

Abstract
Progress in the field of axonal regeneration research has been like the process of axonal growth itself: there is steady progress toward reaching the target, but there are episodes of mis-targeting, mis-guidance along false routes, and connections that must later be withdrawn. This primer will address issues in the study of axonal growth after central nervous system injury in an attempt to provide guidance toward the goal of progress in the field. We address definitions of axonal growth, sprouting and regeneration after injury, and the research tools to assess growth.
Introduction
For decades, it has been recognized that axon regeneration is the only way to restore function after severe spinal cord injuries (SCI) that interrupt the long tracts that mediate motor and sensory function. Indeed, SCI and axon regeneration are so linked in the minds of scientists and the lay public that enabling regeneration after SCI is iconic. Achieving axonal regeneration with recovery of function would truly be an extraordinary achievement.
Despite progress, measured both as a gain in understanding of the molecular, cellular and systems-level underpinnings of axonal growth, and in the number of investigators studying the topic, success has not yet been achieved. Indeed, progress in the field is non-linear, with many instances of premature celebration of success, mistargeting, sidesteps and occasional episodes of withdrawal. The reasons for this are numerous, ranging from lack of clarity in use of terminology related to axonal growth and limitations of experimental methods to a lack of rigor in interpretation.
This primer aims to provide a framework for the study of axonal growth after spinal cord injury. We focus on SCI not only because it is iconic, but also because it exemplifies all of the issues that plague studies of axon regeneration in any CNS region with mixed white and gray matter. We begin by addressing the meaning of different terms used to describe growth after injury, especially the terms “regeneration” and “sprouting”. Inconsistent use of these terms in the scientific literature creates ambiguity or frank error in interpreting experimental findings. We then review several model systems for studying axonal growth after spinal cord injury, highlighting the advantages and limitations of several models. Finally, we will discuss the tools available to study axonal regeneration, and how these might best be applied to reach new levels of insight that will point the way to strategies for improving outcomes after spinal cord injury.
I. Distinctions Between types of Axon Growth: Regeneration and Sprouting
There is enormous inconsistency in the literature in the use of the terms regeneration and sprouting. In part this is because the terms are defined differently by individuals studying differing aspects of axonal regeneration, and are even defined differently by those studying the same aspects of axonal regeneration. Part of the inconsistent use in the field may reflect uncertainty about what is really happening anatomically.
What defines axonal regeneration? At the organ replacement level, regeneration can refer to cellular proliferation to replace tissue. When applied to axons, regeneration refers to re-growth of a transected axon, as in the case of a peripheral axon growing back along the distal stump of a crushed or transected nerve to reinnervate its normal target (Fig 1C). There are nuances in the application of this simple term in several circumstances, based on the features of new axonal growth, including from where along the length of the axon the growth originates, the distance over which an axon grows, and whether the growing axon reaches its normal target. This will be discussed in greater detail below. Most researchers agree that new growth arising from the cut end of a transected axon, and extending beyond the lesion site, represents canonical axon regeneration. As noted above, this can occur after peripheral nerve injury, and nearly entirely fails after central injury.
Figure 1. Regeneration and Sprouting.
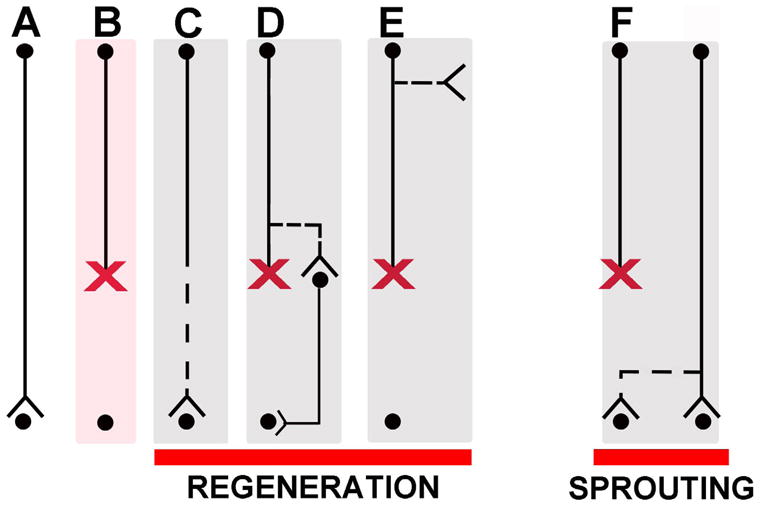
The left side of the panel illustrates several examples of types of regeneration. (A) Intact axon. (B) Transected or crushed axon. (C) Canonical regenerating axon: new growth occurs from tip of transected axon leading to reinnervation of its normal target. (D) Regenerating axon, wherein new growth arises not from tip of transected axon, but from region of axon close to injury site. In some literature, this is referred to as “regenerative sprouting”, a term we avoid because it can generate confusion. (E) Another example of a regenerating axon, wherein new growth arises from a transected axon, but from a region of the axon that is remote from the injury site. This type of growth has been described to arise from CST axons in the cervical spine cord after thoracic level injuries (Bareyre et al., 2004). This has also been referred to as “regenerative sprouting.” (F) An example of canonical sprouting: here damage to one pathway induces compensatory growth of new connections from nearby undamaged axons.
The term “sprouting” has been used in a much more inconsistent way. Ramon y Cajal used the term to refer to early growth from the tip of an injured axon: “The innervation of the peripheral stump of cut nerves (occurs) through the growth, across the scar, of nerve sprouts arising in the central stump…”, (Ramon y Cajal, 1928) p. 223.
In the renaissance of regeneration research, Liu and Chambers (Liu and Chambers, 1958) and McCouch (McCouch et al., 1958) used the term “sprouting” in a new way to refer to growth arising from an axon that was not itself damaged (Fig 1G), specifically growth of the central projections of intact dorsal root ganglion axons after injury to adjoining roots. This usage followed on earlier studies of growth of motor axons following partial denervation of muscle (Causey and Hoffman, 1955; Edds, 1953; Edds and Small, 1951; Hoffman, 1952).
Use of the term “sprouting” in this manner continued in studies of growth after injury in numerous brain structures, especially the hippocampus, throughout the 1970’s. It soon became clear, however, that different growth phenomena were occurring, sometimes involving cut axons and sometimes involving axons that were uninjured. Many different terms were applied loosely, including the term “plasticity” (Raisman, 1969), which is now used in so many ways as to be almost meaningless in an anatomical context.
Moore (1973) tried to bring some order to the terminological chaos, defining two basic phenomena:
“A) In regenerative sprouting, the axons of neurons innervating a structure are severed and the axon distal to the lesion degenerates. The proximal stumps form growth cones and regenerate new axons and terminals. B) In collateral sprouting, part of the innervation to a structure is severed. The distal axons and terminals degenerate and collateral sprouts form from remaining, uninjured axons to reconstitute a terminal plexus.”
As we would use the terms today, the first definition of Moore would constitute canonical axon regeneration. The second is incomplete in that it does not encompass the different growth phenomena that are now known to occur following an injury.
An example of a form of growth that is not encompassed by the definitions above arises from the spinal cord injury field; following a thoracic spinal cord injury, new axonal branches extend out from corticospinal axons several spinal segments above the lesion site; these new axonal branches form contacts with spinal interneurons (Fig. 1D) forming a relay that can restore input to segments beyond the injury (Bareyre et al., 2004). New branches can also emerge at much higher levels of the neuraxis including the brainstem after axons are transected in the spinal cord (Z’Graggen et al., 2000) (Fig 1E). It has not been established whether such novel connections lead to functional relays as in Fig. 1D. The use of the term “sprouting” in this circumstance contradicts the definition of sprouting as growth arising from a spared, intact axon. A more descriptive approach for this phenomenon is cumbersome but clear: “axon branching arising from the proximal region of a transected axon.” Such a description will avoid confusion regarding the terms “regeneration”, “sprouting” or “regenerative sprouting,” to describe new growth arising from a transected axon, well away from the lesion site. It should be noted that the above studies did not show definitively that new branches were from axons that were transected at a lower level. This seems likely, but it cannot be excluded that new branches came from descending axons that terminate above the lesion and were not transected.
Sub-categories of sprouting have been defined based on the distance over which axons grow. For example, in the case of muscle reinnervation following partial peripheral nerve lesions, very short distance growth arising from spared axon terminals in the zone of innervation is referred to as “terminal sprouting.” Reinnervation arising from a spared axon has been called “collateral sprouting.” The latter type of sprouting has been described following partial denervation at multiple levels of the neuraxis including the spinal cord (Rosenzweig, 2009; Weidner, 2001).
There may be even shorter distance growth in which a surviving axon in a denervated zone forms new presynaptic specializations on denervated dendrites. This has been referred to as “reactive synaptogenesis,” a term that may overlap with “terminal collateral sprouting.”
Obviously, the proliferation and inconsistent use of terms leads to lack of clarity. Clarity is improved by simply describing the actual anatomical event to the extent that is possible, even if this is cumbersome.
II. Axon Regeneration After Spinal Cord Injury
There is general agreement that the greatest hope for recovery of function after spinal cord injury involves regeneration of the long tracts that mediate sensory and motor function. But what constitutes “axonal regeneration” and what is the minimal evidence required to make the claim that it has occurred?
We propose that the term axon regeneration should be reserved for: 1) growth of a cut axon, and 2) extension into or beyond a lesion. Regenerating axons can either end abortively (functionally irrelevant), form ectopic connections (could be either beneficial or detrimental to function), or form connections with their normal targets (likely to restore function). Regenerating axons may either extend through a lesion, through something that is implanted (peripheral nerve bridge, cellular graft, or bioengineered scaffold), or around the lesion through surviving white or gray matter. The level of proof for axonal regeneration should be rigorous, and is discussed in the next section.
After a spinal cord injury, there is essentially no re-growth of axons beyond the point of the injury. Instead, damaged axons end in what Ramon y Cajal called “retraction balls”. Recent evidence suggests that these are not static structures, and that there are periods of extension and retraction. In any case, the net result is no extension past the point of the original injury.
There are, however, a number of interventions that cause axons to grow to some extent. For example, axons may grow into a spinal cord lesion site that has been experimentally grafted with cells that provide a matrix permissive for axonal growth, such as sciatic nerve grafts, fibroblasts, marrow stromal cells, neural stem cells or Schwann cells. Because axons are normally completely absent from the center of a lesion, some would refer to axonal growth into the lesion site as “regeneration.” But if the axons growing into the lesion site arise from host axons neighboring the injury that were not transected, then is this growth “sprouting”, “regeneration” or “regenerative sprouting”? There is usually no way to answer to this question definitively, so use of the generic term “axon growth” followed by a description of the location and origin of the growth may be optimal without over-interpreting the findings. If it is shown that axons that grow into a graft originate from intact axons rather than transected axons, “axonal growth arising from spared axons” is accurate. If axons that grow into a graft unequivocally originate from transected axons, this would be bona fide “regeneration.” Regardless of the source of new growth, whether sprouting or regeneration, functional improvement is the ultimate goal of translational work in these model systems. However, imprecise or indiscriminate use of terms poses the risk of misguiding or misrepresenting the findings of an experiment, potentially undermining clear understanding of basic mechanisms influencing axonal growth after injury and ultimately retarding progress in the field.
What are the minimal criteria to establish a claim for axon regeneration? First, it is critical to provide compelling evidence that the axons that extend past a lesion are not spared. Criteria for this have been described (Steward et al., 2003), and are reasonably well accepted by the field. Next, how does one prove that growth involves “regeneration”; that is, that an axon growing into or beyond a lesion site originated from a transected axon?
Evidence Required for Claims of Axon Regeneration
Regeneration can be proven when all of the axons of a projecting system are lesioned (i.e., no axons are spared), and growth of labeled axons from an identified source is observed into or around the lesion site. Usually, this involves tract tracing to identify the origin, course, and termination of axons (Fig 2A-D). Studies in which pathways are labeled by genetically-driven fluorescent markers provide an alternative approach providing that the identity of the labeled axons can be definitively established, and it can be confirmed that the lesions completely interrupt the genetically labeled pathway (more on this below).
