

Abstract
Glutamatergic synapses are located mostly on dendritic spines in the adult nervous system. The spines serve as postsynaptic compartments, containing components that mediate and control the synaptic signal. Early in development, when glutamatergic synapses are initially forming, waves of excitatory activity pass through many parts of the nervous system and are driven in part by a class of heteropentameric β2-containing nicotinic acetylcholine receptors (β2*-nAChRs). These β2*-nAChRs are widely distributed and, when activated, can depolarize the membrane and elevate intracellular calcium levels in neurons. We show here that β2*-nAChRs are essential for acquisition of normal numbers of dendritic spines during development. Mice constitutively lacking the β2-nAChR gene have fewer dendritic spines than do age-matched wild-type mice at all times examined. Activation of β2*-nAChRs by nicotine either in vivo or in organotypic slice culture quickly elevates the number of spines. RNA interference studies both in vivo and in organotypic culture demonstrate that the β2*-nAChRs act in a cell-autonomous manner to increase the number of spines. The increase depends on intracellular calcium and activation of calcium, calmodulin-dependent protein kinase II. Absence of β2*-nAChRs in vivo causes a disproportionate number of glutamatergic synapses to be localized on dendritic shafts, rather than on spines as occurs in wild type. This shift in synapse location is found both in the hippocampus and cortex, indicating the breadth of the effect. Because spine synapses differ from shaft synapses in their signaling capabilities, the shift observed is likely to have significant consequences for network function.
Introduction
Dendritic spines provide critical postsynaptic compartments for excitatory glutamatergic transmission in the mammalian brain. The spines facilitate throughput of the excitatory signal to the dendritic trunk, while at the same time constraining features, such as local calcium influx, to mediate synapse-specific effects (Fiala et al., 1998; Yuste et al., 1999; Sheng and Hoogenraad, 2007; Yuste, 2011). Numerous molecular mechanisms have been shown to promote the formation, localization, and stabilization of dendritic spines (Tolias et al., 2005; Xie et al., 2007; Lai and Ip, 2009; Tran et al., 2009; Han et al., 2010; Misra et al., 2010). Recently, it has been reported that adult mice lacking a major class of nicotinic acetylcholine receptors (nAChRs), namely β2-containing heteropentameric receptors (β2*-nAChRs), have altered numbers of dendritic spines in a variety of cortical regions in brain (Ballesteros-Yáñez et al., 2010). How, when, and where β2*-nAChRs exert these effects are unknown.
Spine synapses form most rapidly and reach adult levels along dendrites during the first 2 weeks of postnatal life (Fiala et al., 1998; Hennou et al., 2002). This is a time when nAChRs, including β2*-nAChRs, reach their highest relative levels (Zhang et al., 1998; Adams et al., 2002) and help drive spontaneous waves of excitation through many regions of the CNS (Bansal et al., 2000; Hanson and Landmesser, 2003; Le Magueresse et al., 2006). The ability of β2*-nAChRs to elevate calcium in neurons provides a possible mechanism for influencing a variety of features, including spine formation (Dajas-Bailador and Wonnacott, 2004; Albuquerque et al., 2009).
We show here that activation of β2*-nAChRs by nicotine quickly induces dendritic spines in a cell-autonomous manner. The induction is calcium dependent and requires functional calcium, calmodulin-dependent protein kinase II (CaMKII). Mice constitutively lacking the β2-nAChR gene (β2KO mice) still form substantial numbers of glutamatergic synapses but acquire an abnormally high proportion of them on dendritic shafts. The spine deficit and corresponding increase in shaft synapses occurs both in the hippocampus and in the cortex, demonstrating the breadth of the effect. Moreover, the changes persist into the adult, indicating that the animal is unable to compensate for the deficit in spine number resulting from the absence of β2*-nAChR signaling. This shift in the distribution of synapses would be expected to have significant effects on network signaling and synaptic plasticity and may account in part for behavioral deficits found in β2KO mice (Picciotto et al., 1995; Levin et al., 2009; Wang et al., 2009).
Materials and Methods
General methods.
Origin of mouse mutant lines, preparation of mouse hippocampal slice cultures and rat hippocampal cell cultures, viral injections, perfusion-fixation to obtain tissue slices, immunohistochemistry, and production and use of short-hairpin RNA (shRNA) constructs for RNA interference (RNAi) were performed as described previously (Lozada et al., 2012). For perfusion-fixation, animals were anesthetized and perfused either with ice-cold or with room-temperature 4% paraformaldehyde; postfixation was performed with 4% paraformaldehyde for 1–2 h. Sindbis viral injection (generous gift from G. Patrick, University of California, San Diego, La Jolla, CA) was performed only 18 h before perfusion-fixation in all cases. For analysis at postnatal day 4 (P4) and P12, the sindbis injections were performed as described previously (Lozada et al., 2012). For analysis at P25 and P40, mice were placed in a Kopf 1430 stereotaxic frame (David Kopf Instruments) and given injections with a microsyringe (5 μl at a rate of 1 μl per minute; Hamilton Company) at the following coordinates: anteroposterior, −2.3; lateral, ±1.5; DV, 1.4. Animals of either sex were used in all experiments.
Fluorescence imaging.
Fluorescence images from brain slices and slice cultures were obtained with an SP5 confocal microscope (Leica) and analyzed by ImageJ. Images from cell and slice cultures were obtained with an Axiovert S100 TV microscope (Zeiss) and Slidebook software (Intelligent, Imaging, Innovations) applied to vertical stacks of contiguous optical sections. Immunostaining, collection of images for slices and cell culture, and subsequent analyses were performed as described previously (Lozada et al., 2012). A mouse anti-drebrin antibody (1:500; clone M2F6; Medical and Biological Laboratories), detected with a donkey anti-mouse antibody (1:500; FITC; Jackson ImmunoResearch Laboratories), was used as an additional method to quantify spines in cell culture. To visualize spines in slice cultures and in hippocampal and cortical slices from perfusion-fixed animals, green fluorescent protein (GFP) was introduced in vivo via lenti- or sindbis-viral constructs. Results were equivalent in the two cases and therefore combined. Some experiments used Thy-1M-GFP mice, which express GFP in hippocampal pyramidal neurons. For CA1 pyramidal neurons in slices, spine counts were performed on the medial portion of the apical dendrite (40–100 μm from the cell body); for layer 5/6 pyramidal neurons in the visual cortex, spine counts were performed on apical dendrites traversing layer 5. In cell culture, spines were counted along 10–20 μm sections of primary and secondary apical dendrites, avoiding areas within 50 μm of the soma. Spine identity and linkage to dendrite were confirmed by systematic analysis of vertical stacks composed of 20–30 optical sections taken at 0.5 μm intervals. Absolute values for spine density varied with temperature of fixation (Kirov et al., 2004), but all experiments included controls and experimentals prepared, fixed, processed, and imaged in the same way so that the results could be directly compared within the experiment. Labeled puncta on spines and shafts were defined as areas containing at least four contiguous pixels with staining above background. Backgrounds were set at 3 SDs above the mean staining intensity of six randomly selected regions. Puncta touching both a shaft and a spine were assigned a spine location. Colocalization of two kinds of puncta required that they have at least 1 pixel in common as revealed by ImageJ applied to the three collapsed optical sections for slice images or as revealed by Slidebook for cell culture images. Quantifications were performed blind to condition.
FM4-64 labeling.
