous particle separation in a microchannel having asymmetrically arranged multiple branches.’ Lab Chip 9: 1638–1639 [DOI] [PubMed] [Google Scholar]
- Andreola G, Rivoltini L, Castelli C, Huber V, Perego P, Deho P, Squarcina P, Accornero P, Lozupone F, Lugini L, et al. 2002. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med 195: 1303–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelucci A, D'Ascenzo S, Festuccia C, Gravina GL, Bologna M, Dolo V, Pavan A 2000. Vesicle-associated urokinase plasminogen activator promotes invasion in prostate cancer cell lines. Clin Exp Metastasis 18: 163–170 [DOI] [PubMed] [Google Scholar]
- Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L 2011. Treatment of HER2-positive breast cancer: Current status and future perspectives. Nat Rev Clin Oncol 9: 16–32 [DOI] [PubMed] [Google Scholar]
- Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO, Skog J 2011. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat Commun 2: 180 doi: 10.1038/ncomms1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baran J, Baj-Krzyworzeka M, Weglarczyk K, Szatanek R, Zembala M, Barbasz J, Czupryna A, Szczepanik A, Zembala M 2009. Circulating tumour-derived microvesicles in plasma of gastric cancer patients. Cancer Immunol Immunother 59: 841–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsmedh A, Szeles A, Henriksson M, Bratt A, Folkman MJ, Spetz AL, Holmgren L 2001. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc Natl Acad Sci 98: 6407–6411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnig G, Quate CF, Gerber C 1986. Atomic force microscope. Phys Rev Lett 56: 930–933 [DOI] [PubMed] [Google Scholar]
- Bobrie A, Colombo M, Raposo G, Thery C 2009. Exosome secretion: Molecular mechanisms and roles in immune responses. Traffic 12: 1659–1668 [DOI] [PubMed] [Google Scholar]
- Brill A, Dashevsky O, Rivo J, Gozal Y, Varon D 2005. Platelet-derived microparticles induce angiogenesis and stimulate post-ischemic revascularization. Cardiovasc Res 67: 30–38 [DOI] [PubMed] [Google Scholar]
- Bucki R, Janmey PA, Vegners R, Giraud F, Sulpice JC 2001. Involvement of phosphatidylinositol 4,5-bisphosphate in phosphatidylserine exposure in platelets: Use of a permeant phosphoinositide-binding peptide. Biochemistry 40: 15752–15761 [DOI] [PubMed] [Google Scholar]
- Buehler MF, Allen TM, Davis EJ 1991. Microparticle raman spectroscopy of multicomponent aerosols. J Colloid Interface Sci 146: 79–89 [Google Scholar]
- Carmeliet P 2005. Angiogenesis in life, disease and medicine. Nature 438: 932–936 [DOI] [PubMed] [Google Scholar]
- Charras GT, Hu C-K, Coughlin M, Mitchison TJ 2006. Reassembly of contractile actin cortex in cell blebs. J Cell Biol 175: 477–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian P, Hankey GJ, Eikelboom JW, Thom J, Baker RI, McQuillan A, Staton J, Yi Q 2003. Endothelial and platelet activation in acute ischemic stroke and its etiological subtypes. Stroke 34: 2132–2137 [DOI] [PubMed] [Google Scholar]
- Choi DS, Park JO, Jang SC, Yoon YJ, Jung JW, Choi DY, Kim JW, Kang JS, Park J, Hwang D, et al. 2011. Proteomic analysis of microvesicles derived from human colorectal cancer ascites. Proteomics 11: 2745–2751 [DOI] [PubMed] [Google Scholar]
- Cocucci E, Racchetti G, Meldolesi J 2009. Shedding microvesicles: Artefacts no more. Trends Cell Biol 19: 43–51 [DOI] [PubMed] [Google Scholar]
- Combes V, Taylor TE, Juhan-Vague I, Mège JL, Mwenechanya J, Tembo M, Grau GE, Molyneux ME 2004. Circulating endothelial microparticles in Malawian children with severe falciparum malaria complicated with coma. JAMA 291: 2542–2544 [DOI] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA 2009. The impact of retrotransposons on human genome evolution. Nat Rev Genet 10: 691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce CM 2009. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 10: 704–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA 2005. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 106: 1604–1611 [DOI] [PubMed] [Google Scholar]