Figure 2. Anterograde or Retrograde Labeling to Identify New Axonal Growth.
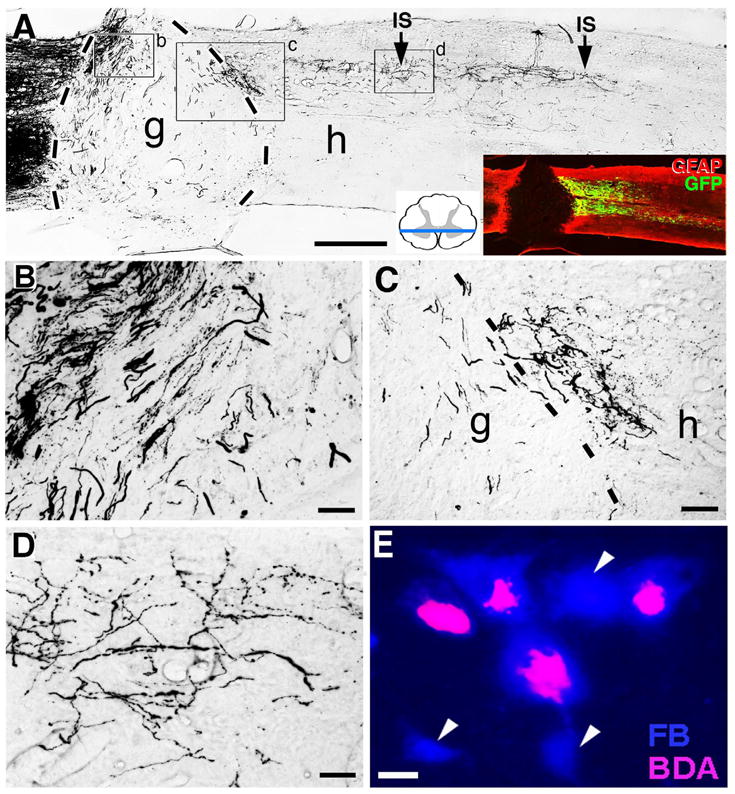
An example of anterograde axonal labeling to identify new axonal growth and putative regeneration is shown in panels A–D (from (Lu P, 2012)). Double retrograde labeling as a means of demonstrating new axonal growth and putative regeneration is shown in panel E (from Wolfram Tetzlaff, Univ. British Columbia). (A) After T3 complete transection, darkly labeled reticulospinal axons approach the lesion site from the left (rostral) direction in these 35μm-thick horizontal sections of the spinal cord. A graft (g) of bone marrow stromal cells is present in the lesion site. The border between the host spinal cord and the graft in the lesion site is indicated by black dashed lines. Darkly labeled axons are seen penetrating the graft (B), and some of these axons have grown to the caudal aspect of the graft occupying the lesion site, and beyond this point into the host spinal cord below the lesion site (C–D). The inset indicates one plane of the complete transection site by GFAP immunolabeling. Green indicates expression of Brain Derived Neurotrophic Factor (BDNF) and a reporter gene expressed by a lentiviral vector injected into the spinal cord below the lesion to attract growing axons; IS indicates vector injections sites. (B) In the graft adjacent to the rostral host/graft interface, numerous host axons have penetrated the graft. (C) At the distal graft (g) / host (h) interface, darkly labeled axons cross from the graft into the spinal cord below. Dashed lines indicate the border between graft and host. Note the irregular morphology and trajectories of axons growing beyond the lesion site. Axons are not in tightly fasciculated bundles typical of intact axons, but instead exhibit highly varied and branched morphologies, are dispersed from one another, and turn in various directions. (D) 1.5mm caudal to the lesion site, darkly labeled axons remain detectable and continue to exhibit varying trajectories. This continues 4mm caudal to the lesion. (E) Red nucleus in the brainstem of the rat. Rats were injected with the retrograde tracer Fast Blue at cervical segment 8 to back label intact rubrospinal neurons. The spinal cord was lesioned at the 4th cervical segment eight days later. After a delay, injured rats were treated with a sciatic nerve graft in the lesion site and BDNF infusions. Two months later, a second retrograde tracer was placed at the free end of the sciatic nerve segment in the lesion site, and the brainstem was subsequently examined. Some cells in the red nucleus were single labeled for Fast Blue (arrowheads), and some were double labeled for Fast Blue and BDA (visualized with the fluorophore Avidic-CY3). Double-labeled cells represented neurons that grew axons into the peripheral nerve graft; single labeled cells represented neurons that did not grow axons into the lesion site. Scale bar A, 1mm; A inset, 0.8mm; B, 70μm; C, 150μm; D, 70μm; E, 3μm.
Somewhat less satisfying, but still reasonably compelling evidence of regeneration can be obtained through a combination of double retrograde tracing. For example, in the case of studies of regeneration of descending pathways after SCI, a retrograde tracer is injected before the lesion (Fig 2E) to identify the cells of origin of a pathway that will subsequently be lesioned. After the lesion is performed and sufficient time has passed to allow potential axonal regeneration, a second (different) retrograde tracer is injected at the site of the original tracer injection. Hypothetically, an axon that has regenerated below a complete lesion of the system will exhibit labeling of the neuronal somata with both tracers (Fig 2E). A shortcoming of this approach is that it is not possible to determine the point of origin of the axons that grow or the course of the axons past the lesion.
For all assessments, it is critical to confirm that the experimental lesion completely transects the pathway being studied. Important evidence in this regard can be obtained by an analysis of axon distribution at different times post-injury. Long distance axon regeneration will take some time, including the time required for: 1) recovery from the axonal injury; 2) molecular changes required for a shift to a growth mode; and 3) elongation of the axon. Ramon y Cajal provided estimates of the timing of growth of regenerating peripheral nerves that sound quite plausible today: 1) Preparation of the dividing phase and growth of sprouts within the central stump (proximal to the injury): 2–5 days; 2) Growth through the scar (velocity of 0.25mm per day); elongation within the supportive environment of the peripheral stump (2.64mm/day) (Ramon y Cajal, 1928). Even under “regeneration enabled” circumstances, the rate of elongation may be slower in the CNS.
Studies of Regeneration in Specific Spinal Systems
A spinal cord injury creates a particularly hostile environment for regenerating axons. Astrocytes surrounding the lesion become reactive and extend processes. In most species including humans, the phagocytosis of degenerating neural tissue leads to the formation of large cystic cavities. If the lesion is complete, regenerating axons must grow into and beyond the lesion to reconnect with their normal targets. If the lesion is incomplete, some axons may extend along surviving bridges of white or gray matter. Depending on the lesion model and the axonal projection under study, new growth can occur into, or around, the lesion. We will now consider different axonal systems in the study of spinal cord injury, together with issues in assuring lesion completeness and establishing that regeneration has occurred.
Dorsal Column Sensory Axons
When performed properly, lesions of the dorsal spinal cord transect all ascending dorsal column sensory axons. This represents a model that can unequivocally demonstrate central axonal regeneration without requiring transection of the entire spinal cord (Fig 3). Rats and mice can readily survive this type of lesion with minimal challenges to survival. Lesion completeness can be established by confirming an absence of sensory axon terminals in the nucleus gracilis, for example by tracing ascending projections arising from the sciatic nerve (Fig 3F,G)(Lu et al., 2004; Taylor et al., 2006). Confirmation of lesion completeness by examination of the nucleus gracilis assumes that regenerating axons did not reach the nucleus gracilis, an assumption that is reasonable unless lesions are placed in close proximity to the nucleus (e.g., C1 level (Alto et al., 2009; Bonner et al., 2011)).
Figure 3. Spinal Cord Dorsal Column Sensory Regeneration Model.
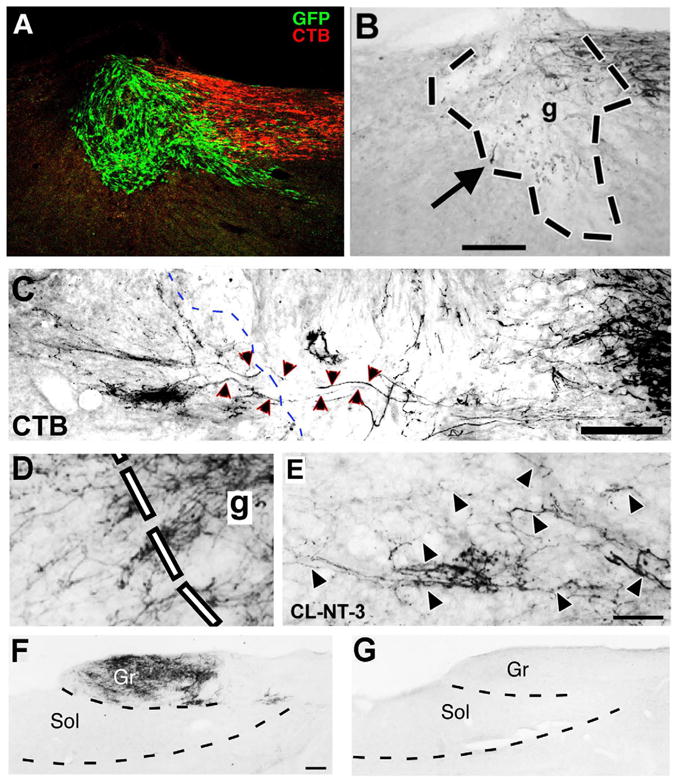
The dorsal columns of the spinal cord contain axons that ascend from the lumbar region to the nucleus gracilis in the medulla. These axons can be traced as they ascend the spinal cord by injections of the transganglionic tracer Cholera Toxin B subunit (CTB) into the sciatic nerve. (A) In animals that undergo C3 dorsal column transection lesions, CTB labeled dorsal column sensory axons approach the lesion site, but few penetrate a graft of bone marrow stromal cells placed in the lesion site; grafted cells express the reporter gene GFP (from (Lu et al., 2004). Sagittal, 35μm-thick section. (B) A higher magnification view at the light level shows the approach of axons to the lesion site (upper right), and rare axons in the graft (graft outlined within black lines). Arrow indicates a single axon that reaches the rostral graft/host interface, but does not regenerate beyond the lesion site. (C) Following combinatorial therapy with a conditioning lesion of the sciatic nerve, a marrow stromal cell graft in the lesion cavity, and lentiviral NT-3 growth factor delivery rostral to the lesion, numerous axons penetrate the graft and regenerate beyond it. Rostral aspect of graft is outlined by dashed lines. Regenerating axons within and beyond the graft are indicated by arrowheads. (D) At the rostral host-graft interface, numerous CTB-labeled axons regenerate out of the graft and into host white matter beyond the lesion. Axons exhibit irregular trajectories, make abrupt turns, and are generally dispersed, features typical of regenerating axons. (E) At a distance of 2mm rostral to the lesion site, regenerating host axons are present in adult white matter. Arrowheads indicate individual axons. From Alto et al., (2007) and Blesch et al., (2012). (F) Lesion completeness in this model can be confirmed by examination of the medulla. In intact animals, dense reaction product is evident in nucleus gracilis after injection of CTB into the sciatic nerve. Gr, gracilis; Sol, soleus. Transverse 35μm-thick section. (G) In contrast, the nucleus gracilis is devoid of CTB labeling in animals that have undergone complete C4 dorsal column lesions. Scale bar = A, 250μm; B, 500μm; C, 1mm; D-E, 20μm; F-G, 200μm.
Lesion completeness can be further assessed by injecting retrograde tracers into the nucleus gracilis after a dorsal column lesion and observing an absence of tracer in the dorsal root ganglia. There is a caveat about such negative findings however, because absence of evidence is not compelling evidence of absence. For example, there is always a possibility of technical failure of retrograde transport.
The dorsal column lesion model is helpful for understanding mechanisms underlying central axonal regeneration and identifying experimental effects of candidate therapies for enhancement of axonal regeneration. Functional sensory deficits can be assessed, but to restore sensory function, therapies must lead to axonal regeneration all the way to the nucleus gracilis. So far, sensory axon regeneration back to the dorsal column nuclei has only been seen following lesions at high cervical levels (Alto et al., 2009; Bonner et al., 2011).