FM4-64 staining was performed in cell culture after successive washes to remove nicotine, incubating 1 h in 50 nm methyllycaconitine (MLA) to block α7-nAChRs and 1 μm dihydro-β-erythroidine (DHβE) to block β2*-nAChRs and prevent any residual nicotinic activity, and then staining with DiI and incubating 60 s with 5 μm FM4-64 in the absence or presence of 50 mm KCl before rinsing and imaging as described previously (Pyle et al., 1999; Lozada et al., 2012). Alternatively, spines were visualized in culture by infecting cells with lenti-GFP 5 d before analysis. Counts of FM4-64-labeled puncta along 20 μm segments of dendrites and spines were performed blind to condition. Puncta abutting any portion of a spine were scored as spine-like synapses; puncta contacting the surface of a dendritic shaft but not a spine were scored as dendritic shaft synapses. Puncta lying centrally over a dendritic shaft were not scored.
Statistical analysis.
Data represent means ± SEMs. Unless otherwise indicated, statistical significance was assessed with the Student's t test for unpaired values and with one-way ANOVA followed by the Bonferroni post hoc test for multiple values. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Results
Nicotine induction of dendritic spines via β2*-nAChRs
To test the effects of nicotinic activity on spine number, we first used organotypic cultures prepared from mouse hippocampus. Slices obtained at P0–P1 were maintained in culture for 5 d, with drugs being applied for the last 4 d. This corresponds to a time in vivo when pyramidal neurons in the hippocampus begin to generate substantial numbers of dendritic spines (Fiala et al., 1998). The cultures had little, if any, endogenous cholinergic input because they lacked the septum, which provides most, if not all, of the cholinergic innervation of the hippocampus in vivo (Lewis et al., 1967). Cultures were treated with low levels of nicotine (0.5–1 μm), concentrations equivalent to peak serum levels in smokers (Matta et al., 2007). After 4–6 d, the slices were infected with sindbis–GFP to visualize spines and analyzed 10 h later. Quantification indicated a significant increase in pyramidal spines in the CA1 region (Fig. 1A,B). The increase did not require activation of the other major class of nAChRs, homopentamers containing α7 subunits (α7-nAChRs); the specific α7-nAChR antagonist MLA had no effect, and slices from mice lacking α7-nAChRs (α7KO mice) responded to nicotine in full. The increase did depend on β2*-nAChRs, however, because the effect was blocked by DHβE, and nicotine could not increase the number of spines in β2KO slices. [The finding that spine numbers in β2KO slices were no lower than those in the untreated control wild-type (WT) slices is consistent with these organotypic cultures having little, if any, endogenous nicotinic cholinergic activity.] Morphological analysis of the projections using previously described criteria (Tyler and Pozzo-Miller, 2003) indicated that the nicotinic effect was confined to spines, principally type III spines that displayed a 260 ± 30% increase (p ≤ 0.05; n = 12 cells, 3 cultures per condition) compared with controls. A nominal increase was seen in type I spines; no change occurred in type II spines or filopodia. The results clearly indicate that stimulation of β2*nAChRs, but not α7-nAChRs, increases spine numbers.
Figure 1.

Nicotinic stimulation of β2*-nAChRs increases dendritic spine numbers on pyramidal neurons. A, P0–P1 mouse hippocampal slices in culture 5 d with the indicated compounds for the last 4 d, infected with sindbis–GFP the last 10 h before fixation, and imaged. Scale bar, 5 μm. B, Quantification of spines. Con, Control, no additives; Nic, nicotine, 0.5 or 1 μm; MLA, 50 nm MLA (block α7-nAChRs); DHβE, 10 μm DHβE (block β2*-nAChRs); α7KO, α7KO slices instead of wild types; β2KO, β2KO slices instead of wild types. C, Slices as in A but treated with the indicated compounds only for the last hour before imaging. D, Quantification of spines after the 1 h treatment with the indicated compounds. Nic, 1 μm nicotine; Blkrs, 50 μm APV, 20 μm NBQX, 20 μm gabazine, 0.5 mm phaclofen, and 250 μm (RS)-MCPG for NMDA, AMPA, GABAA, GABAB, and metabotropic glutamate receptors, respectively; TTX, 1 μm TTX; Bapta, 100 μm BAPTA-AM; KN62, 10 μm KN62; KN93, 10 μm; KN92, 10 μm; Con, no additives. E, Absence of effects on spine number by the indicated antagonists applied for 1 h as in D but in the absence of nicotine, compared with control and nicotine. F, Reversibility: incubation with 1 μm nicotine for the last hour (1 h) or for 4 d (4d) in the absence or presence of MLA, then washed 1 h (1 h) or 4 h in MLA and DHβE (4 h plus blkrs) before counting spines (3–5 cells per culture; 6 cultures per condition).
The nicotinic effect was rapid and prominent for developing neurons. Incubating the slice cultures with 1 μm nicotine for only the last hour of the culture period induced the full increase in spine numbers (Fig. 1C,D). The increase was not prevented by a battery of antagonists for ionotropic and metabotropic glutamate and GABA receptors or by tetrodotoxin (TTX) to block activity. It was blocked, however, by preincubating the slices with BAPTA-AM, indicating that intracellular calcium was required. Furthermore, CaMKII seemed to be the target of the calcium signaling because the specific CaMKII blockers KN62 and KN93 (but not the inactive analog KN92) each prevented the increase (Fig. 1D). The blockers alone had no effect on control levels of spines (Fig. 1E). The initial effect was reversible: when slices were treated with nicotine for 1 h and then rinsed for 1 hour, the increase in spine formation was abolished (Fig. 1F). When slices were treated with nicotine for 4 d, however, increased spine numbers remained 4 h after the nicotine was replaced with nAChR blockers. As in the case of the initial increase, this multihour stabilization of the increment was unaffected by MLA, indicating that α7-nAChR activation was not required.
Endogenous actions of β2*-nAChRs to drive spine formation in vivo
To determine whether endogenous nicotinic cholinergic signaling through β2*-nAChRs contributes to spine numbers in vivo, we compared spine numbers in WT mice with those in β2KOs, mice lacking the α7-nAChR gene (α7KOs), and mice lacking both β2- and α7-nAChR genes [double knockouts (dKOs)]. We visualized spines by intracranially injecting viral constructs expressing GFP. Spine numbers were quantified per unit length dendrite. We found that dendritic spines on pyramidal neurons both in the hippocampal CA1 region and layer 5/6 of the visual cortex were substantially reduced in number at P12 in β2KOs and dKOs compared with wild types; α7KOs, in contrast, were indistinguishable from wild types in this respect (Fig. 2A,B). Similar results were obtained when the labeling was achieved either by injecting a lentiviral construct (lenti-GFP) at P1 and examining it at P12, or by injecting a sindbis viral construct (sindbis–GFP) the night before brain fixation. The spine decrements could be detected as early as P4 and persisted as long as P40, the oldest time analyzed (Fig. 2C). The results demonstrate that the β2-nAChR gene product is necessary to generate and/or maintain normal numbers of dendritic spines and that compensation does not occur over this time frame despite the constitutive nature of the KO.
Figure 2.