- Dieckmann Y, Colfen H, Hofmann H, Petri-Fink A 2009. Particle size distribution measurements of manganese-doped ZnS nanoparticles. Anal Chem 81: 3889–3895 [DOI] [PubMed] [Google Scholar]
- Di Vizio D, Kim J, Hager MH, Morello M, Yang W, Lafargue CJ, True LD, Rubin MA, Adam RM, Beroukhim R, et al. 2009. Oncosome formation in prostate cancer: Association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res 69: 5601–5609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doeuvre L, Plawinski L, Toti F, Anglés-Cano E 2009. Cell-derived microparticles: A new challenge in neuroscience. J Neurochem 110: 457–468 [DOI] [PubMed] [Google Scholar]
- Donaldson JG 2003. Multiple roles for Arf6: Sorting, structuring, and signaling at the plasma membrane. J Biol Chem 278: 41573–41576 [DOI] [PubMed] [Google Scholar]
- Dragovic RA, Gardiner C, Brooks AS, Tannetta DS, Ferguson DJ, Hole P, Carr B, Redman CW, Harris AL, Dobson PJ, et al. 2011. Sizing and phenotyping of cellular vesicles using nanoparticle tracking analysis. Nanomedicine 7: 780–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza-Schorey C, Chavrier P 2006. ARF proteins: Roles in membrane traffic and beyond. Nat Rev Mol Cell Biol 7: 347–358 [DOI] [PubMed] [Google Scholar]
- Eken C, Gasser O, Zenhaeusern G, Oehri I, Hess C, Schifferli JA 2008. Polymorphonuclear neutrophil-derived ectosomes interfere with the maturation of monocyte-derived dendritic cells. J Immunol 180: 817–824 [DOI] [PubMed] [Google Scholar]
- Flanagan J, Middeldorp J, Sculley T 2003. Localization of the Epstein-Barr virus protein LMP 1 to exosomes. J Gen Virol 84: 1871–1879 [DOI] [PubMed] [Google Scholar]
- Fleissner F, Goerzig Y, Haverich A, Thum T 2012. Microvesicles as novel biomarkers and therapeutic targets in transplantation medicine. Am J Transplant 12: 289–297 [DOI] [PubMed] [Google Scholar]
- Friedl P, Wolf K 2003. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat Rev Cancer 3: 362–374 [DOI] [PubMed] [Google Scholar]
- Friedl P, Wolf K 2010. Plasticity of cell migration: A multiscale tuning model. J Cell Biol 188: 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friend C, Marovitz W, Henie G, Henie W, Tsuei D, Hirschhorn K, Holland JG, Cuttner J 1978. Observations on cell lines derived from a patient with Hodgkin's disease. Cancer Res 38: 2581–2591 [PubMed] [Google Scholar]
- Gadea G, de Toledo M, Anguille C, Roux P 2007. Loss of p53 promotes Rho–ROCK-dependent cell migration and invasion in 3D matrices. J Cell Biol 178: 23–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser O, Hess C, Miot S, Deon C, Sanchez JC, Schifferli JA 2003. Characterisation and properties of ectosomes released by human polymorphonuclear neutrophils. Exp Cell Res 285: 243–257 [DOI] [PubMed] [Google Scholar]
- Gelderman MP, Simak J 2008. Flow cytometric analysis of cell membrane microparticles. Methods Mol Biol 484: 79–93 [DOI] [PubMed] [Google Scholar]
- Ghiran I, Kuo WP 2010. Examining the role of microvesicles to develop prognostic and diagnostic assays. J Appl Oral Sci 18: i doi: org/10.1590/S1678-77572010000500001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Secreto CR, Knox TR, Ding W, Mukhopadhyay D, Kay NE 2010. Circulating microvesicles in B-cell chronic lymphocytic leukemia can stimulate marrow stromal cells: Implications for disease progression. Blood 115: 1755–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb EA, Brown CJ, Lam WL 2011. The functional role of long non-coding RNA in human carcinomas. Mol Cancer 10: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebel B, Wodarz A 2006. Tumor suppressors: Control of signaling by endocytosis. Curr Biol 16: R91–R92 doi: 10.1016/j.cub.2006.01.022 [DOI] [PubMed] [Google Scholar]
- Ginestra A, La Placa MD, Saladino F, Cassara D, Nagase H, Vittorelli ML 1998. The amount and proteolytic content of vesicles shed by human cancer cell lines correlates with their in vitro invasiveness. Anticancer Res 18: 3433–3437 [PubMed] [Google Scholar]