Corticospinal Axons
The study of corticospinal tract (CST) projections is important in spinal cord injury models, as this motor projection is critical for human voluntary motor function. However, because CST axons descend in several different tracts, it is particularly challenging to distinguish regeneration from sprouting of spared axons, and to detect inadvertently spared axons.
In species used most extensively for experimental studies, corticospinal axons originate primarily from neurons in layer V in the sensorimotor cortex. It is important to note, however, that other cortical areas also contribute, including the dorso-medial frontal cortex. Most CST axons decussate in the pyramidal decussation and then descend through the spinal cord in three tracts: a dorsal tract in the ventral part of the dorsal column (the main tract in rodents), a dorsolateral tract (the main tract in primates), and a ventral tract that is sparse in most species and is not detected in some strains of mice (Fig 4). The dorsal and dorsolateral CST contain axons from the contralateral cortex whereas axons in the ventral CST are from the ipsilateral cortex. Our impression, based on assessment of labeling in hundreds of rats and mice, is that the parcellation of axons between the two minor tracts varies even across individuals within the same species.
Figure 4. The Corticospinal Tract (CST).
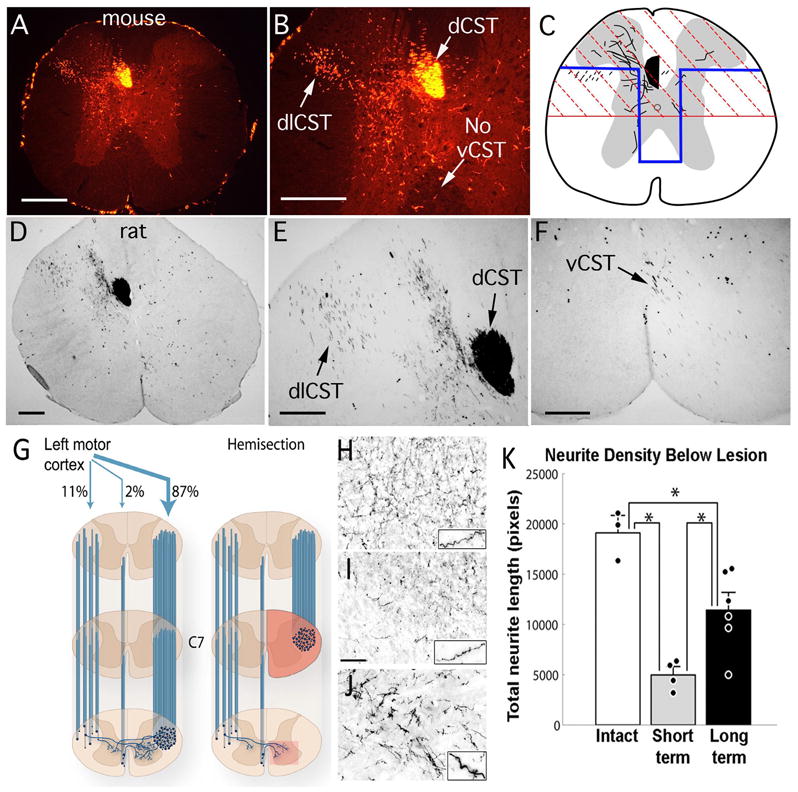
(A, B) Corticospinal axons in a mouse are traced here by making placing four injections of mini-ruby BDA into the right sensorimotor cortex. CST axons from the right hemisphere are labeled. In mice, CST axons descend primarily in two tracts: the dorsal CST, in the ventral part of the dorsal column (dCST), and the dorsolateral CST (dlCST) in the dorsal part of the lateral column. Note the absence of labeled axons in the ventral column on the right side; in rats, some axons are present in this region. Note also the presence of a few labeled axons in the dorsal column on the right in the same location as the dCST. This illustrates the fact that there are a small number of axons that descend in the spinal cord ipsilateral to the cortex of origin. In this case, labeled CST axon arbors are found mainly in the ventral horn gray matter. The distribution of axonal arbors depends on whether the injections target mainly the sensory vs. motor divisions of the sensorimotor cortex. Injections that mainly target the primary motor cortex preferentially label CST axon arbors in the ventral horn whereas injections that mainly target the sensory cortex preferentially label CST axon arbors in the dorsal horn. (C) Schematic illustration of partial transection lesions that are commonly used for assessing CST regeneration in mice and rats. The red region denotes a dorsal hemisection; the blue region indicates a “T” lesion, which is intended to include the ventral CST in rats (modified from Zheng et al., 2006). (D, E) Corticospinal axons in a rat are traced here by making six injections of mini-ruby BDA into the right sensorimotor cortex. Labels are as in A, B. In this case, labeled axonal arbors are found mainly in the dorsal horn gray matter. (F) higher magnification view of BDA-labeled axons in the ventral column (vCST). Scale bars = 250μm. (G, K) Sprouting of CST axons below a hemisection injury in rhesus monkeys. (G) The schematic on the left illustrates the organization of the CST in rhesus monkeys. Descending CST axons from the left motor cortex are shown. The main component of CST axons descend in the lateral column, as in humans. About 87% descend on the side contralateral to the cortex of origin; 11% descend through the lateral column on the ipsilateral side; the ventral CST contains about 2% of the total number of labeled axons. The schematic on the right illustrates the lesion model, a hemisection at C7, and the CST axons that would survive such lesions. The box indicates the CST arbors in the gray matter that arise from axons decussating across the spinal cord midline. (H) Density of BDA-labeled CST axon arbors in the gray matter in intact monkeys (which includes labeled axons from the main tract and the crossing axons); (I) arbors of surviving crossed CST axons at early time points after a C7 hemisection; (J) arbors of crossed CST axons traced 8 months after a hemisection injury, when extensive compensatory sprouting has occurred. (K) Quantitative assessment of the density of crossed CST axons in the gray matter in intact monkeys, at short intervals after the injury, and at long post-injury intervals. By eight months post-lesion, there is a substantial reconstitution of corticospinal axons below the lesion site, due to sprouting of spared contralateral axons. Panels G-K are from Rosenzweig et al., 2010. Scale bar in “I” = 100μm, and applies to H–J.
Regarding the use of rodent models for spinal cord injury studies in general and CST regeneration in particular, it is noteworthy that most CST axons in rodents are located in the spinal cord dorsal white matter; this is a key distinction from humans, where the main CST descends in the lateral columns.
CST axon collaterals leave the main tract and terminate mainly on the side contralateral to the cortex of origin. A few CST axons re-cross the midline at segmental levels to terminate ipsilaterally (Fig. 4). Re-crossing axons are sparse in rats, somewhat more common in mice, and are more prominent in primates. The extent of re-crossing in humans is not known.
Several publications have reported regeneration of CST axons after spinal cord injury in rodents, but many of these studies leave doubts. Unless the spinal cord is transected completely, lesions usually spare axons in one or the other of the component pathways, so that axons observed below the lesion site could be due to sprouting from spared axons. Complete transections can solve this problem, but are difficult to create and are extremely disabling to the animals. Many early claims of CST regeneration after complete transection have not stood the test of time and replication, based on later evidence that axons were actually spared. Most often, spared axons lie within the most ventral and lateral aspects of the lesion site. Also, complete transections create an environment that is an extraordinary barrier because the two stumps pull apart leaving a fluid filled space that can be many millimeters in length. Even when filled with a transplant or a growth-promoting substrate, a large lesion represents a very challenging barrier for regenerating axons. In our view, no study to date has convincingly demonstrated regeneration of CST axons across a complete spinal cord transection site, and this remains a key goal of spinal cord regeneration research.
Although complete transection, properly done, may be the “gold standard” for demonstrating regeneration, achieving regeneration in this model may be a higher bar than is needed to identify a meaningful therapeutic advance. Indeed, the bar is even higher than most cases of human spinal cord injury because human injuries are most often crush injuries due to vertebral displacement or contusion injuries. There is often at least some spared rim of white matter even in severe human injuries.
Because a complete lesion is technically difficult, disabling for the animal, and creates a substantial barrier to regeneration, most contemporary studies of CST regeneration use partial injury models. One commonly used model is a dorsal hemisection (Fig. 4C), which in rats, spares the ventral CST. When the ventral CST is spared after removal of all dorsal projections, ventral projections can exhibit remarkable branching and ramification that support partial functional improvement (Rosenzweig et al., 2010; Weidner, 2001).
Contusion injuries created by impactors can completely destroy the dorsal CST, but usually spare both the dorsolateral and ventral CST, which can be a source of sprouting below the injury. Given the extent and variability of the contusion lesion, it is very difficult to determine whether CST axons caudal to the injury are the result of sprouting from spared axons or regeneration. The former is far more likely.
A “T-lesion” has been used in rats (Fig. 4C) in an attempt to eliminate all dorsal, dorsolateral and ventral CST axons (Liebscher et al., 2005), but these lesions are technically very challenging and, potentially, of variable accuracy. Also, as typically performed, the lesions can spare the dorsal part of the lateral column, potentially sparing axons of the dorsolateral CST.
Lateral hemisections have also been used to examine corticospinal growth after destroying CST axons traveling on one side of the spinal cord. In rodents, it is difficult to selectively destroy CST axons on one side because the main component of CST axons in the dorsal column is adjacent to the midline. Often, the lesions spare axons near the midline, or extend across the midline to involve the contralateral, “intact” system. Accordingly, the lateral hemisection model in rodents is vulnerable to uncertainties both with regard to regeneration and sprouting. Moreover, some corticospinal tract axons decussate across the spinal cord midline; these spinal-decussating axons are sparse in normal rats, but are present in mice and common in primates (Rosenzweig et al., 2009). Indeed, following a lateral hemisection in primates, corticospinal axons that normally decussate across the spinal cord midline sprout exuberantly and reconstitute up to 50% of axon terminals lost after lateral hemisection, a remarkable degree of anatomical plasticity (Fig 4G-I) (Rosenzweig et al., 2010).
Spinal cord “crush” models (even “complete” crush) can spare tracts of white matter and are difficult to create consistently. Sparing of ventrally-located axons is particularly problematic. Careful histological analyses can address some of these concerns, with rigorous documentation of lesion extent and serial tracing of axons across different planes of sampling (Fig. 5). Functional analyses are compromised unless thorough histological analyses are carried out on every animal to confirm lesion completeness.
Figure 5. CST Tract Reconstruction.
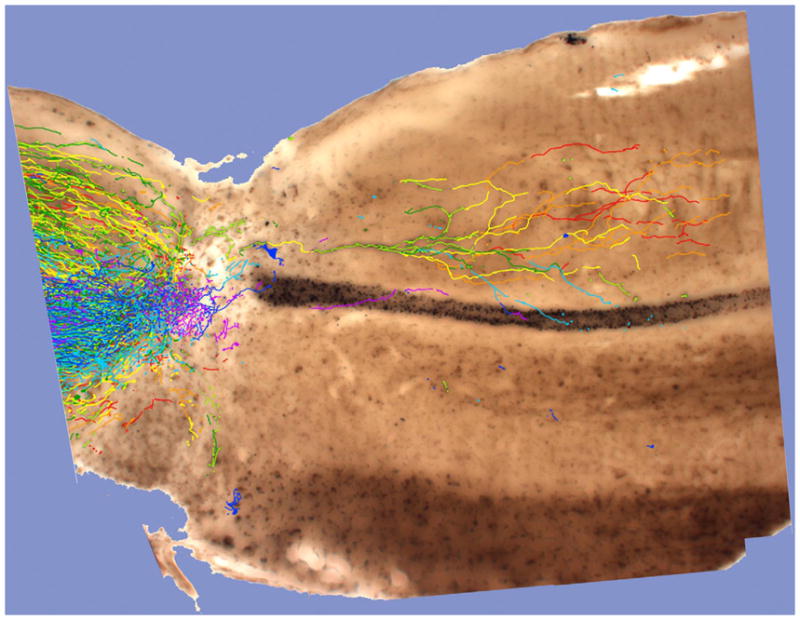
CST axons were anterogradely traced with BDA in a PTEN-deleted mouse that survived for a total of 12 weeks following a complete spinal cord crush at T8. The image illustrates BDA-labeled axons in 50 μm thick serial sagittal sections. Labeled axons in each image were traced in Adobe® Photoshop®, and the tracings were stacked and superimposed onto a light micrograph of one section containing the central canal. Axon segments are rainbow color-coded according to depth of each of the 8 serial tissue sections, thus color-coding relative depth in 50 μm increments through 400 μm of tissue. Order of colors is red, orange, yellow, yellow green, green, cyan, blue, purple. This image is from the case described in Liu et al., 2010 in which sections were embedded and sectioned for electron microscopy in order to locate BDA labeled synapses. (Image courtesy of Rafer Willenberg, UC Irvine.)