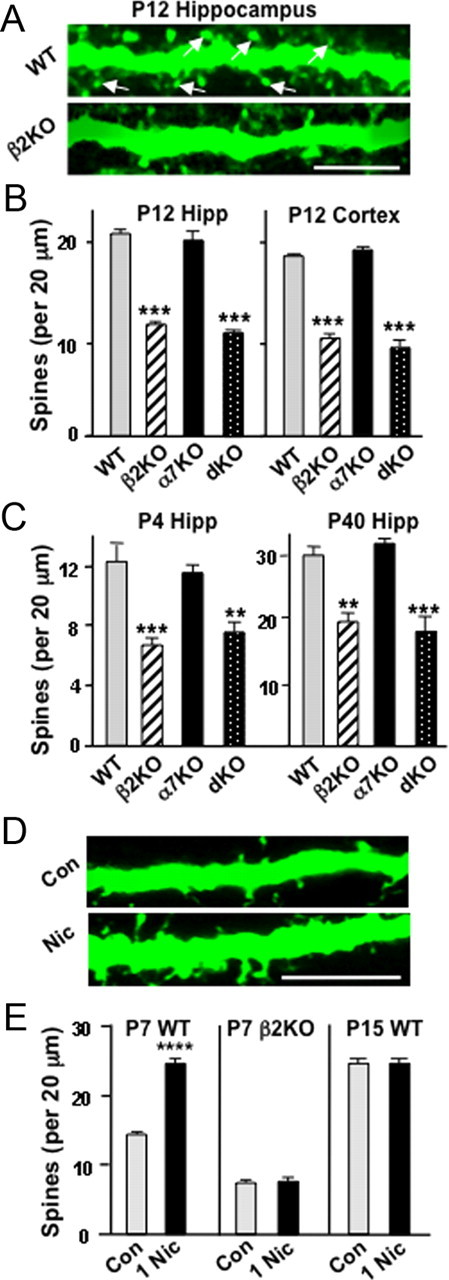
Role of β2*-nAChRs in supporting dendritic spines in vivo. A, Images showing dendrites of cells labeled by intracranially injecting lenti-GFP into WT (top) or β2KO (bottom) mice on P1–P2 and imaging fixed slices on P12 (arrows indicate examples of spines). Scale bar, 5 μm. B, Spine counts per 20 μm dendritic length of WT and KO pyramidal neurons in the CA1 (left, hippocampus) and layer 5/6 pyramidal neurons in the visual cortex (right, cortex). C, Spine counts along sindbis–GFP dendrites of CA1 pyramidal neurons at P4 or P40 (5–10 cells per animal; 3–5 animals per genotype). D, Images of dendrites from P7 Thy-1M-GFP mouse pups, which received stereotaxic intracranial hippocampal injections of PBS (Con) or 1 μm nicotine (Nic) and were perfusion fixed 1 h later. Scale bar, 10 μm. E, Quantification of spine numbers on CA1 pyramidal neuron apical dendrites for P7 WT Thy-1M-GFP, P7 β2KO (sindbis–GFP-labeled), and P15 WT Thy-1M-GFP mice given injections of nicotine (n = 3–5 animals, 4 cells per animal). Hipp, Hippocampus.
To test whether exogenous nicotine can elevate the number of spines in vivo over and above that produced by endogenous cholinergic input, we stereotaxically injected nicotine or vehicle directly into the hippocampus of P7 Thy-1M-GFP mouse pups, which express GFP in CA1 pyramidal neurons (Feng et al., 2000). One hour after receiving 1 μm nicotine, animals were perfusion fixed, and hippocampal slices were analyzed for spine incidence (Fig. 2D). The nicotine treatment caused a significant increase in the number of dendritic spines on CA1 pyramidal neurons in Thy-1M-GFP mice (Fig. 2E). No increase was observed with P7 β2KO mice compared with WT littermates (labeled by sindbis–GFP injection), consistent with the β2-nAChR gene being required for the nicotinic effect. No increase was seen with P15 Thy-1M-GFP mouse pups, although dye coinjection confirmed nicotine delivery to the appropriate location in all cases. The ability of nicotine to enhance spine numbers over and above that produced by endogenous nicotinic cholinergic activity may be confined to early postnatal times when neurons are most actively engaged in generating dendritic spines and forming synapses de novo (Fiala et al., 1998; Hennou et al., 2002).
RNAi demonstration of cell-autonomous β2*-nAChR actions
To determine whether the β2*-nAChR requirement is cell autonomous, we used RNAi using shRNAs. Mice were given injections intracranially on P1–P2 with lentiviral constructs (Adesnik et al., 2008) encoding shRNAs specific for mouse β2-nAChR transcripts (β2-RNAi) or a scrambled control RNA (β2-Scr) having the same composition but different sequence (Avale et al., 2008). We also included shRNAs for mouse α7-nAChR transcripts (α7-RNAi) and a scrambled sequence control (α7-Scr) and performed immunostaining at P12. Expression of β2-RNAi substantially reduced the number of dendritic spines on apical dendrites of pyramidal neurons in the hippocampal CA1 region (Fig. 3A,B). No decrement was caused by the α7-RNAi construct compared with scrambled controls. Both the α7-RNAi and β2-RNAi constructs have previously been shown to be effective (Avale et al., 2008; Campbell et al., 2010). The β2-RNAi was specific in that it produced no off-target effects as judged by expression in the cognate KO (Fig. 3C).
Figure 3.

RNAi demonstration that β2*-nAChRs act cell-autonomously to elevate spine numbers. A, Labeled dendrites of cells expressing either β2-Scr (top) or β2-RNAi (bottom), injected intracranially on P1–P2 and imaged on P12 (arrows, spine examples). Scale bar, 10 μm. B, Spine counts per 20 μm dendrite of CA1 pyramidal neurons expressing the indicated RNAi constructs (n = 3–8 animals, 4–16 neurons per animal). C, Quantification of spines per dendritic length in β2KOs expressing the indicated construct. D, P4 slices infected with viral vectors expressing β2-RNAi or scRNAi and maintained for 7 d in culture before treating 1 h with 1 μm nicotine (Nic) or buffer (Buf) and quantifying spines (3–5 cells per culture; 6 cultures per condition).
Similar results were obtained with nicotine treatment in slice culture. The β2-RNAi construct prevented the nicotine-induced increase in spine number, reducing spine levels to those seen in controls expressing β2-Scr and receiving vehicle instead of nicotine (Fig. 3D). The results demonstrate that β2*-nAChRs act in a cell-autonomous manner to increase spine number.
Blockade of β2*-nAChRs causes shift in synaptic location
The reduced numbers of dendritic spines seen in β2KOs and in nicotine-treated cultures when β2*-nAChRs are blocked strongly suggest that fewer spine synapses will be present under these conditions. Both the incidence of spontaneous synaptic activity and the number of glutamatergic synapses revealed by immunostaining, however, previously failed to show any decrements in β2KOs or in nicotine-treated cultures when β2*-nAChRs were pharmacologically inhibited (Lozada et al., 2012). These results raised the possibility that in the absence of β2*-nAChR signaling, neurons accept additional glutamatergic synapses onto their dendritic shafts, thereby compensating at least partially for a decrement in spines.
To test this possibility, we first examined mouse hippocampal slices in organotypic culture, distinguishing glutamatergic shaft versus spine synapses. Nicotine treatment increased the number of glutamatergic synapses both on spines and on shafts, as judged either by the increased numbers of VGluT and PSD-95 colocalized puncta (Fig. 4A,B) or by the similar increases for VGluT and GluR1 puncta (Fig. 4C,D). Immunostaining for presynaptic and postsynaptic markers in this manner has frequently been used to assess the number of glutamatergic synapses (Christopherson et al., 2005; Kayser et al., 2006; Stevens et al., 2007; Eroglu et al., 2009). Selective blockade of α7-nAChRs by MLA produced decrements in both classes of synapses, consistent with the overall decrements seen in α7KOs in vivo and in nicotine-treated cultures with α7-nAChRs blocked (Lozada et al., 2012). In contrast, selective blockade of β2*-nAChRs decreased the number of spine synapses in nicotine-treated cultures while increasing the number of shaft synapses (Fig. 4B,D). The results reveal a shift in the overall pattern, consistent with fewer spines being available for innervation when β2*-nAChRs are dysfunctional or blocked.