- Giusti I, D'Ascenzo S, Millimaggi D, Taraboletti G, Carta G, Franceschini N, Pavan A, Dolo V 2008. Cathepsin B mediates the pH-dependent proinvasive activity of tumor-shed microvesicles. Neoplasia 10: 481–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC, Tetta C, Bussolati B, Camussi G 2011. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res 71: 5346–5356 [DOI] [PubMed] [Google Scholar]
- Hakulinen J, Sankkila L, Sugiyama N, Lehti K, Keski-Oja J 2008. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J Cell Biochem 105: 1211–1218 [DOI] [PubMed] [Google Scholar]
- Hayer A, Stoeber M, Ritz D, Engel S, Meyer HH, Helenius A 2010. Caveolin-1 is ubiquitinated and targeted to intralumenal vesicles in endolysosomes for degradation. J Cell Biol 191: 615–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera MB, Fonsato V, Gatti S, Deregibus MC, Sordi A, Cantarella D, Calogero R, Bussolati B, Tetta C, Camussi G 2010. Human liver stem cell-derived microvesicles accelerate hepatic regeneration in hepatectomized rats. J Cell Mol Med 14: 1605–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt CE, Bullock SL 2009. Subcellular mRNA localization in animal cells and why it matters. Science 326: 1212–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda A, Nogami M, Yokozeki T, Yamazaki M, Nakamura H, Watanabe H, Kawamoto K, Nakayama K, Morris AJ, Frohman MA, et al. 1999. Phosphatidylinositol 4-phosphate 5-kinase α is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell 99: 521–532 [DOI] [PubMed] [Google Scholar]
- Hong BS, Cho JH, Kim H, Choi EJ, Rho S, Kim J, Kim JH, Choi DS, Kim YK, Hwang D, et al. 2009. Colorectal cancer cell-derived microvesicles are enriched in cell cycle-related mRNAs that promote proliferation of endothelial cells. BMC Genomics 10: 556 doi: 10.1186/1471-2164-10-556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoo C, Starostin N, West P, Mecartney M 2008. A comparison of atomic force microscopy (AFM) and dynamic light scattering (DLS) methods to characterize nanoparticle size distributions. J Nanopart Res 10: 89–96 [Google Scholar]
- Huang LR, Cox EC, Austin RH, Sturm JC 2004. Continuous particle separation through deterministic lateral displacement. Science 304: 987–990 [DOI] [PubMed] [Google Scholar]
- Huber V, Fais S, Iero M, Lugini L, Canese P, Squarcina P, Zaccheddu A, Colone M, Arancia G, Gentile M, et al. 2005. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: Role in immune escape. Gastroenterology 128: 1796–1804 [DOI] [PubMed] [Google Scholar]
- Hung MC, Lau YK 1999. Basic science of HER-2/neu: A review. Semin Oncol 26: 51–59 [PubMed] [Google Scholar]
- Jiang A, Lehti K, Wang X, Weiss SJ, Keski-Oja J, Pei D 2001. Regulation of membrane-type matrix metalloproteinase 1 activity by dynamin-mediated endocytosis. Proc Nat Acad Sci 98: 13693–13698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladavac K, Kasza K, Grier DG 2004. Sorting mesoscopic objects with periodic potential landscapes: Optical fractionation. Phys Rev E Stat Nonlin Soft Matter Phys 70: 010901 doi: 10.1103/PhysRevE.70.010901 [DOI] [PubMed] [Google Scholar]
- Lawrie AS, Albanyan A, Cardigan RA, Mackie IJ, Harrison P 2009. Microparticle sizing by dynamic light scattering in fresh-frozen plasma. Vox Sang 96: 206–212 [DOI] [PubMed] [Google Scholar]
- Lee TH, D'Asti E, Magnus N, Al-Nedawi K, Meehan B, Rak J 2011. Microvesicles as mediators of intercellular communication in cancer—the emerging science of cellular ‘debris.’ Semin Immunopathol 33: 455–467 [DOI] [PubMed] [Google Scholar]
- Li B, Antonyak MA, Zhang J, Cerione RA 2012. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene doi: 10.1038/onc.2011.636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima LG, Chammas R, Monteiro RQ, Moreira ME, Barcinski MA 2009. Tumor-derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine-dependent manner. Cancer Lett 283: 168–175 [DOI] [PubMed] [Google Scholar]
- Losche W, Scholz T, Temmler U, Oberle V, Claus RA 2004. Platelet-derived microvesicles transfer tissue factor to monocytes but not to neutrophils. Platelets 15: 109–115 [DOI] [PubMed] [Google Scholar]