In all of these partial lesion models, strong supportive evidence can mitigate concerns about sparing. If the axons take a course that is not seen in un-injured animals, the claim for regeneration can be persuasive. For example, deletion of the tumor suppressor gene “phosphatase and tensin homolog” (PTEN) in mice after either dorsal hemisection or severe crush lesion results in bilateral extension of CST axons below the lesion that originate from a single hemisphere (Liu et al., 2010). Such bilateral projections are extremely rare in controls, and their abundance in PTEN deleted mice is evidence for regeneration.
Even when it can be established that axons have re-grown past the lesion, it is usually not possible to conclude with certainty whether these axons originate from transected axons or from sprouts of spared CST axons that ordinarily terminate rostral to the injury. This requires complete reconstruction of the origin and course of the axons, which in turn requires sparse labeling.
Absence of CST growth into grafts and transplants
Many studies have assessed whether grafts or transplants can support CST growth. Implanted matrices have included Schwann cells, astrocytes, neural stem cells, fibroblasts, oligodendrocyte precursor cells, bone marrow stromal cells or other substances (Blesch and Tuszynski, 2009). While these matrices support the growth of other motor systems, including raphespinal, rubrospinal and reticulospinal projections after injury, it is noteworthy that none of these matrices support CST axon growth. The only matrix to date that supports CST growth are grafts of fetal spinal cord (Coumans et al., 2001), and even then, growth is modest. Also, fetal spinal cord grafts are of limited practical usefulness because the grafted cells exhibit variable survival and rarely fill the lesion site (Coumans et al., 2001). A major unmet challenge in the field of spinal cord injury research remains the identification of a substrate or matrix that will enable CST axon growth into a cystic lesion site.
Although the CST has been relatively refractory to most therapeutic manipulations, other descending systems including raphespinal, cerulospinal, reticulospinal, rubrospinal and propriospinal axons are somewhat more responsive (Blesch, 2009). These systems mediate functions (locomotion, posture, balance, autonomic control) that would be important to comprehensively improve functional outcomes in people with SCI (Anderson et al., 2008).
Serotonergic systems
Serotonergic projections to the spinal cord from the brainstem raphe nuclei modulate the activity of spinal motor systems, and preservation or restoration of serotonergic input improves locomotor function (Courtine et al., 2009; Rossignol and Dubuc, 1994; Thompson et al., 2011). Raphespinal axons arise from cells in the midline raphe (Fig 6) and travel caudally through the spinal cord as dispersed bundles of axons neighboring the central gray matter (Fig 6). Complete lesions of raphespinal axons require extensive bilateral lesions that extend ventrally well below the central canal. Accordingly, the most reliable model for examining regeneration of this system is a complete spinal cord transection or crush (Fig. 6C). While there has been some question regarding the existence of intrinsic serotonin-containing neurons with the spinal cord that would complicate the assessment of axonal regeneration even below a complete transection site, routine serotonin immunohistochemistry with an antibody to 5-hydroxytyptamine (5HT) does not detect residual neuronal or axonal labeling below a complete injury (Fig 6C). Although there are few reports of regeneration after complete lesions (Coumans et al., 2001), the extent of regeneration reported is modest.
Figure 6. Raphe-Spinal Pathways.
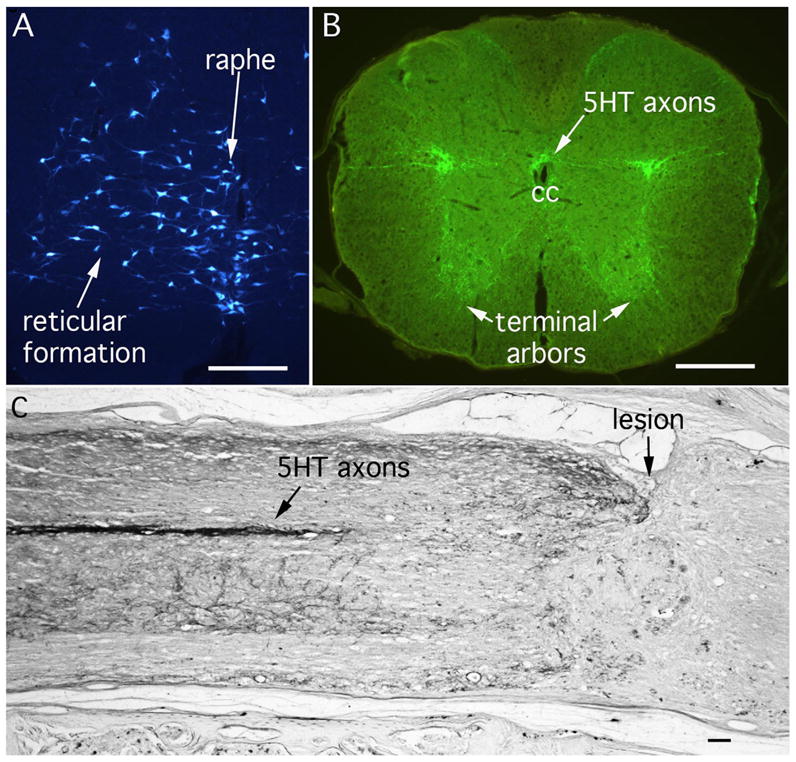
(A) Retrogradely labeled neurons in the midline raphe and reticular formation after injections of true blue into the spinal cord of a mouse. Neurons in the raphe nucleus give rise to serotonergic (5HT) axons that project to the spinal cord. (B) Immunofluorescence for 5HT in the thoracic spinal cord. CC = central canal. (C) Immunolabeling for 5HT after a “complete crush” injury in mice. The lesion site is indicated. Scale bars = 250μm.
Many previous studies report treatment-related increases in serotonergic axons below an injury and growth of serotonergic axons into partial spinal cord lesion sites containing cell grafts (Lu et al., 2003). Such growth could result either from regeneration of transected axons or sprouting of neighboring axon terminals that were spared by the lesion. Distinguishing between these is probably impossible, so “increase in serotonergic axon density” or “axon growth into the lesion site” are the most appropriate phrases for describing these forms of axon growth.
Rubrospinal axons
Rubrospinal projections are considered to be rudimentary in humans although this point is not entirely settled (Nathan and Smith, 1982; ten Donkelaar, 1988). In rodents, rubrospinal axons arise from the magnocellular division of the red nucleus (Fig 7A), cross the midline, and project through the dorsal part of the lateral column of the spinal cord and modulate motor function. (Kuchler et al., 2002; Morris et al., 2011). Rubrospinal axons can be labeled by making tracer injections into the brainstem (Fig. 7D&E show the pathway after injections in a mouse). The rubrospinal tract can be completely transected by lateral funicular lesions, which therefore represent an attractive model system for the study of mechanisms underlying motor axon regeneration, albeit with the important caveat that the projection is of limited importance in humans. Rubrospinal axons exhibit a greater capacity to regenerate than CST axons (Liu et al., 1999). This system, like all others, is also subject to the caveat that growth into or beyond a lesion site can arise from either sprouting of spared axons or regeneration of transected axons unless it can be confirmed by complete reconstruction of axons extending past the lesion that growth originated from an axon that was unequivocally cut. In the absence of such confirmation, studies of this system should apply the term “axonal growth” in sub-total lesion models.
Figure 7. Brainstem Motor Pathways: Rubrospinal, Reticulospinal and Vestibulospinal tracts.
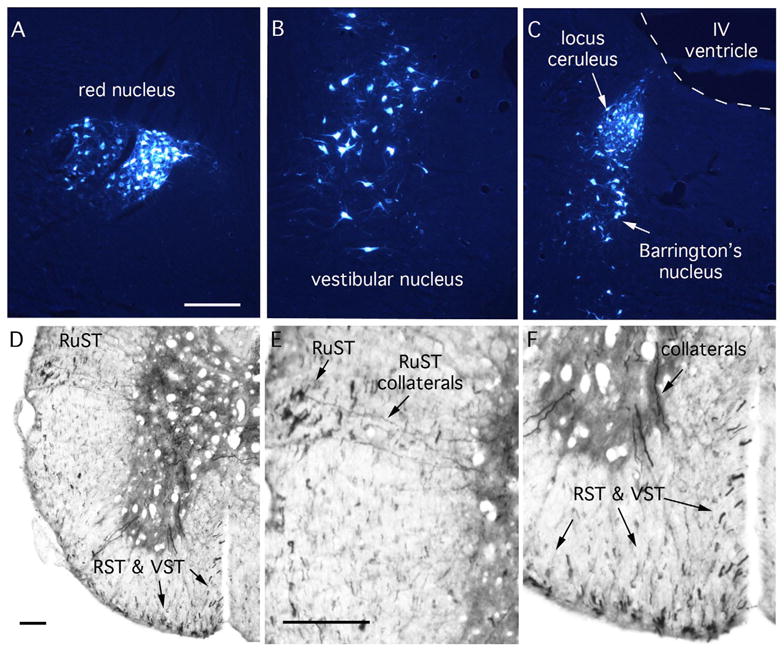
A–C: Retrogradely labeled neurons in the brainstem after injections of true blue into the spinal cord of a mouse. (A) red nucleus; (B) vestibular nucleus; (C) locus ceruleus and Barrington’s nucleus. D–F illustrate BDA labeled axons after a large injection of BDA into the brainstem of a mouse. RuST, rubrospinal axons; RST, reticulospinal axons; VST, vestibulospinal axons. Note collaterals extending from the RuST into the gray matter in E. Collaterals in F are likely from the RST or VST. Scale bars = 250μm.
Reticulospinal axons
Reticulospinal projections are the principal motor pathways in lower vertebrates that lack a cortex. In limbless animals, reticulospinal pathways control trunk musculature to mediate swimming and crawling. In vertebrates with limbs, reticulospinal pathways activate spinal motor neuron pools involved in a variety of functions including locomotion and postural maintenance (Alstermark et al., 1983; Shapovalov and Gurevitch, 1970; ten Donkelaar et al., 1980; Wilson and Yoshida, 1968).
Several brainstem nuclei give rise to reticulospinal projections, with the greatest density arising from the pontine gigantocellular reticular nucleus. Reticulospinal axons can be labeled by injecting anterogradely transported tracers into the brainstem (Figs. 2, 7); but tracer injections may also label other spinally-projecting brainstem axonal systems, including vestibulospinal, rubrospinal, cerulospinal and raphespinal tracts. Axons that are labeled in the spinal cord as a result of tracer injections targeting the reticulospinal pathway are widely dispersed in the spinal cord, but are predominantly located in the ventral column (Figure 7D&F). Because descending axons are dispersed, complete spinal cord transections are the best model to unequivocally assess whether axons of this system have regenerated. Reticulospinal axons grow into cellular matrices placed within partial spinal cord lesion sites (Blesch and Tuszynski, 2009; Jin et al., 2002), and, as with other systems described above, this growth may arise either from regeneration of transected axons or sprouting of neighboring, intact axons. Unless there is compelling evidence that ingrowing axons arise from an axon that has definitively been cut, the term “axon growth” should be used when referring to axons that extend into a lesion. When interventions increase axon number below a lesion, “increase in reticulospinal axon number” is the most appropriate phrase.