Figure 4.
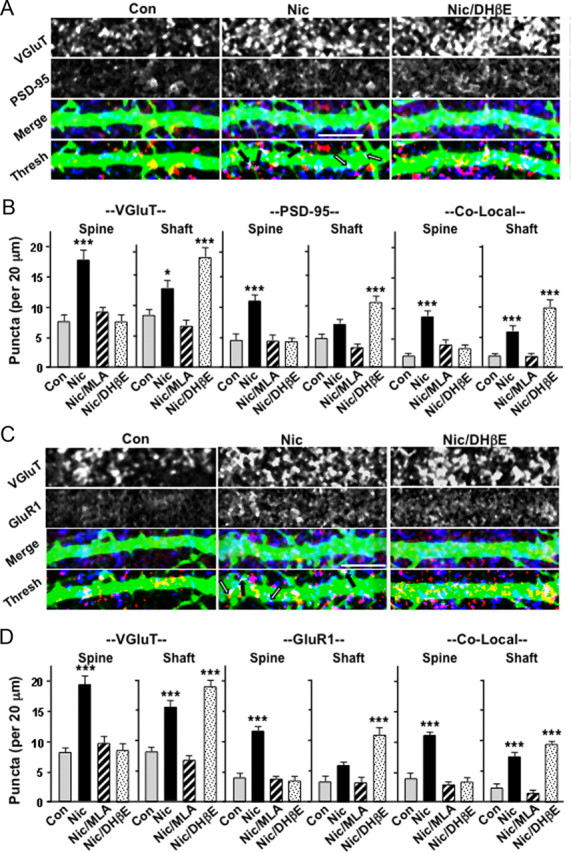
In the absence of functional β2*-nAChRs, nicotine-induced glutamatergic synapse formation forces more synapses to be located on dendritic shafts. A, Sindbis–GFP-labeled dendrites (green) of neurons in the CA1 region of P0–P1 hippocampal slices maintained in culture for 5 d and treated with the indicated compounds for the last 4 d, before immunostaining for VGluT (blue) and PSD-95 (red). Images are shown individually (VGluT, PSD-95), merged (Merge), and thresholded (Thresh); arrows indicate examples of colocalizations on dendritic spines (black) and shafts (white). B, Quantification of VGluT (left) and PSD-95 (middle) and colocalization of puncta (right) on dendritic spines (Spine) and shafts (Shaft). C, Images collected and shown as in A but immunostained for GluR1 (red) instead of PSD-95. D, Quantification of the puncta as in B (3–5 cells per culture; 6 cultures per condition). Scale bars, 10 μm. Con, Control; Nic, nicotine.
Similar conclusions were reached with different techniques applied to rat hippocampal cells in culture. Long-term nicotine exposure again increased the number of spines per unit length dendrite, and did so in a way that was dependent on β2*-nAChRs (blocked by DHβE) but not α7-nAChRs because it was not prevented by the specific antagonist αBungarotoxin (αBgt; Fig. 5A–C). The same pattern was obtained when spines were identified by immunostaining for drebrin after a 1 week exposure to nicotine, and either αBgt or MLA was used to block α7-nAChRs or DHβE was used to block β2*-nAChRs (Fig. 5D).
Figure 5.

Nicotinic stimulation in cell culture in the absence of functional β2*-nAChRs increases the number of functional glutamatergic synapses that form on dendritic shafts, as opposed to dendritic spines. A, Neurites of rat hippocampal neurons transfected to express GFP and incubated with (Nic) or without (Con) 1 μm nicotine for 20 d and imaged. B, Immunostaining cultures for PSD-95 (red) overlaid with GFP (green) in a nicotine-treated culture as in A. C, Spine counts on control cells or cells treated with nicotine, 100 nm αBgt, and 10 μm DHβE. D, Quantification of spines in cultures grown 10 d with the indicated compounds present for the last 7 d. Drebrin immunostaining was used to visualize spines. Drug concentrations are as in C; 50 nm MLA. E, Confocal images of FM4-64-labeled neurons (red) expressing lenti-GFP (green). Arrows and asterisks indicate examples of FM4-64 puncta on spines and dendritic shafts, respectively (puncta lying centrally over shafts were not scored). F, Quantification of FM4-64 puncta on spines (Spine) and shafts (Shaft) for the conditions indicated, scored as in E (3–5 cultures; 5 cells per culture). Scale bars: A, E, 5 μm; B, 10 μm. Con, Control; Nic, nicotine.
Use of cell culture enabled us to apply a second test for synapses, namely the assay of KCl-stimulated dye uptake that reflects synaptic vesicle recycling. This has been used previously to quantify the number of active synapses (Pyle et al., 1999; Lozada et al., 2012). Cultures were incubated with nicotine for 7 d followed by nicotinic antagonists for 1 h to terminate acute effects and then assayed for KCl-dependent FM4-64 uptake in a 1 min period (Fig. 5E). Independently quantifying synapses identified in this manner on dendritic shafts versus those on dendritic spines indicated that the nicotine treatment prominently increased the latter (Fig. 5F). However, blockade of β2*-nAChRs with DHβE during the nicotine treatment resulted in a substantial increase in shaft synapses with no change in spine synapses (Fig. 5F). The results indicate that nicotinic stimulation of neurons with inactive β2*-nAChRs still drives synapse formation, apparently by activating α7-nAChRs as described recently (Lozada et al., 2012), but the additional synapses must form on dendritic shafts because spine numbers are reduced in the absence of β2*-nAChR signaling.
Persistent requirement for β2*-nAChRs in vivo for glutamatergic spine synapses
To test whether β2KOs display a shift in the location of their glutamatergic synapses in vivo as predicted by the culture experiments, we compared wild types and KOs with respect to glutamatergic synapses on dendritic spines versus shafts. Glutamatergic synapses on dendrites were visualized both in the hippocampal CA1 region and in the visual cortex layer 5/6 by injecting sindbis–GFP stereotaxically into the respective locations and subsequently immunostaining tissue slices for VGluT and PSD-95 (Fig. 6A,C). Quantification of puncta was confined to edges of dendrites so that spine and shaft locations could be clearly distinguished. This prevented quantification of total synaptic number per dendrite but did provide a good basis for detecting differences in the ratio of spine versus shaft synapses as a function of mouse type. Analyzing pyramidal apical dendrites at P12 either in the CA1 or in layer 5/6 revealed that β2KOs had fewer spine synapses than do wild types and a trend toward more shaft synapses (Fig. 6B,D). Both tissues showed statistically significant decreases in the numbers of VGluT puncta, PSD-95 puncta, and their colocalization on spines at this age. Layer 5/6 also showed statistically significant increases in all three for shaft synapses, whereas the CA1 showed increased VGluT puncta on shafts at P12 and a trend toward increases in PSD-95 puncta and colocalization. This was clearly different from α7KOs, which had reduced numbers of VGluT and PSD-95 puncta and reduced colocalization both on spines and on shafts, consistent with fewer glutamatergic synapses overall as reported previously (Lozada et al., 2012). Numbers in dKOs were consistent with the individual KOs.
Figure 6.