- Loutherback K, Chou K, Newman J, Puchalla J, Austin R, Sturm J 2010. Improved performance of deterministic lateral displacement arrays with triangular posts. Microfluidics and Nanofluidics 9: 1143–1149 [Google Scholar]
- MacDonald MP, Spalding GC, Dholakia K 2003. Microfluidic sorting in an optical lattice. Nature 426: 421–424 [DOI] [PubMed] [Google Scholar]
- Maguire CA, Balaj L, Sivaraman S, Crommentuijn MHW, Ericsson M, Mincheva-Nilsson L, Baranov V, Gianni D, Tannous BA, Sena-Esteves M, et al. 2012. Microvesicle-associated AAV vector as a novel gene delivery system. Mol Ther 20: 960–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H, Sakuma S, Yamanishi Y, Arai F 2010. Size-dependent filtration and trapping of microparticle in a microfluidic chip using centrifugal force and a graduated mechanical gap. J Robotics and Mechatronics 22: 280–285 [Google Scholar]
- Mause SF, Weber C 2010. Microparticles: Protagonists of a novel communication network for intercellular information exchange. Circ Res 107: 1047–1057 [DOI] [PubMed] [Google Scholar]
- Millimaggi D, Mari M, D'Ascenzo S, Carosa E, Jannini EA, Zucker S, Carta G, Pavan A, Dolo V 2007. Tumor vesicle-associated CD147 modulates the angiogenic capability of endothelial cells. Neoplasia 9: 349–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne G, Rhodes D, MacDonald M, Dholakia K 2007. Fractionation of polydisperse colloid with acousto-optically generated potential energy landscapes. Opt Lett 32: 1144–1146 [DOI] [PubMed] [Google Scholar]
- Mohamed H, McCurdy LD, Szarowski DH, Duva S, Turner JN, Caggana M 2004. Development of a rare cell fractionation device: Application for cancer detection. IEEE Trans Nanobioscience 3: 251–256 [DOI] [PubMed] [Google Scholar]
- Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G, D'Souza-Schorey C 2009. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol 19: 1875–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan-Chari V, Clancy JW, Sedgwick A, D'Souza-Schorey C 2010. Microvesicles: Mediators of extracellular communication during cancer progression. J Cell Sci 123: 1603–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel W 2005. Unconventional secretory routes: Direct protein export across the plasma membrane of mammalian cells. Traffic 6: 607–614 [DOI] [PubMed] [Google Scholar]
- Ohshima K, Inoue K, Fujiwara A, Hatakeyama K, Kanto K, Watanabe Y, Muramatsu K, Fukuda Y, Ogura S, Yamaguchi K, et al. 2010. Let-7 microRNA family is selectively secreted into the extracellular environment via exosomes in a metastatic gastric cancer cell line. PLoS ONE 5: e13247 doi: 10.1371/journal.pone.0013247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondrej V, Lukasova E, Falk M, Kozubek S 2007. The role of actin and microtubule networks in plasmid DNA intracellular trafficking. Acta Biochim Pol 54: 657–663 [PubMed] [Google Scholar]
- Paluch E, Sykes C, Prost J, Bornens M 2006. Dynamic modes of the cortical actomyosin gel during cell locomotion and division. Trends Cell Biol 16: 5–10 [DOI] [PubMed] [Google Scholar]
- Pawelczyk M, Baj Z, Chmielewski H, Kaczorowska B, Klimek A 2009. The influence of hyperlipidemia on platelet activity markers in patients after ischemic stroke. Cerebrovasc Dis 27: 131–137 [DOI] [PubMed] [Google Scholar]
- Peng J, Wallar BJ, Flanders A, Swiatek PJ, Alberts AS 2003. Disruption of the diaphanous-related formin Drf1 gene encoding mDia1 reveals a role for Drf3 as an effector for Cdc42. Curr Biol 13: 534–545 [DOI] [PubMed] [Google Scholar]
- Pilzer D, Gasser O, Moskovich O, Schifferli JA, Fishelson Z 2005. Emission of membrane vesicles: Roles in complement resistance, immunity and cancer. Springer Semin Immunopathol 27: 375–387 [DOI] [PubMed] [Google Scholar]
- Pinner S, Sahai E 2008. PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat Cell Biol 10: 127–137 [DOI] [PubMed] [Google Scholar]
- Plouffe BD, Mahalanabis M, Lewis LH, Klapperich CM, Murthy SK 2012. Clinically relevant microfluidic magnetophoretic isolation of rare-cell populations for diagnostic and therapeutic monitoring applications. Anal Chem 84: 1336–1344 [DOI] [PubMed] [Google Scholar]