Cerulospinal axons
Noradrenergic inputs to the spinal cord arise from the locus ceruleus (Fig 7C) just dorsal to another important nucleus called Barrington’s nucleus that is a key regulator of bladder function (Fig. 7C). Cerulospinal axons modulate the activity of intraspinal circuitry including motor systems (White and Neuman, 1980). These projections travel in dispersed bundles of axons predominantly in lateral spinal cord white matter, and can be identified by immunolabeling for tyrosine hydroxylase (TH) or dopamine beta hydroxylase (DBH) (Tuszynski et al., 1994) (Fig. 7). The same general issues apply with this system as for the other pathways in terms of documenting regeneration and distinguishing regeneration and sprouting.
Propriospinal axons
Propriospinal neurons project up and down the spinal cord to coordinate spinal circuitry, including inter-limb coordination (Kostyuk and Vasilenko, 1978) (Alstermark et al., 1984; Courtine et al., 2008). There are no specific means of identifying these projections, as the neuronal somata are located in spinal cord gray matter and are difficult to selectively target with tracer injections. Thus, these projections have been difficult to study in the context of spinal cord injury. By injecting retrograde tracers into the stumps of sciatic nerve grafts or tubes placed in sites of complete spinal cord transection it has been shown that intraspinal neurons extend axons into permissive matrices placed in lesion sites (Xu et al., 1997). New advances involving genetic labeling of defined neuron types hold the potential to make this population of neurons amenable to experimental study (more on this below).
Distinguishing New Axonal Growth From Incomplete Lesions
No discussion of axonal growth after spinal cord injury, whether resulting from regeneration or sprouting, is complete without reference to the problem of “false resurrections.” This refers to the risk of mistaking an unintentionally spared axon for a newly growing axon. This issue in spinal cord regeneration research is no less important – or problematic – today than when it was addressed in detail in 2003 (Steward et al., 2003). Few additional comments can be added to the original commentary. It remains vitally important that any description of new axonal growth avoid this major pitfall, which can divert the field for years in pursuit of ephemeral notions that ultimately fail the test of replication.
Other Pitfalls
Two other potential sources of error in judging axonal growth after injury merit discussion. Depending on the type of spinal cord lesion created, and particularly in the case of compressive/contusive type injuries, the lesion gradually expands over several weeks into an oval or cigar-shaped cavity extending along the rostral-caudal spinal cord axis (Gruner et al., 1996). Thus, what begins as a small lesion can become an enlarged, elongated lesion. In judging axonal growth into and beyond this type of lesion, it is critical to define the boundaries of the expanded lesion so that one does not mistakenly assume axons have regenerated beyond a lesion when in fact they remain within a (larger) lesion. Immunostaining for GFAP provides one way to define lesion margins, and immunostaining for vimentin, nestin or NG2 can also be useful (Fitch, 2008). A second issue to consider in judging the effect of an experimental manipulation on axonal growth is the “dying back” phenomenon (Ramon y Cajal, 1928), wherein lesioned axons typically retract from the site of injury. Myelinated axons often retract approximately one myelinated segment to a Node of Ranvier proximal to the lesion site. If an experimental therapy reduces axonal dieback, then it is possible to mistakenly interpret this as new axonal growth up to the lesion margin. This error can be avoided by sampling several time points shortly after the lesion, to ensure that axonal dieback followed by new growth has actually occurred.
Summary of Guidelines for Demonstrating Axonal Regeneration
Evidence to support a claim that an injured axon has regenerated into or beyond a lesion should ideally include:
Use of a model in which regenerated axons can be definitively distinguished from spared axons. Options include using models in which all axons of the projection system have been cut; or a partial lesion or crush/compression models in which individual “regenerating” axons can be unequivocally traced to their point of origin in the lesioned tract (either strict serial section reconstruction or analytical techniques using unsectioned spinal cord (Erturk et al., 2012), Fig. 8). Documentation of lesion extent is critical. A single photomicrograph showing a “complete” lesion in one 40μm-thick section is not evidence of a complete lesion in the remaining 3000μm of spinal cord; systematic sampling and documentation of lesion extent through the full width of the spinal cord should be provided.
A demonstration that the morphology of putatively growing axons is consistent with new axonal growth: strictly linear axons in normal tracts strongly suggest axon sparing whereas regenerated axons will more likely exhibit an irregular growth trajectory (Lu et al., 2004).
A time course analysis confirming that axons actively extend over time starting from the point of injury, extending into or around a lesion site, then gradually beyond the lesion over clear serial time points.
-
Where possible, a confirmation of loss of innervation of targets.
Residual labeling likely represents an incomplete lesion unless compelling evidence is presented to the contrary. In complete transection models, the absence of detectable axons at long distances beyond the lesion suggests a complete lesion; in dorsal column sensory axon transection models, sectioning of the medulla should confirm an absence of axons in the target (Lu et al., 2004).
A demonstration of an absence of similar labeling in convincing controls, with adequate numbers of subjects.
Figure 8. Three Dimensional Imaging of the Unsectioned Spinal Cord.

Using tetrahydrofuran-based methods, the unsectioned adult rat spinal cord can be “cleared,” supporting visualization of the course of individual, fluorescently labeled axons through a spinal cord lesion site (Erturk et al., 2012). This supports tracing the origin and course of individual, lesioned axons. (A) The whole-mounted spinal cord in a 3D representation, showing axons labeled in transgenic M mice (Feng et al., 2000) expressing GFP in sparse neuronal populations (Erturk et al., 2012). (B) Reconstruction of a plane of section from the same spinal cord demonstrating GFP-labeled sensory axons (arrows) approaching and growing within a lesion site (lesion margins indicated by dashed lines). Rats underwent peripheral “conditioning” lesions of the sciatic nerve to enhance regeneration. Caudal is to the right, rostral to the left; the direction of axonal regeneration is right to left. (Courtesy of A Erturk and F Bradke.)
Supportive evidence, in addition to the preceding, to support a claim of regeneration:
Demonstration that axons are located in ectopic locations, outside the normal topography of axon distribution for the system under study, reflecting new growth.
If axons are growing through an implant of some type placed in the lesion site, visualizing axon growth not only from the most ventral, dorsal or lateral aspects of the lesion site, where spared axons are most likely to be present, but through the central regions of the implant.
Finally, independent replication of a reported experimental effect lends confidence. One clear example of successful replication in spinal cord injury research is the growth-enhancing effect of conditioning lesions of the sciatic nerve on centrally-projecting sensory axons (Bisby and Pollock, 1983; McQuarrie et al., 1977; Neumann and Woolf, 1999; Oudega et al., 1994). Moreover, efficacy in different models of SCI further confirms the biological validity of a presumed mechanism related to regeneration.
III. SCI vs. Other Models for Studies of Axonal Regeneration
We have focused on spinal cord injury because it exemplifies the problems that arise in studies of axon regeneration in most areas of the CNS. There is an extensive literature on axon regeneration in the olfactory nerve and optic nerve, but these CNS structures differ in important respects from the spinal cord or other CNS areas. The olfactory nerve is a special case because olfactory receptors undergo continuous turnover, so there is naturally occurring axon growth in the nerve. This may reflect the fact that the olfactory nerve contains a special type of glial cell, olfactory ensheathing glia (OEG), that either support or are at least permissive for olfactory axon growth.
The optic nerve is also a CNS structure; it contains the central processes of retinal ganglion cells, which are axonal in nature and are indistinguishable anatomically from other CNS axons. The glial environment of the optic nerve consists of oligodendrocytes and astrocytes, replicating inhibitory features at sites of injury consisting of astrocytic “scar” formation and the presence of myelin-associated growth inhibition (nogo, MAG, OMgp, others; (Benson et al., 2005; Bray et al., 1991; Cao et al., 2010; Giger et al., 2010; Keirstead et al., 1989; Low et al., 2008; Schwab et al., 2006)). Like other central axons, optic nerve axons fail to regenerate beyond this inhibitory milieu (Benowitz and Yin, 2008; Bray et al., 1991; Park et al., 2008). The optic nerve differs from other CNS areas in several respects, however. First, it is a pure axonal tract (no gray matter). Second, in distinction to most spinal axons, the vast majority (>99%) of retinal ganglion cells die after optic nerve transection, a far greater proportion than the number of degenerating neuronal cell bodies that give rise to axons traversing a spinal cord lesion site. This raises the possibility that a unique biological feature of a subset of surviving retinal ganglion axons is the actual subject of study. The simplicity of the optic projection to thalamic and collicular targets is a virtue: the nerve consists essentially of a single projection to few targets. If an optic nerve lesion is complete, then there is little question that regeneration has occurred. However, its simplicity is also a drawback: the optic nerve model poorly replicates the diverse and complex nature of a spinal cord injury, which by virtue of containing both gray and white matter results in hemorrhagic necrosis, extensive inflammation, and secondary cell death and cavitation. Moreover, the complex circuitry of the spinal cord presents a diversity of inappropriate targets through which growing axons must hypothetically navigate before restoring useful function. Thus, the primary strength of the optic nerve model may lie in understanding fundamental mechanisms underlying axonal degeneration and regeneration, leading to the identification of targets that can then be tested in models of SCI (Kurimoto et al., 2010; Park et al., 2008). The model is discussed in more detail in other reviews (Benowitz and Yin, 2008; Maclaren and Taylor, 1997).
Peripheral Nerve Injury
Studies of peripheral nerve injury have been invaluable in identifying neural mechanisms that underlie successful regeneration (Griffin et al., 2010; Longo et al., 1984; Ma et al., 2011; Ramon y Cajal, 1928); peripheral nerve injury models continue to yield important findings in the field (Ma et al., 2011; Mantuano et al., 2011). The difference in perception between investigators studying central versus peripheral axonal regeneration can be amusing, as peripheral nerve investigators highlight the incompleteness and limitations in axonal regeneration after injury, whereas spinal cord investigators relish the day that growth of central axons will begin to approach the intrinsic capabilities of peripherally injured axons. As noted early in this monograph, there is also often a gulf in the use of the terms “growth”, “sprouting” and “regenerationa” as applied in the peripheral nerve literature and the CNS. A review of peripheral nerve models is beyond the scope of this Primer and interested readers are referred to recent reviews (Griffin et al., 2010; Zochodne, 2012).
IV. Why Are Controls Sometimes Inadequate?
Comparison of an experimental treatment group to an untreated or inactive-drug control group would seemingly compensate for several potential errors addressed in preceding sections. Yet this is often not the case. Several classes of errors can account for mistaken findings despite the use of control groups.
A common error is under-powering of studies. This topic has been addressed in detail in a recent monograph (Scott, 2008). Conceptually, inadequate numbers of study subjects would most commonly lead to the mistaken conclusion that a treatment has no effect (a Type II statistical error) when in fact greater numbers of subjects are required to demonstrate the effect of a smaller yet biologically significant effect. The problem is that under-powered studies with negative results are not generally published. Consequently, under-powered studies that yield statistically significant results (a Type I statistical error) may be over-represented in the literature. Indeed, there have been several reports in the field of spinal cord injury research where early suggestions of treatment effects evaporate when larger numbers of subjects are examined. The problem of preferential publication of studies with Type I statistical errors has been called the “file drawer problem” (Kennedy, 2004): journals are the likely repository of the 5% of the studies with Type I errors while file drawers contain the 95% of the studies in which differences do not reach statistical significance.
This and other problems of reproducibility have been highlighted by the FORE-SCI Project sponsored by the National Institutes of Neurological Disorders and Stroke. The Program funded contracts that supported replication of promising reports related to neuroprotection or regeneration. Of 11 published replications, only one (a study involving a neuroprotective strategy) has fully confirmed the findings in the original report [for a review, see (Steward et al., 2011)]. Mistaken conclusions retard progress in the field and drain resources; greater efforts are required to avoid these miscues. Efforts by experimentalists to gain training in models of spinal cord injury, together with the use of proper controls, blinded treatments and assessments, and true observer objectivity, will reduce, but not always eliminate, the risk of errors.