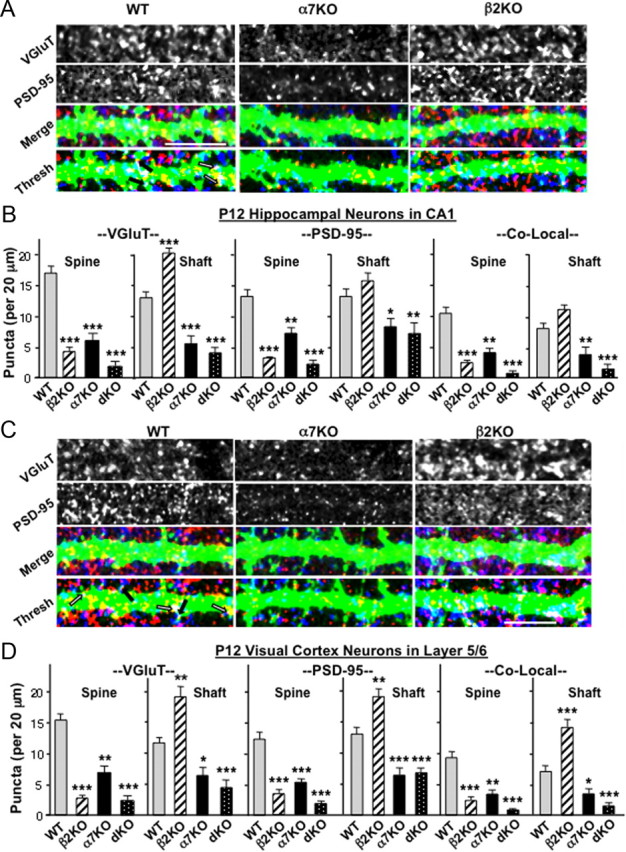
Deleting the β2-nAChR gene shifts more synapse formation to a dendritic shaft location in vivo. A, Representative images of apical dendrites of CA1 pyramidal neurons in P12 WT (left), α7KO (middle), and β2KO (right) mice visualized by in vivo labeling with sindbis–GFP, immunostained for VGluT (blue) and PSD-95 (red) after fixation, and shown individually (top two rows), merged (Merge), and thresholded (Thresh). Arrows indicate examples of colocalization on dendritic spines (black) and shafts (white). B, Quantification of VGluT (left), PSD-95 (middle), and co-localization of puncta (right, Co-Local) on dendritic spines (Spine) and shafts (Shaft) in P12 CA1. C, Dendrites extending from pyramidal neurons in cortical layer 5/6 of P12 WT (left), α7KO (middle), and β2KO (right) mice visualized with sindbis–GFP and immunostained fixed slices as in A. D, Quantification of VGluT, PSD-95, and colocalized puncta (Co-Local) on cortical dendritic spines and shafts as in B (3–4 cells per animal; 4–12 animals per genotype). Scale bars, 10 μm.
At later times in development, a greater proportion of glutamatergic synapses normally appear on dendritic spines (Fiala et al., 1998; Yuste and Bonhoeffer, 2004; Sheng and Hoogenraad, 2007). To test whether the shift of glutamatergic synapses away from spines and onto dendritic shafts seen in early β2KO mice remains subsequently, we examined P25 hippocampus. Applying the same labeling and counting strategies previously used at P12 showed that P25 wild types have a greater proportion of glutamatergic synapses located on dendritic spines than on shafts, as expected from previous work (Fig. 7). Nonetheless, the reverse distribution seen in P12 β2KOs persists: by these criteria (Eroglu et al., 2009), the number of shaft synapses at P25 in the CA1 is significantly greater than seen in wild types, and the number of spine synapses is significantly smaller than in wild types. The deficits seen in glutamatergic synapses both on dendritic shafts and on spines in α7KOs at P12 remain at P25 (Fig. 7).
Figure 7.

The synaptic shift detected in P12 β2KOs remains at P25, despite the increasing counter trend in wild types. A, Apical dendrites of CA1 pyramidal neurons in P25 WT (left), α7KO (middle), and β2KO (lower) mice immunostained for VGluT (blue) and PSD-95 (red) and shown separately, merged (Merge), and thresholded (Thresh). Arrows and scale bars are as in Figure 6. B, Quantification of VGluT (left), PSD-95 (middle), and colocalized puncta (right, Co-Local) on dendritic spines (Spine) and shafts (Shaft) in P25 CA1 (3–4 cells per animal, 3–4 animals per genotype).
As in the case of spine numbers, the effects of β2*-nAChRs on the location of glutamatergic synapses are cell autonomous. This was shown by distinguishing spine versus shaft synapses on neurons expressing either β2-RNA or β2-Scr in vivo for up to 12 d. As in the case of β2KOs, expression of β2-RNAi in WT neurons decreased the numbers of spines and decreased the numbers of VGluT and PSD-95 puncta and their colocalization on spines, compared with that seen on WT neurons expressing β2-Scr (Fig. 8). Conversely, β2-RNA expression increased the incidence of VGluT and PSD-95 puncta and their colocalization on dendritic shafts, compared with β2-Scr expression. The results indicate that cell-autonomous actions of β2*-nAChRs enhance dendritic spine number and, as a result, enable more glutamatergic synapses to be located on spines.
Figure 8.

RNAi demonstration that β2*-nAChRs act cell-autonomously to elevate the number of synapses formed on dendritic spines in vivo. A, Apical dendrites of CA1 pyramidal cells expressing either β2-Scr (left) or β2-RNAi (right), injected intracranially on P1–P2 and immunostained on P12 for VGluT (blue) and PSD-95 (red) shown separately, merged (Merge), and thresholded (Thresh). Arrows and scale bars are as in Figure 6. B, Quantification of VGluT (left), PSD-95 (middle), and colocalized puncta (right, Co-Local) on dendritic spines (Spine) and shafts (Shaft) of P12 CA1 pyramidal neurons (2–3 cells per animal, 3 animals per genotype, 2 litters per condition).
Discussion
The comparison of β2KOs with age-matched wild types shows unambiguously that the β2-nAChR gene product is necessary to acquire normal levels of dendritic spines. The ability of nicotine to drive spine formation both in vivo and in organotypic culture in a manner dependent on β2*-nAChRs further documents the capability of the receptors in this process. Spine dependence on β2*-nAChRs extends into early adulthood and can be seen not only in the hippocampus but also in the cortex, revealing the breadth of the phenomenon. In the absence of β2*-nAChRs, neurons continue to generate normal numbers of glutamatergic synapses but apparently are then forced to position more of them on dendritic shafts, given the reduced number of spines available. Because spine synapses differ from shaft synapses with respect to signaling properties and plasticity (Yuste and Bonhoeffer, 2004; Aoto et al., 2007; Harvey and Svoboda, 2007; Sheng and Hoogenraad, 2007; Yuste, 2011), a shift of this nature in the distribution of synapses would be expected to have significant effects on network function and plasticity.
The present studies revealed no compensatory change in the aberrant distribution of synapses in β2KOs over the time period examined. The abnormally high fraction of glutamatergic synapses found on dendritic shafts at P12 remained at P25, and the deficits in spine numbers quantified as early as P4 remained as late as P40. Although compensation has been inferred for some cholinergic traits in other paradigms (Besson et al., 2007), constitutive removal of the β2-nAChR gene does not apparently induce relevant compensatory changes in the levels of other nAChR subtypes (Albuquerque et al., 2009; Gotti et al., 2009; Changeux, 2010). The persistence of spine deficits in β2KOs is consistent with a recent report that adult β2KOs have reduced spine numbers in several cortical regions and altered dendritic trees as well, although not all areas are affected equally (Ballesteros-Yáñez et al., 2010). A previous study using a toxin to generate cholinergic deficits also found reductions in spine numbers as a result (Robertson et al., 1998). The consistent deficits reported in the present study were obtained by examining defined regions (hippocampal CA1 and sensory cortex layer 5/6) across experimental conditions and focusing on consistent regions of the dendritic tree that could be readily distinguished across neuronal populations.