- Pluskota E, Woody NM, Szpak D, Ballantyne CM, Soloviev DA, Simon DI, Plow EF 2008. Expression, activation, and function of integrin αMβ2 (Mac-1) on neutrophil-derived microparticles. Blood 112: 2327–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poste G, Nicolson GL 1980. Arrest and metastasis of blood-borne tumor cells are modified by fusion of plasma membrane vesicles from highly metastatic cells. Proc Natl Acad Sci 77: 399–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pully VV, Lenferink ATM, Otto C 2011. Time-lapse Raman imaging of single live lymphocytes. J Raman Spectrosc 42: 167–173 [Google Scholar]
- Rabinowits G, Gercel-Taylor C, Day JM, Taylor DD, Kloecker GH 2009. Exosomal microRNA: A diagnostic marker for lung cancer. Clin Lung Cancer 10: 42–46 [DOI] [PubMed] [Google Scholar]
- Radisic M, Iyer RK, Murthy SK 2006. Micro- and nanotechnology in cell separation. Int J Nanomedicine 1: 3–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, Ratajczak MZ 2006a. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: Evidence for horizontal transfer of mRNA and protein delivery. Leukemia 20: 847–856 [DOI] [PubMed] [Google Scholar]
- Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ 2006b. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 20: 1487–1495 [DOI] [PubMed] [Google Scholar]
- Robert S, Poncelet P, Lacroix R, Arnaud L, Giraudo L, Hauchard A, Sampol J, Dignat-George F 2009. Standardization of platelet-derived microparticle counting using calibrated beads and a Cytomics FC500 routine flow cytometer: A first step towards multicenter studies? J Thromb Haemost 7: 190–197 [DOI] [PubMed] [Google Scholar]
- Rood IM, Deegens JK, Merchant ML, Tamboer WP, Wilkey DW, Wetzels JF, Klein JB 2010. Comparison of three methods for isolation of urinary microvesicles to identify biomarkers of nephrotic syndrome. Kidney Int 78: 810–816 [DOI] [PubMed] [Google Scholar]
- Sabeh F, Shimizu-Hirota R, Weiss SJ 2009. Protease-dependent versus -independent cancer cell invasion programs: Three-dimensional amoeboid movement revisited. J Cell Biol 185: 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Marshall CJ 2003. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol 5: 711–719 [DOI] [PubMed] [Google Scholar]
- Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ 2008. Rac activation and inactivation control plasticity of tumor cell movement. Cell 135: 510–523 [DOI] [PubMed] [Google Scholar]
- Saraste A, Pulkki K 2000. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res 45: 528–537 [DOI] [PubMed] [Google Scholar]
- Schorey JS, Bhatnagar S 2008. Exosome function: From tumor immunology to pathogen biology. Traffic 9: 871–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scita G, Di Fiore PP 2010. The endocytic matrix. Nature 463: 464–473 [DOI] [PubMed] [Google Scholar]
- Shim S, Gascoyne P, Noshari J, Stemke Hale K 2011. Dynamic physical properties of dissociated tumor cells revealed by dielectrophoretic field-flow fractionation. Integr Biol 3: 850–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siedlecki CA, Wen Wang I, Higashi JM, Kottke-Marchant K, Marchant RE 1999. Platelet-derived microparticles on synthetic surfaces observed by atomic force microscopy and fluorescence microscopy. Biomaterials 20: 1521–1529 [DOI] [PubMed] [Google Scholar]
- Simons M, Raposo G 2009. Exosomes–vesicular carriers for intercellular communication. Curr Opin Cell Biol 21: 575–581 [DOI] [PubMed] [Google Scholar]
- Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky AM, Breakefield XO 2008. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 10: 1470–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RL, Spalding GC, Dholakia K, MacDonald MP 2007. Colloidal sorting in dynamic optical lattices. J Opt 9: S134 doi: 10.1088/1464-4258/9/8/S05 [Google Scholar]
- Starostina N, Brodsky M, Prikhodko S, Hoo CM, Mecartney ML, West P 2008. AFM capabilities in characterization of particles and surfaces: From angstroms to microns. J Cosmet Sci 59: 225–232 [PubMed] [Google Scholar]