An adequate sample size to determine the effect of an experimental treatment varies by the potential effect size of the treatment, and the variability of the measures used to assess the outcomes. For example, when using a complete spinal cord transection model, control groups exhibit no detectable supraspinal axons below the lesion. If a treatment actually causes regeneration, relatively few animals (less than 6 per group) would provide reliable anatomical outcome data because all values in the control group would be “0”. In partial lesion models, it is more difficult to achieve consistency, so variability in outcomes usually increases, and greater sample sizes are needed.
When function is the outcome measure, there can be considerable variability arising from several sources. First, behavioral performance is influenced by multiple factors other than the anatomical substrate (motivation, concurrent illness, various medications, etc.). Second, functional outcome is related to final lesion size, and many physiological factors contribute to final lesion volume, only some of which are under experimental control (for example, extent of hemorrhage). Accordingly, functional outcome studies may require dozens of animals per group to reach reliable conclusions in partial lesion models. Rarely are studies of such size performed, however. Moreover, studies with a large “n” can only be performed by staging over time, which creates other ambiguities.
Another error that can lead to misinterpretation of experimental outcome is the use of controls from previous studies in a new set of experiments (historical controls), or combining of animals into single groups from experiments conducted at different time points (Sharp et al., 2010). Some of the variables that drift over time include techniques of surgery, post-operative care, data collection (especially in functional assessment), and even the routine handling by vivarium staff. All of these variables are directly related to personnel, and even if the same people are involved, skill level changes over time. Variables unrelated to personnel include time of year and genetic constituency of the study subjects (particularly inbred animal strains). When the need to control variability is high, as with small effect size, drift over time can influence experimental outcome independently of the effect of a controlled variable (e.g., a therapeutic experimental manipulation). This drift can even occur within the time frame of a single experiment. We are familiar with a case in which an investigator performed “complete” spinal cord lesions on a group of animals that received an experimental therapy in the morning, then performed complete transections on the entire “control” (untreated) group in the afternoon. There was a significant difference in functional outcome and axonal “regeneration” between groups. However, independent inspection of the lesions revealed that all lesions were incomplete in the experimental (morning) group and were more complete in the control (afternoon) group. Apparently, the investigator, who did not have much experience in performing spinal cord lesions, gained greater skill and experience in performing lesions over the operative day. This highlights the need to intersperse “control” and “experimental” subjects continually, to generally utilize similar numbers of control and experimental subjects, and to perform studies in a blinded manner.
V. Methods of Studying Axonal Growth
The methods used to study axonal growth after spinal cord injury depend on the axonal system under study and the experimental hypothesis.
Immunolabeling
For pathways that contain unique proteins, immunolabeling is often used. For example, 5-hydroxytryptamine (5HT) labeling is a satisfactory method for identifying serotonergic projections; dopamine beta hydroxlase (DBH) immunolabeling can identify cerulospinal projections; and Substance P, IB4, and CGRP immunolabeling can identify different subclasses of primary afferents from dorsal root ganglion cells.
Some immunolabels have been used to label specific axonal systems, but lack specificity leading to potential confounds in data interpretation. For example, choline acetyltransferase is expressed by alpha motor neurons and pregangionic sympathetic neurons. Protein kinase C – gamma (PKC-gamma) labeling has been used to identify CST axons, but this label is not specific and cannot be used to detect growth responses of CST systems. PKC-gamma is mainly useful for detecting the loss of axons in the CST following lesions. Similarly, growth-associated protein 43 (GAP-43) labeling has been used by some investigators as an indicator of growing axons, but in fact, GAP43 is expressed constitutively by some spinal cord systems including the CST. Thus the presence of GAP43-labeled axons after a lesion is not a useful indicator of new growth. The study of growth of CST projections, and many other systems, requires tracers or genetic labels.
Tract Tracing
Tract tracing has been the gold standard for studying new growth from axonal systems that lack specific immunolabels, including corticospinal, rubrospinal, reticulospinal and some sensory systems. Many anterograde tracers are available that provide exquisite axonal morphology, including dextran amines, phytohemagglutinin (PHA), and fluorogold. Mini-ruby BDA provides the additional advantage that its fluorescence can be directly visualized, without amplification by immunolabeling. A particularly useful tracer for central sensory projections is the transganglionic tracer cholera toxin B (CTB). This tracer can be very simply injected into the sciatic nerve, and it will fill central dorsal column axonal projections at all levels up to the nucleus gracilis.
A great benefit of anterograde tracing methods is their system specificity and degree of anatomical detail. There can be artifacts, however. For example, tracers that leak into the CSF can be taken up in unexpected ways after lesions, leading to misinterpretation of findings (Steward et al., 2007). Anterograde tracers are typically injected into the site of greatest concentration of cell bodies projecting axons to the spinal cord, or into multiple locations. For example, the Tuszynski lab routinely utilizes 24 injections into the rat motor cortex to label CST axons projecting to cervical and lumbar spinal cord segments, in an effort to label as many axons as possible. One consequence, however, is that because so many axons are labeled, detecting the origin and course of individual axons around a lesion site is very difficult. An alternative method is to map the motor cortex using intracortical microstimulation to label CST projections to a specific spinal segment, then limit tracer injections to this identified region.
Retrograde tracing methods are also useful in studies of spinal cord injury. Injections of a retrograde tracer like fluorogold into the intact spinal cord can label essentially all of the neurons that project to that site. If injected below a complete transection, a retrograde tracer can label neurons arising from different parts of the nervous system that may have grown caudal to the lesion. The relative efficacy of regeneration can be quantified by counting retrogradely labeled cell bodies in spinal segments, brainstem and cortex. An important limitation of retrograde tracing techniques is that it is not possible to trace regenerating axons. Also, there can be spread of the tracer through spinal fluid pathways or into a matrix placed in the lesion site, leading to misinterpretation of regeneration. Thus, low volume tracer injections should be made at a slow rate of infusion, and histological analysis should include confirmation that tracer has not spread into the leptomeninges or the lesion site.
Genetic tracing
Transgenic animals that express fluorescent proteins in specific neuronal subsets provide potentially powerful tools for the study of regeneration. One strategy involves expressing fluorescent proteins under the control of neuron type-specific promoters (Bareyre et al., 2005). Another approach involves the use of Bacterial Artificial Chromosome (BAC) mice (Gong et al., 2003; Heiman et al., 2008). Heintz and colleagues developed transgenic mice that express the GFP reporter in highly restricted subsets of neurons, including corticospinal, raphespinal and dorsal root ganglion neurons. Genetic labels can provide specificity in axonal labeling that is hypothetically independent of tracer transport (Konzack et al., 2007). Moreover, BAC mice bearing GFP-tagged polyribosomes (BAC-TRAP mice) provide an exceptional opportunity to identify potential regeneration-associated transcriptional events in a cell-type specific manner (Heiman et al., 2008).
Although approaches using genetic labeling offer compelling promise, there are caveats, in our experience. First, it is essential to control for the possibility that injury alters the selectivity in the pattern of expression by particular neuron types (that is, labeling is no longer completely specific to particular axon types). We have seen evidence of this after spinal cord injury in mice in which the CST is genetically labeled. Second, genetic labeling leads to bilateral labeling, but it is often important to determine laterality in studies of regeneration. For example, a useful criterion for identifying axons as regenerated is that they extend along the “wrong” side (Liu et al., 2010; Steward et al., 2008). Laterality can only be determined when the pathway is labeled unilaterally. Finally, genetically-labeled axons undergoing Wallerian degeneration continue to be fluorescent for a surprisingly long period of time. This complicates analysis of early growth responses.
Fluorescent protein-expressing viral vectors have provided new tools for the study of regeneration (Fig. 9). Several serotypes of AAV vectors nearly exclusively infect neurons, allowing neuronal infection with fluorescent protein as a tracer that fills the axons and dendritic trees, providing exquisite anatomical resolution (Low et al., 2010). A hypothetical advantage of using GFP as a neuronal tracer rather than transported dyes is the fact that GFP reputedly moves through the cell through passive diffusion rather than axonal transport, and is not accordingly vulnerable to artifacts associated with injury-related changes in axonal transport. That is, rates of axonal transport increase after neural injury, and greater tracer labeling in an axon may reflect accelerated transport rather than true structural change; GFP may not be subject to this potential artifact.
Figure 9. Genetic Tract Labeling.
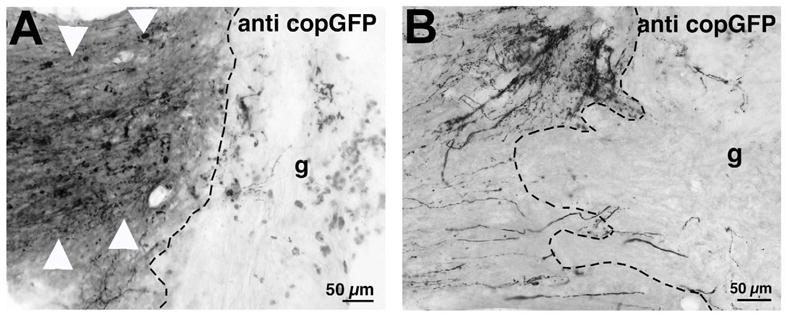
In vivo gene delivery vectors with dual promoters can identify specific neurons in vivo that express a candidate regeneration gene of interest, and can label the axonal projections of these neurons exclusively. In this case, AAV2 vectors expressing a candidate regeneration gene and the copGFP gene have been injected into the rat motor cortex. The axonal projections of these neurons in the spinal cord exhibit copGFP labeling (Low et al., 2010). (A) Corticospinal axons expressing copGFP are labeled with a light-level antibody as they approach a C3 dorsal column lesion site; bone marrow stromal cells have been grafted (g) into the lesion. (B) In a different field, individual axons that have incorporated the copGFP reporter are shown with fine morphological detail.
Viral vectors expressing GFP may also be employed elegantly to study the effects of genetic manipulation of axonal growth. For example, we have utilized an AAV vector coding for a candidate regeneration-associated gene that also expresses the GFP reporter; a neuron that incorporates the AAV vector will both express the candidate gene and label that neuron’s axon with GFP. This allows specific assessment of a gene effect on growth only in transduced neurons, potentially enhancing the sensitivity to detect an effect on growth (Low et al., 2010).
Genetic Animal Models of Regeneration
Transgenic mice can be a very useful model for examining the role of specific genes in axonal growth after adult injury. Several points must be considered when interpreting results from these models, however. First, genes that are deleted in neural development may perturb development of spinal pathways, leading to uncertainties regarding interpretation of results after adult injury. For example, early post-natal deletion of PTEN enhanced CST growth after spinal cord injury (Liu et al., 2010); however, deletion at this stage, while the CST is developing, could have altered the anatomy of its spinal projections with the result that partial lesion models in the adult failed to remove aberrant axon projections. Accordingly, a precise survey of the anatomy of the CST projection in adult unlesioned PTEN-deletion mice was required to confirm that axons were not in locations that would be inadvertently spared (Liu et al., 2010). Another caveat of transgenic mouse models in regeneration research is the possibility that developmental compensation may occur for loss of the targeted gene, leading to erroneous conclusions regarding the role of the deleted gene.
Finally, a caveat to studies of axon regeneration in mice is a unique wound healing response that occurs at the lesion site, which results in a contracted, cell rich lesion (Zhang et al., 1996) rather than a large, cystic lesion cavity. Accordingly, it remains to be seen whether manipulations that enable axon growth in mice will also be effective in other species.