The RNAi experiments demonstrate that the β2*-nAChR effect is cell autonomous, requiring the postsynaptic cell to express the receptors if normal numbers of dendritic spines are to be achieved. The cell-autonomous feature extends to the β2*-nAChR effect on synaptic distribution, with the receptors causing more glutamatergic synapses to be located on spines. Off-target effects of the RNAi construct were excluded by the experiments showing that it had no effect on spine number in β2KOs. The initial effect of the receptors was quick, requiring no more than 1 h for nicotinic stimulation to increase spine numbers by one-half or more both in vivo and in organotypic culture. Stabilization took longer, perhaps requiring protein synthesis and anchoring of the cytoskeleton. Notably, the number of spines in organotypic cultures lacking cholinergic input was similar to that seen in early postnatal wild types in vivo where cholinergic input would have been normal. This likely reflects, in part, the reported phenomenon that slice preparation and transfer to culture can increase spine number (Kirov et al., 1999). It is also likely, however, that the β2*-nAChR effect on spines becomes prominent only after the first few days of postnatal life; spines do form in the absence of β2*-nAChRs as found in β2KOs. The finding that nicotine elevated spine numbers in vivo when injected at P7 but not at P15 suggests that endogenous nicotinic activity may achieve maximal rates by P15.
The initial mechanism underlying spine induction by β2*-nAChRs involved intracellular calcium and the activation of CaMKII. Likely calcium sources include influx through voltage-gated channels and release from internal stores (Dajas-Bailador and Wonnacott, 2004; Albuquerque et al., 2009). A prominent calcium-dependent pathway that is known to induce and stabilize dendritic spines uses CaMKII activation of either kalirin-7 or Tiam1 to mediate downstream events that rearrange the actin cytoskeleton (Tolias et al., 2005; Xie et al., 2007). These and other pathways (Gu et al., 2008; Wang et al., 2008; McClelland et al., 2010; Saneyoshi et al., 2010; Kwon and Sabatini, 2011; Murakoshi et al., 2011) offer candidate mechanisms for the cell-autonomous action. Although β2*-nAChRs are expressed by pyramidal neurons, precise dendritic/somatic locations for the receptors have yet to be identified. Understanding the mechanisms controlling β2*-nAChR location will become important if β2*-nAChR effects on spines are highly localized along the dendrite. In this case, endogenous nicotinic signaling acting through β2*-nAChRs could determine the precise location of spine synapses, thereby influencing their relative contributions to summation and neuronal excitation.
A complementary role is played by α7-nAChRs. As indicated previously (Lozada et al., 2012) and in the present study, α7-nAChRs are required for normal numbers of glutamatergic synapses to form during development. This effect is independent of β2*-nAChR function. Both in vivo analysis and culture experiments demonstrate that activation of α7-nAChRs in the absence of β2*-nAChRs causes a disproportionate number of glutamatergic synapses to form on dendritic shafts as opposed to spines. Like β2*-nAChRs, α7-nAChRs are widely expressed early in development and participate in the spontaneous waves of excitation seen at those times. Also like β2*-nAChRs, α7-nAChRs depolarize the membrane and elevate intracellular calcium levels, but do so by different mechanisms. Notably, α7-nAChRs have a high relative permeability to calcium and also recruit calcium from internal stores (Bertrand et al., 1993; Séguéla et al., 1993; Dajas-Bailador and Wonnacott 2004; Fayuk and Yakel, 2007; Albuquerque et al., 2009; Shen and Yakel, 2009). Other differences between α7-nAChRs and β2*-nAChRs include their sensitivities to agonist, their rates of desensitization, and, probably most important, their locations on the membrane and tethering to specific components that mediate signal transduction (McGehee et al., 1995; Fabian-Fine et al., 2001; Albuquerque et al., 2009; Gotti et al., 2009; Changeux, 2010). This latter feature most likely accounts for the differences between α7-nAChRs and β2*-nAChRs in their effects on glutamatergic synapse formation. One reflection of this is the distinctive ability of α7-nAChRs, via associated components, to alter gene expression in a calcium-dependent manner (Chang and Berg, 2001; Hu et al., 2002; Albuquerque et al., 2009). Such components include the membrane calcium pump PMCA2, tethered to α7-nAChRs via the scaffold protein PSD-95 in a way that permits calcium-dependent control of α7-nAChR location and function on the neuronal surface (Gomez-Varela et al., 2012).
The finding that β2*-nAChRs and α7-nAChRs exert independent and complementary effects on glutamatergic synapse formation has multiple implications. First, it underscores the fact that the presence of spines does not ensure the presence of synapses. Activation of β2*-nAChRs without α7-nAChR participation elevated the number of spines without increasing the number of morphological or functional synaptic sites. Spines lacking synapses under these conditions may be relatively unstable, retracting and reextending over a relatively short time frame. It is known that spines usually form before innervation in vivo and may exist as such for up to several days (Knott et al., 2006).
A second implication of the changes seen in synaptic distribution is that the relative numbers of synapses on dendritic spines versus shafts are not predetermined but rather can be altered by extrinsic mechanisms. Previous studies documented an increase in the proportion of glutamatergic synapses on spines (vs dendritic shafts) as development proceeds (Fiala et al., 1998), whereas pruning decreases the total number of synapses in general, and shaft synapses in particular, during maturation (Harris et al., 1992; Fiala et al., 1998; Hua and Smith, 2004). The analysis performed here did not indicate the total number of synapses in each category but did permit quantitative comparisons across conditions. Selective loss of β2*-nAChRs produced a clear shift toward more glutamatergic synapses being located on dendritic shafts. The shift was apparent even up through early adolescence when spine synapses in wild types vastly outnumbered shaft synapses (Yuste and Bonhoeffer, 2004). There are precedents for independent regulation of spine and shaft synapses (Aoto et al., 2007).
A third implication is that repeated exposure to exogenous agonists such as nicotine in vivo may alter the balance of spontaneous activity through β2*-nAChRs and α7-nAChRs either by selectively desensitizing or preferentially activating one class of nAChRs (Albuquerque et al., 2009; Gotti et al., 2009; Changeux, 2010). This could shift the distribution of synapses formed and produce long-term consequences. Perhaps reflecting these kinds of effects, exposure to nicotine during gestation and early postnatal life is well known to produce lasting behavioral changes in the adult (Heath and Picciotto, 2009). The complementary roles of β2*-nAChRs and α7-nAChRs in shaping the formation of glutamatergic networks are also likely to explain, as least in part, the many behavioral deficits attributed to mice lacking one or the other of these receptors (Picciotto et al., 1995; Grubb et al., 2003; Myers et al.,2005; Hanson and Landmesser, 2006; Hoyle et al., 2006; Le Magueresse et al., 2006; Young et al., 2007; Levin et al., 2009; Wang et al., 2009).
Footnotes
This work was supported by grants from the National Institutes of Health (NS012601, NS034569, and DA237482) and the Tobacco-Related Disease Research Program (17FT-0053 and 19XT-0072). We thank Uwe Maskos and Andrew W. Halff for the β2- and α7-RNAi constructs, respectively; Xiao-Yun Wang and Jeff Schoellerman for expert technical assistance; and Gentry Patrick and Stevan Djakovic for stocks of sindbis–GFP.