- Takagi J, Yamada M, Yasuda M, Seki M 2005. Continuous particle separation in a microchannel having asymmetrically arranged multiple branches. Lab Chip 5: 778–784 [DOI] [PubMed] [Google Scholar]
- Tang DG, Conti CJ 2004. Endothelial cell development, vasculogenesis, angiogenesis, and tumor neovascularization: An update. Semin Thromb Hemost 30: 109–117 [DOI] [PubMed] [Google Scholar]
- Taraboletti G, D'Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V 2002. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am J Pathol 160: 673–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraboletti G, D'Ascenzo S, Giusti I, Marchetti D, Borsotti P, Millimaggi D, Giavazzi R, Pavan A, Dolo V 2006. Bioavailability of VEGF in tumor-shed vesicles depends on vesicle burst induced by acidic pH. Neoplasia 8: 96–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery C, Amigorena S, Raposo G, Clayton A 2006. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol 30: 3.22.1–3.22.29 doi: 10.1002/0471143030.cb0322s30 [DOI] [PubMed] [Google Scholar]
- Thery C, Ostrowski M, Segura E 2009. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 9: 581–593 [DOI] [PubMed] [Google Scholar]
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO 2007. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9: 654–659 [DOI] [PubMed] [Google Scholar]
- Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, Corbelli A, Fais S, Parmiani G, Rivoltini L 2006. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-β-mediated suppressive activity on T lymphocytes. Cancer Res 66: 9290–9298 [DOI] [PubMed] [Google Scholar]
- Valenti R, Huber V, Iero M, Filipazzi P, Parmiani G, Rivoltini L 2007. Tumor-released microvesicles as vehicles of immunosuppression. Cancer Res 67: 2912–2915 [DOI] [PubMed] [Google Scholar]
- van der Pol E, Hoekstra AG, Sturk A, Otto C, Van Leeuwen TG, Nieuwland R 2010. Optical and non-optical methods for detection and characterization of microparticles and exosomes. J Thromb Haemost 8: 2596–2607 [DOI] [PubMed] [Google Scholar]
- van Doormaal FF, Kleinjan A, Di Nisio M, Buller HR, Nieuwland R 2009. Cell-derived microvesicles and cancer. Neth J Med 67: 266–273 [PubMed] [Google Scholar]
- Wysoczynski M, Ratajczak MZ 2009. Lung cancer secreted microvesicles: Underappreciated modulators of microenvironment in expanding tumors. Int J Cancer 125: 1595–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Bai O, Yuan J, Chibbar R, Slattery K, Wei Y, Deng Y, Xiang J 2009. Tumor apoptotic bodies inhibit CTL responses and antitumor immunity via membrane-bound transforming growth factor-β1 inducing CD8+ T-cell anergy and CD4+ Tr1 cell responses. Cancer Res 69: 7756–7766 [DOI] [PubMed] [Google Scholar]
- Yamada M, Seki M 2006. Microfluidic particle sorter employing flow splitting and recombining. Anal Chem 78: 1357–1362 [DOI] [PubMed] [Google Scholar]
- Yao Z, Yi W, Qiao L 2010. A microfluidic device for continuous-flow magnetically controlled capture and isolation of microparticles. J Microelectromech Syst 19: 743–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuana Y, Oosterkamp TH, Bahatyrova S, Ashcroft B, Garcia Rodriguez P, Bertina RM, Osanto S 2010. Atomic force microscopy: A novel approach to the detection of nanosized blood microparticles. J Thromb Haemost 8: 315–323 [DOI] [PubMed] [Google Scholar]
- Zheng T, Yu H, Alexander C, Beebe D, Smith L 2007. Lectin-modified microchannels for mammalian cell capture and purification. Biomed Microdevices 9: 611–617 [DOI] [PubMed] [Google Scholar]
- Zhiwei Z, Soohyun L, Ahn CH 2008. A polymer microfluidic chip with interdigitated electrodes arrays for simultaneous dielectrophoretic manipulation and impedimetric detection of microparticles. IEEE Sens J 8: 527–535 [Google Scholar]
- Zhou Z 2007. New phosphatidylserine receptors: Clearance of apoptotic cells and more. Dev Cell 13: 759–760 [DOI] [PubMed] [Google Scholar]
- Zwicker JI, Liebman HA, Neuberg D, Lacroix R, Bauer KA, Furie BC, Furie B 2009. Tumor-derived tissue factor-bearing microparticles are associated with venous thromboembolic events in malignancy. Clin Cancer Res 15: 6830–6840 [DOI] [PMC free article] [PubMed] [Google Scholar]