VI. The Value of Functional Data
There has been a sense in the spinal cord injury literature that a finding is not of major importance unless there is a functional “benefit” from a therapeutic manipulation. This perspective is astonishingly naïve. Even among the most impressive reports of axonal growth to date, the overall restitution of axon number is far below normal innervation density. Extensive restoration of function may require restitution of neural circuitry to pre-lesion patterns that, during development, formed as a result of a precise orchestration of genetic and epigenetic events sequentially over time. This collective set of developmental events included both intracellular mechanisms in the neuron and environmental expression of diffusible guidance cues, extracellular matrix molecules and cell adhesion molecules in precise temporal and spatial gradients. Moreover, remyelination of every new axon segment may be required to overcome conduction block. This set of restorative events is unlikely to occur after adult injury. Accordingly, the extent to which non-directed or partially directed growth can be functionally beneficial, as opposed to deleterious (causing spasticity or cause pain), remains to be determined. We have only recently reached the point that this question can even be addressed because, finally, there are manipulations that produce at least some growth past the lesion.
Directed rehabilitation, trophic gradients and other means may be required to shape the nature of circuit reformation, but even under these circumstances, will the number, topography and remyelination of newly growing axons be sufficient to improve function? Moreover, we must also ask whether our most commonly used functional measures are relevant to humans. For example, is restoration of walking ability in a quadrupedal rodent relevant to the bipedal locomotion of humans that requires fine control of posture and balance?
Nonetheless, partial improvements in behavior (often optimistically referred to as “functional recovery” in the literature) can be meaningful and informative regarding cellular and systems-level mechanisms that are required to improve function. Screening tools such as the Basso-Beattie-Bresnahan (BBB) scale (Basso et al., 1995) provide a convenient starting point, but quantifiable ordinate measures that are directly related to particular axon systems are needed to definitively relate axon growth with recovery.
The requirement that experiments pass the criterion of demonstrating “functional benefit” to be considered of major importance in the spinal cord injury field should be soundly rejected by investigators, reviewers and journal editors. We remain at a stage of spinal cord injury research in which discovery of fundamental mechanisms contributing to new axonal growth is critical: from new mechanistic discoveries that lead to significant axonal sprouting and regeneration, we will sequentially amplify the number of growing axons, the distance over which they grow, and their guidance to and connection with appropriate targets. Moreover, greater mechanistic understanding of the interaction of growing axons with motor and sensory activation (electrically or behaviorally, i.e., rehabilitation) will then be required to shape systems output to functionally beneficial outcomes. At this point, we simply don’t know what is reasonable to expect in terms of the functional consequence of a given degree of regenerative axon growth. Thus, a “reset” of functional expectations is reasonable.
Publishing Discoveries on Axon Regeneration in the Contemporary Literature
Throughout this primer, we have highlighted the need for rigor in studies of axon regeneration in the study of spinal cord injury. Axon regeneration is inherently anatomical, and studies of regeneration require details of methodology and adequate presentation of that detail in published works. Yet this compelling need counters modern publishing trends. Today’s most attractive venues for publishing science frequently do not allow full presentation of methods or relevant control data, including full documentation of lesion extent. Indeed, economic pressures facing journals are leading to presentation of fewer details, especially in the print version. Moreover, some journals prohibit supplementary figures, precluding desirable documentation. A lack of full documentation increases the likelihood that errors or mis-interpretations will go undetected by reviewers and readers. Failures to replicate published findings continue to plague the field of spinal cord injury research, especially on the topic of axon regeneration. It is daunting that every report of a treatment that produced dramatic regeneration and recovery of function after spinal cord injury has failed to stand the test of time and scrutiny. Studies of regeneration after spinal cord injury require highly compelling data and in depth scrutiny to avoid leading the field in false directions.
VII. Conclusion
This is a golden era of neuroscience research with significant potential to impact future human therapy, including spinal cord injury. We have moved beyond an overly simplistic view of the organization and function of neural systems, and in parallel with this, have emerged from an overly simplistic view that we simply need to “grow axons” to restore function. Further progress in the field will be enhanced by accurately describing the biological phenomena we are attempting to understand, and by using models and interpreting the data they generate in a truly objective and realistic manner.
Acknowledgments
Supported by the NIH, the Veterans Administration, the Craig H. Neilsen Foundation, and the Bernard and Anne Spitzer Charitable Trust.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alstermark B, Lundberg A, Sasaki S. Integration in descending motor pathways controlling the forelimb in the cat. 12. Interneurones which may mediate descending feed-forward inhibition and feed-back inhibition from the forelimb to C3-C4 propriospinal neurones. Exp Brain Res; Exper Hirnfor Experim Cereb. 1984;56:308–322. doi: 10.1007/BF00236286. [DOI] [PubMed] [Google Scholar]
- Alstermark B, Pinter M, Sasaki S. Brainstem relay of disynaptic pyramidal EPSPs to neck motoneurons in the cat. Brain Res. 1983;259:147–150. doi: 10.1016/0006-8993(83)91078-8. [DOI] [PubMed] [Google Scholar]
- Alto LT, Havton LA, Conner JM, Hollis ER, II, Blesch A, Tuszynski MH. Chemotropic guidance facilitates axonal regeneration and synapse formation after spinal cord injury. Nat Neurosci. 2009;12:1106–1113. doi: 10.1038/nn.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K, Aito S, Atkins M, Biering-Sorensen F, Charlifue S, Curt A, Ditunno J, Glass C, Marino R, Marshall R, et al. Functional recovery measures for spinal cord injury: an evidence-based review for clinical practice and research. J Spinal Cord Med. 2008;31:133–144. doi: 10.1080/10790268.2008.11760704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Misgeld T, Sanes JR. Transgenic labeling of the corticospinal tract for monitoring axonal responses to spinal cord injury. Nat Med. 2005;11:1355–1360. doi: 10.1038/nm1331. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Raineteau O, Mettenleiter TC, Weinmann O, Schwab ME. The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat Neurosci. 2004;7:269–277. doi: 10.1038/nn1195. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. Journal of neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Benowitz L, Yin Y. Rewiring the injured CNS: lessons from the optic nerve. Exp Neurol. 2008;209:389–398. doi: 10.1016/j.expneurol.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson MD, Romero MI, Lush ME, Lu QR, Henkemeyer M, Parada LF. Ephrin-B3 is a myelin-based inhibitor of neurite outgrowth. Proc Nat Acad Sci. 2005;102:10694–10699. doi: 10.1073/pnas.0504021102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisby MA, Pollock B. Increased regeneration rate in peripheral nerve axons following double lesions: enhancement of the conditioning lesion phenomenon. J Neurobiol. 1983;14:467–472. doi: 10.1002/neu.480140607. [DOI] [PubMed] [Google Scholar]
- Blesch A, Tuszynski MH. Spinal cord injury: plasticity, regeneration and the challenge of translational drug development. Trends in neurosciences. 2009;32:41–47. doi: 10.1016/j.tins.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Blesch A, Tuszynski MH. Spinal cord injury: Plasticity, regeneration and the challenge of translational drug development. TINS. 2009;32:41–47. doi: 10.1016/j.tins.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Bonner JF, Connors TM, Silverman WF, Kowalski DP, Lemay MA, Fischer I. Grafted neural progenitors integrate and restore synaptic connectivity across the injured spinal cord. J Neurosci. 2011;31:4675–4686. doi: 10.1523/JNEUROSCI.4130-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray GM, Villegas-Perez MP, Vidal-Sanz M, Carter DA, Aguayo AJ. Neuronal and nonneuronal influences on retinal ganglion cell survival, axonal regrowth, and connectivity after axotomy. Ann NY Acad Sci. 1991;633:214–228. doi: 10.1111/j.1749-6632.1991.tb15613.x. [DOI] [PubMed] [Google Scholar]
- Cao Z, Gao Y, Deng K, Williams G, Doherty P, Walsh FS. Receptors for myelin inhibitors: Structures and therapeutic opportunities. Mol Cell Neurosci. 2010;43:1–14. doi: 10.1016/j.mcn.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Causey G, Hoffman H. Axon sprouting partially deneurotized nerves. Brain. 1955;78:661–668. doi: 10.1093/brain/78.4.661. [DOI] [PubMed] [Google Scholar]
- Coumans JV, Lin TT, Dai HN, MacArthur L, McAtee M, Nash C, Bregman BS. Axonal regeneration and functional recovery after complete spinal cord transection in rats by delayed treatment with transplants and neurotrophins. J Neurosci. 2001;21:9334–9344. doi: 10.1523/JNEUROSCI.21-23-09334.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtine G, Gerasimenko Y, van den Brand R, Yew A, Musienko P, Zhong H, Song B, Ao Y, Ichiyama RM, Lavrov I, et al. Transformation of nonfunctional spinal circuits into functional states after the loss of brain input. Nat Neurosci. 2009;12:1333–1342. doi: 10.1038/nn.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtine G, Song B, Roy RR, Zhong H, Herrmann JE, Ao Y, Qi J, Edgerton VR, Sofroniew MV. Recovery of supraspinal control of stepping via indirect propriospinal relay connections after spinal cord injury. Nat Med. 2008;14:69–74. doi: 10.1038/nm1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edds MV., Jr Collateral nerve regeneration. Q Rev Biol. 1953;28:260–276. doi: 10.1086/399699. [DOI] [PubMed] [Google Scholar]
- Edds MV, Jr, Small WT. The behavior of residual axons in partially denervated muscles of the monkey. J Exp Med. 1951;93:207–216. doi: 10.1084/jem.93.3.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erturk A, Mauch CP, Hellal F, Forstner F, Keck T, Becker K, Jahrling N, Steffens H, Richter M, Hubener M, et al. Three-dimensional imaging of the unsectioned adult spinal cord to assess axon regeneration and glial responses after injury. Nat Med. 2012;18:166–171. doi: 10.1038/nm.2600. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Silver J. Glial Cells, Inflammation and CNS Trauma. In: Kordower J, Tuszynski MH, editors. CNS Regeneration: Basic Science and Clinical Advances. San Diego: Elsvier; 2008. [Google Scholar]
- Giger RJ, Hollis ER, 2nd, Tuszynski MH. Guidance molecules in axon regeneration. Cold Spring Harbor Perspec Biol. 2010;2:001867. doi: 10.1101/cshperspect.a001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Griffin JW, Pan B, Polley MA, Hoffman PN, Farah MH. Measuring nerve regeneration in the mouse. Exp Neurol. 2010;223:60–71. doi: 10.1016/j.expneurol.2009.12.033. [DOI] [PubMed] [Google Scholar]
- Gruner JA, Yee AK, Blight AR. Histological and functional evaluation of experimental spinal cord injury: evidence of a stepwise response to graded compression. Brain Res. 1996;729:90–101. [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman H. Acceleration and retardation of the process of axon-sprouting in partially devervated muscles. Aust J Exp Biol Med Sci. 1952;30:541–566. doi: 10.1038/icb.1952.52. [DOI] [PubMed] [Google Scholar]