The authors declare no competing financial interests.
References
- Adams CE, Broide RS, Chen Y, Winzer-Serhan UH, Henderson TA, Leslie FM, Freedman R. Development of the α7 nicotinic cholinergic receptor in rat hippocampal formation. Dev Brain Res. 2002;139:175–187. doi: 10.1016/s0165-3806(02)00547-3. [DOI] [PubMed] [Google Scholar]
- Adesnik H, Li G, During MJ, Pleasure SJ, Nicoll RA. NMDA receptors inhibit synapse unsilencing during brain development. Proc Natl Acad Sci U S A. 2008;105:5597–5602. doi: 10.1073/pnas.0800946105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EFR, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Phys Revs. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Ting P, Maghsoodi B, Xu N, Henkemeyer M, Chen L. Postsynaptic EphrinB3 promotes shaft glutamatergic synapse formation. J Neurosci. 2007;27:7508–7519. doi: 10.1523/JNEUROSCI.0705-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avale ME, Faure P, Pons S, Robledo P, Deltheil T, David DJ, Gardier AM, Maldonado R, Granon S, Changeux JP, Maskos U. Interplay of β2* nicotinic receptors and dopamine pathways in the control of spontaneous locomotion. Proc Natl Acad Sci U S A. 2008;105:15991–15996. doi: 10.1073/pnas.0807635105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros-Yáñez I, Benavides-Piccione R, Bourgeois J-P, Changeux JP, DeFelipe J. Alterations of cortical pyramidal neurons in mice lacking high affinity nicotinic receptors. Proc Natl Acad Sci U S A. 2010;107:11567–11572. doi: 10.1073/pnas.1006269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal A, Singer JH, Hwang BJ, Xu W, Beaudet A, Feller MB. Mice lacking specific nAChR subunits exhibit dramatically altered spontaneous activity patterns and reveal a limited role for retinal waves in forming ON/OFF circuits in the inner retina. J Neurosci. 2000;20:7672–7681. doi: 10.1523/JNEUROSCI.20-20-07672.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D, Galzi JL, Devillers-Thiéry A, Bertrand S, Changeux JP. Mutations at two distinct sites within the channel domain M2 alter calcium permeability of neuronal α7 nicotinic receptor. Proc Natl Acad Sci U S A. 1993;90:6971–6975. doi: 10.1073/pnas.90.15.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson M, Granon S, Mameli-Engvall M, Cloëz-Tayarani I, Maubourguet N, Cormier A, Cazala P, David V, Changeux JP, Faure P. Long-term effects of chronic nicotine exposure on brain nicotinic receptors. Proc Natl Acad Sci U S A. 2007;104:8155–8160. doi: 10.1073/pnas.0702698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell NR, Fernandes CC, Halff AW, Berg DK. Endogenous signaling through alpha7-containing nicotinic receptors promotes maturation and integration of adult-born neurons in the hippocampus. J Neurosci. 2010;30:8734–8744. doi: 10.1523/JNEUROSCI.0931-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KT, Berg DK. Endogenous signaling through alpha7-containing nicotinic receptors promotes maturation and integration of adult-born neurons in the hippocampus. Neuron. 2001;32:855–865. doi: 10.1523/JNEUROSCI.0931-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux JP. Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat Rev Neurosci. 2010;11:389–401. doi: 10.1038/nrn2849. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador F, Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. TIPS. 2004;25:317–324. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor α2δ-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, Fine A. Ultrastructural distribution of the α7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci. 2001;21:7993–8003. doi: 10.1523/JNEUROSCI.21-20-07993.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayuk D, Yakel JL. Dendritic Ca2+ signalling due to activation of alpha 7-containing nicotinic acetylcholine receptors in hippocampal neurons. J Physiol. 2007;582:597–611. doi: 10.1113/jphysiol.2007.135319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, Harris KM. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci. 1998;18:8900–8911. doi: 10.1523/JNEUROSCI.18-21-08900.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Varela D, Schmidt M, Schoellerman J, Peters EC, Berg DK. PMCA2 via PSD-95 controls calcium signaling by α7-containing nicotinic acetylcholine receptors on aspiny interneurons. J Neurosci. 2012;32:6894–6905. doi: 10.1523/JNEUROSCI.5972-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I, Moretti M, Pedrazzi P, Pucci L, Zoli M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol. 2009;78:703–711. doi: 10.1016/j.bcp.2009.05.024. [DOI] [PubMed] [Google Scholar]
- Grubb MS, Rossi FM, Changeux JP, Thompson ID. Abnormal functional organization in the dorsal lateral geniculate nucleus of mice lacking the β2 subunit of the nicotinic acetylcholine receptor. Neuron. 2003;40:1161–1172. doi: 10.1016/s0896-6273(03)00789-x. [DOI] [PubMed] [Google Scholar]
- Gu J, Firestein BL, Zheng JQ. Microtubules in dendritic spine development. J Neurosci. 2008;28:12120–12124. doi: 10.1523/JNEUROSCI.2509-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Nam J, Li Y, Kim S, Cho SH, Cho YS, Choi SY, Choi J, Han K, Kim Y, Na M, Kim H, Bae YC, Choi SY, Kim E. Regulation of dendritic spines, spatial memory, and embryonic development by TANC family of PSD-95-interacting proteins. J Neurosci. 2010;30:15102–15112. doi: 10.1523/JNEUROSCI.3128-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT. Characterization of the circuits that generate spontaneous episodes of activity in the early embryonic mouse spinal cord. J Neurosci. 2003;23:587–600. doi: 10.1523/JNEUROSCI.23-02-00587.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT. Increasing the frequency of spontaneous rhythmic activity disrupts pool-specific axon fasciculation and pathfinding of embryonic spinal motoneurons. J Neurosci. 2006;26:12769–12780. doi: 10.1523/JNEUROSCI.4170-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450:1195–1200. doi: 10.1038/nature06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath CH, Picciotto MR. Nicotine-induced plasticity during development: modulation of the cholinergic system and long-term consequences for circuits involved in attention and sensory processing. Neuropharmacology. 2009;56:254–262. doi: 10.1016/j.neuropharm.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennou S, Khalilov I, Diabira D, Ben-Ari Y, Gozlan H. Early sequential formation of functional GABAA and glutamatergic synapses on CA1 interneurons of the rat foetal hippocampus. Eur J Neurosci. 2002;16:197–208. doi: 10.1046/j.1460-9568.2002.02073.x. [DOI] [PubMed] [Google Scholar]
- Hoyle E, Genn RF, Fernandes C, Stolerman IP. Impaired performance of alpha7 nicotinic receptor knockout mice in the five-choice serial reaction time task. Psychopharmacology. 2006;189:211–223. doi: 10.1007/s00213-006-0549-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Liu QS, Chang KT, Berg DK. Endogenous signaling through alpha7-containing nicotinic receptors promotes maturation and integration of adult-born neurons in the hippocampus. Mol Cell Neurosci. 2002;21:616–625. doi: 10.1523/JNEUROSCI.0931-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua JY, Smith SJ. Neural activity and the dynamics of central nervous system development. Nat Neurosci. 2004;7:327–332. doi: 10.1038/nn1218. [DOI] [PubMed] [Google Scholar]
- Kayser MS, McClelland AC, Hughes EG, Dalva MB. Intracellular and trans-synaptic regulation of glutamatergic synaptogenesis by EphB receptors. J Neurosci. 2006;26:12152–12164. doi: 10.1523/JNEUROSCI.3072-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirov SA, Sorra KE, Harris KM. Slices have more synapses than perfusion-fixed hippocampus from both young and mature rats. J Neurosci. 1999;19:2876–2886. doi: 10.1523/JNEUROSCI.19-08-02876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirov SA, Petrak LJ, Fiala JC, Harris KM. Dendritic spines disappear with chilling but proliferate excessively upon rewarming of mature hippocampus. Neuroscience. 2004;127:69–80. doi: 10.1016/j.neuroscience.2004.04.053. [DOI] [PubMed] [Google Scholar]
- Knott GW, Holtmaat A, Wilbrecht L, Welker E, Svoboda K. Spine growth precedes synapse formation in the adult neocortex in vivo. Nat Neurosci. 2006;9:1117–1124. doi: 10.1038/nn1747. [DOI] [PubMed] [Google Scholar]
- Kwon HB, Sabatini BL. Glutamate induces de novo growth of functional spines in developing cortex. Nature. 2011;474:100–104. doi: 10.1038/nature09986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai KO, Ip NY. Synapse development and plasticity: roles of ephrin/Eph receptor signaling. Curr Opin Neurobiol. 2009;19:275–283. doi: 10.1016/j.conb.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Le Magueresse C, Safiulina V, Changeux JP, Cherubini E. Nicotinic modulation of network and synaptic transmission in the immature hippocampus investigated with genetically modified mice. J Physiol. 2006;576:533–546. doi: 10.1113/jphysiol.2006.117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Petro A, Rezvani AH, Pollard N, Christopher NC, Strauss M, Avery J, Nicholson J, Rose JE. Nicotinic α7- or β2-containing receptor knockout: effects on radial-arm maze learning and long-term nicotine consumption in mice. Behav Brain Res. 2009;196:207–213. doi: 10.1016/j.bbr.2008.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PR, Shute CC, Silver A. Confirmation from choline acetylase analyses of a massive cholinergic innervation to the rat hippocampus. J Physiol. 1967;191:215–224. doi: 10.1113/jphysiol.1967.sp008246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozada AF, Wang X, Gounko NV, Massey KA, Duan J, Liu X, Berg DK. Glutamatergic synapse formation is promoted by α7-containing nicotinic acetylcholine receptors. J Neurosci. 2012 doi: 10.1523/JNEUROSCI.6246-11.2012. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, Gardiner PS, Grady SR, Heberlein U, Leonard SS, Levin ED, Lukas RJ, Markou A, Marks MJ, McCallum SE, Parameswaran N, et al. Guidelines on nicotine dose selection for in vivo research. Pyschopharmacology. 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- McClelland AC, Hruska M, Coenen AJ, Henkemeyer M, Dalva MB. Trans-synaptic EphB2-ephrin-B3 interaction regulates excitatory synapse density by inhibition of postsynaptic MAPK signaling. Proc Natl Acad Sci U S A. 2010;107:8830–8835. doi: 10.1073/pnas.0910644107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- Misra C, Restituito S, Ferreira J, Rameau GA, Fu J, Ziff EB. Regulation of synaptic structure and function by palmitoylated AMPA receptor binding protein. Mol Cell Neurosci. 2010;43:341–352. doi: 10.1016/j.mcn.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi H, Wang H, Yasuda R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature. 2011;472:100–104. doi: 10.1038/nature09823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CP, Lewcock JW, Hanson MG, Gosgnach S, Aimone JB, Gage FH, Lee KF, Landmesser LT, Pfaff SL. Cholinergic input is required during embryonic development to mediate proper assembly of spinal locomotor circuits. Neuron. 2005;46:37–49. doi: 10.1016/j.neuron.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Léna C, Bessis A, Lallemand Y, Le Novère N, Vincent P, Pich EM, Brûlet P, Changeux JP. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature. 1995;374:65–67. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Choi S, Tsien RW. Visualization of synaptic activity in hippocampal slices with FM1-43 enabled by fluorescence quenching. Neuron. 1999;24:803–808. doi: 10.1016/s0896-6273(00)81028-4. [DOI] [PubMed] [Google Scholar]
- Robertson RT, Gallardo KA, Claytor KJ, Ha DH, Ku KH, Yu BP, Lauterborn JC, Wiley RG, Yu J, Gall CM, Leslie FM. Neonatal treatment with 192 IgG-saporin produces long-term forebrain cholinergic deficits and reduces dendritic branching and spine density of neocortical pyramidal neurons. Cereb Cortex. 1998;8:142–155. doi: 10.1093/cercor/8.2.142. [DOI] [PubMed] [Google Scholar]
- Saneyoshi T, Fortin DA, Soderling TR. Regulation of spine and synapse formation by activity-dependent intracellular signaling pathways. Curr Opin Neurobiol. 2010;20:108–115. doi: 10.1016/j.conb.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JX, Yakel JL. Nicotinic acetylcholine-receptor mediated calcium signaling in the nervous system. Acta Pharmacol Sin. 2009;30:673–680. doi: 10.1038/aps.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45:525–538. doi: 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Tran TS, Rubio ME, Clem RL, Johnson D, Case L, Tessier-Lavigne M, Huganir RL, Ginty DD, Kolodkin AL. Secreted semaphorins control spine distribution and morphogenesis in the postnatal CNS. Nature. 2009;462:1065–1069. doi: 10.1038/nature08628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller L. Miniature synaptic transmission and BDNF modulate dendrite spine growth and form in rat CA1 neurones. J Physiol. 2003;553:497–509. doi: 10.1113/jphysiol.2003.052639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Rangarajan KV, Lawhn-Heath CA, Sarnaik R, Wang BS, Liu X, Cang J. Direction-specific disruption of subcortical visual behavior and receptor fields in mice lacking the β2 subunit of nicotinic acetylcholine receptor. J Neurosci. 2009;29:12909–12918. doi: 10.1523/JNEUROSCI.2128-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Edwards JG, Riley N, Provance DW, Jr, Karcher R, Li XD, Davison IG, Ikebe M, Mercer JA, Kauer JA, Ehlers MD. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135:535–548. doi: 10.1016/j.cell.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Srivastava DP, Photowala H, Kai L, Cahill ME, Woolfrey KM, Shum CY, Surmeier DJ, Penzes P. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. 2007;56:640–656. doi: 10.1016/j.neuron.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JW, Crawford N, Kelly JS, Kerr LE, Marston HM, Spratt C, Finlayson K, Sharkey J. Impaired attention is central to the cognitive deficits observed in alpha 7 deficient mice. Eur Neuropsychopharm. 2007;17:145–155. doi: 10.1016/j.euroneuro.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Yuste R. Dendritic spines and distributed circuits. Neuron. 2011;71:772–781. doi: 10.1016/j.neuron.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat Rev Neurosci. 2004;5:24–34. doi: 10.1038/nrn1300. [DOI] [PubMed] [Google Scholar]
- Yuste R, Majewska A, Cash SS, Denk W. Mechanisms of calcium influx into hippocampal spines: heterogeneity among spines, coincidence detection by NMDA receptors, and optical quantal analysis. J Neurosci. 1999;19:1976–1987. doi: 10.1523/JNEUROSCI.19-06-01976.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Liu C, Miao H, Gong ZH, Nordberg A. Postnatal changes in nicotinic acetylcholine receptor α2, α3, α4, α7 and β2 subunits genes expression in rat brain. Int J Dev Neurosci. 1998;16:507–518. doi: 10.1016/s0736-5748(98)00044-6. [DOI] [PubMed] [Google Scholar]