- Jin Y, Fischer I, Tessler A, Houle JD. Transplants of fibroblasts genetically modified to express BDNF promote axonal regeneration from supraspinal neurons following chronic spinal cord injury. Exp Neurol. 2002;177:265–275. doi: 10.1006/exnr.2002.7980. [DOI] [PubMed] [Google Scholar]
- Keirstead SA, Rasminsky M, Fukuda Y, Carter D, Aguayo AJ. Electrophysiologic responses in hamster superior colliculus evoked by regenerating retinal axons. Science. 1989;246:255–258. doi: 10.1126/science.2799387. [DOI] [PubMed] [Google Scholar]
- Kennedy D. The old file-drawer problem. Science. 2004;305:451. doi: 10.1126/science.305.5683.451. [DOI] [PubMed] [Google Scholar]
- Konzack S, Thies E, Marx A, Mandelkow EM, Mandelkow E. Swimming against the tide: mobility of the microtubule-associated protein tau in neurons. J Neurosci. 2007;27:9916–9927. doi: 10.1523/JNEUROSCI.0927-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyuk PG, Vasilenko DA. Propriospinal neurones as a relay system for transmission of cortico-spinal influences. J de Physiol. 1978;74:247–250. [PubMed] [Google Scholar]
- Kuchler M, Fouad K, Weinmann O, Schwab ME, Raineteau O. Red nucleus projections to distinct motor neuron pools in the rat spinal cord. J Comp Neurol. 2002;448:349–359. doi: 10.1002/cne.10259. [DOI] [PubMed] [Google Scholar]
- Kurimoto T, Yin Y, Omura K, Gilbert HY, Kim D, Cen LP, Moko L, Kugler S, Benowitz LI. Long-distance axon regeneration in the mature optic nerve: contributions of oncomodulin, cAMP, and pten gene deletion. J Neurosci. 2010;30:15654–15663. doi: 10.1523/JNEUROSCI.4340-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebscher T, Schnell L, Schnell D, Scholl J, Schneider R, Gullo M, Fouad K, Mir A, Rausch M, Kindler D, et al. Nogo-A antibody improves regeneration and locomotion of spinal cord-injured rats. Ann Neurol. 2005;58:706–719. doi: 10.1002/ana.20627. [DOI] [PubMed] [Google Scholar]
- Liu CN, Chambers WW. Intraspinal sprouting of dorsal root axons; development of new collaterals and preterminals following partial denervation of the spinal cord in the cat. AMA Arch Neurol Psych. 1958;79:46–61. [PubMed] [Google Scholar]
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci. 2010;13:1075–1081. doi: 10.1038/nn.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kim D, Himes BT, Chow SY, Schallert T, Murray M, Tessler A, Fischer I. Transplants of fibroblasts genetically modified to express BDNF promote regeneration of adult rat rubrospinal axons and recovery of forelimb function. J Neurosci. 1999;19:4370–4387. doi: 10.1523/JNEUROSCI.19-11-04370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo FM, Hayman EG, Davis GE, Ruoslahti E, Engvall E, Manthorpe M, Varon S. Neurite-promoting factors and extracellular matrix components accumulating in vivo within nerve regeneration chambers. Brain Res. 1984;309:105–117. doi: 10.1016/0006-8993(84)91014-x. [DOI] [PubMed] [Google Scholar]
- Low K, Blesch A, Herrmann J, Tuszynski MH. A dual promoter lentiviral vector for the in vivo evaluation of gene therapeutic approaches to axon regeneration after spinal cord injury. Gene Ther. 2010;17:577–591. doi: 10.1038/gt.2010.14. [DOI] [PubMed] [Google Scholar]
- Low K, Culbertson M, Bradke F, Tessier-Lavigne M, Tuszynski MH. Netrin-1 is a novel myelin-associated inhibitor to axon growth. J Neurosci. 2008;28:1099–1108. doi: 10.1523/JNEUROSCI.4906-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PBA, Graham L, Wang W, Samara R, Banos K, Haringer V, Havton L, Weishaup N, Bennett D, Fouad K, Tuszynski MH. Motor axonal regeneration after partial and complete spinal cord transection. J Neurosci. 2012 doi: 10.1523/JNEUROSCI.0308-12.2012. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Jones LL, Snyder EY, Tuszynski MH. Neural stem cells constitutively secrete neurotrophic factors and promote extensive host axonal growth after spinal cord injury. Exp Neurol. 2003;181:115–129. doi: 10.1016/s0014-4886(03)00037-2. [DOI] [PubMed] [Google Scholar]
- Lu P, Yang H, Jones LL, Filbin MT, Tuszynski MH. Combinatorial therapy with neurotrophins and cAMP promotes axonal regeneration beyond sites of spinal cord injury. J Neurosci. 2004;24:6402–6409. doi: 10.1523/JNEUROSCI.1492-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CH, Omura T, Cobos EJ, Latremoliere A, Ghasemlou N, Brenner GJ, van Veen E, Barrett L, Sawada T, Gao F, et al. Accelerating axonal growth promotes motor recovery after peripheral nerve injury in mice. J Clin Inves. 2011;121:4332–4347. doi: 10.1172/JCI58675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclaren RE, Taylor JS. Regeneration in the developing optic nerve: correlating observations in the opossum to other mammalian systems. Prog Neurobiol. 1997;53:381–398. doi: 10.1016/s0301-0082(97)00041-5. [DOI] [PubMed] [Google Scholar]
- Mantuano E, Henry K, Yamauchi T, Hiramatsu N, Yamauchi K, Orita S, Takahashi K, Lin JH, Gonias SL, Campana WM. The unfolded protein response is a major mechanism by which LRP1 regulates Schwann cell survival after injury. J Neurosci. 2011;31:13376–13385. doi: 10.1523/JNEUROSCI.2850-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCouch G, Austin GM, Liu CN, Liu CY. Sprouting as a cause of spasticity. J Neurophysiol. 1958;21:205–216. doi: 10.1152/jn.1958.21.3.205. [DOI] [PubMed] [Google Scholar]
- McQuarrie IG, Grafstein B, Gershon MD. Axonal regeneration in the rat sciatic nerve: effect of a conditioning lesion and of dbcAMP. Brain Res. 1977;132:443–453. doi: 10.1016/0006-8993(77)90193-7. [DOI] [PubMed] [Google Scholar]
- Morris R, Tosolini AP, Goldstein JD, Whishaw IQ. Impaired arpeggio movement in skilled reaching by rubrospinal tract lesions in the rat: a behavioral/anatomical fractionation. J Neurotrauma. 2011;28:2439–2451. doi: 10.1089/neu.2010.1708. [DOI] [PubMed] [Google Scholar]
- Nathan PW, Smith MC. The rubrospinal and central tegmental tracts in man. Brain. 1982;105:223–269. doi: 10.1093/brain/105.2.223. [DOI] [PubMed] [Google Scholar]
- Neumann S, Woolf CJ. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron. 1999;23:83–91. doi: 10.1016/s0896-6273(00)80755-2. [DOI] [PubMed] [Google Scholar]
- Oudega M, Varon S, Hagg T. Regeneration of adult rat sensory axons into intraspinal nerve grafts: promoting effects of conditioning lesion and graft predegeneration. Exp Neurol. 1994;129:194–206. doi: 10.1006/exnr.1994.1161. [DOI] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisman G. Neuronal plasticity in the septal nuclei of the adult rat. Brain Res. 1969;14:25–48. doi: 10.1016/0006-8993(69)90029-8. [DOI] [PubMed] [Google Scholar]
- Ramon y Cajal S. Degeneration and Regeneration of the Nervous System. London: Oxford University Press; 1928. [Google Scholar]
- Rosenzweig ES, Brock JH, Culbertson MD, Lu P, Moseanko R, Edgerton VR, Havton LA, Tuszynski MH. Extensive spinal decussation and bilateral termination of cervical corticospinal projections in rhesus monkeys. J Comp Neurol. 2009;513:151–163. doi: 10.1002/cne.21940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig ES, Brock JH, Edgerton ER, Tuszynski MH. Extensive spontaneous corticospinal tract plasticity after spinal cord injury in the primate spinal cord. J Comp Neurol. 2009;513:151–163. doi: 10.1002/cne.21940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig ES, Courtine G, Jindrich DL, Brock JH, Ferguson AR, Strand SC, Nout YS, Roy RR, Miller DM, Beattie MS, et al. Extensive spontaneous plasticity of corticospinal projections after primate spinal cord injury. Nat Neurosci. 2010;13:1505–1510. doi: 10.1038/nn.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol S, Dubuc R. Spinal pattern generation. Curr Opin Neurobiol. 1994;4:894–902. doi: 10.1016/0959-4388(94)90139-2. [DOI] [PubMed] [Google Scholar]
- Schwab JM, Brechtel K, Mueller CA, Failli V, Kaps HP, Tuli SK, Schluesener HJ. Experimental strategies to promote spinal cord regeneration--an integrative perspective. Prog Neurobiol. 2006;78:91–116. doi: 10.1016/j.pneurobio.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Scott S. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler 9:4–15. Amyotroph Lateral Scler. 2008;9:4–15. doi: 10.1080/17482960701856300. [DOI] [PubMed] [Google Scholar]
- Shapovalov AI, Gurevitch NR. Monosynaptic and disynaptic reticulospinal actions on lumbar motoneurons of the rat. Brain Res. 1970;21:249–263. doi: 10.1016/0006-8993(70)90366-5. [DOI] [PubMed] [Google Scholar]
- Sharp KG, Flanagan LA, Yee KM, Steward O. A re-assessment of a combinatorial treatment involving Schwann cell transplants and elevation of cyclic AMP on recovery of motor function following thoracic spinal cord injury in rats. Exp Neurol. 2010 doi: 10.1016/j.expneurol.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Popovich PG, Dietrich WD, Kleitman N. Replication and reproducibility in spinal cord injury research. Exp Neurol. 2011;233:597–605. doi: 10.1016/j.expneurol.2011.06.017. [DOI] [PubMed] [Google Scholar]
- Steward O, Zheng B, Banos K, Yee KM. Response to: Kim et al., “Axon regeneration in young adult mice lacking Nogo-A/B”. Neuron. 2007;54:191–195. doi: 10.1016/j.neuron.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Steward O, Zheng B, Tessier-Lavigne M. False resurrections: distinguishing regenerated from spared axons in the injured central nervous system. J Comp Neurol. 2003;459:1–8. doi: 10.1002/cne.10593. [DOI] [PubMed] [Google Scholar]
- Steward O, Zheng B, Tessier-Lavigne M, Hofstadter M, Sharp K, Yee KM. Regenerative growth of corticospinal tract axons via the ventral column after spinal cord injury in mice. J Neurosci. 2008;28:6836–6847. doi: 10.1523/JNEUROSCI.5372-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor L, Jones L, Tuszynski MH, Blesch A. Neurotrophin-3 gradients established by lentiviral gene delivery promote short-distance axonal bridging beyond cellular grafts in the injured spinal cord. J Neurosci. 2006;26:9713–9721. doi: 10.1523/JNEUROSCI.0734-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Donkelaar HJ. Evolution of the red nucleus and rubrospinal tract. Behav Brain Res. 1988;28:9–20. doi: 10.1016/0166-4328(88)90072-1. [DOI] [PubMed] [Google Scholar]
- ten Donkelaar HJ, Kusuma A, de Boer-Van Huizen R. Cells of origin of pathways descending to the spinal cord in some quadrupedal reptiles. J Comp Neurol. 1980;192:827–851. doi: 10.1002/cne.901920413. [DOI] [PubMed] [Google Scholar]
- Thompson CK, Jayaraman A, Kinnaird C, Hornby TG. Methods to quantify pharmacologically induced alterations in motor function in human incomplete SCI. J Vis Exper. 2011;50:1–15. doi: 10.3791/2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuszynski MH, Peterson DA, Ray J, Baird A, Nakahara Y, Gage FH. Fibroblasts genetically modified to produce nerve growth factor induce robust neuritic ingrowth after grafting to the spinal cord. Exp Neurol. 1994;126:1–14. doi: 10.1006/exnr.1994.1037. [DOI] [PubMed] [Google Scholar]
- Weidner N, Ner A, Salimi N, Tuszynski MH. Spontaneous corticospinal axonal plasticity and functional recovery after adult CNS injury. Proc Nat Acad Sci. 2001;98:3513–3518. doi: 10.1073/pnas.051626798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SR, Neuman RS. Facilitation of spinal motoneurone excitability by 5-hydroxytryptamine and noradrenaline. Brain Res. 1980;188:119–127. doi: 10.1016/0006-8993(80)90561-2. [DOI] [PubMed] [Google Scholar]
- Wilson VJ, Yoshida M. Vestibulospinal and reticulospinal effects on hindlimb, forelimb, and neck alpha motoneurons of the cat. Proc Nat Acad Sci. 1968;60:836–840. doi: 10.1073/pnas.60.3.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XM, Chen A, Guenard V, Kleitman N, Bunge MB. Bridging Schwann cell transplants promote axonal regeneration from both the rostral and caudal stumps of transected adult rat spinal cord. J Neurocytol. 1997;26:1–16. doi: 10.1023/a:1018557923309. [DOI] [PubMed] [Google Scholar]
- Z’Graggen WJ, Fouad K, Raineteau O, Metz GA, Schwab ME, Kartje GL. Compensatory sprouting and impulse rerouting after unilateral pyramidal tract lesion in neonatal rats. J Neurosci. 2000;20:6561–6569. doi: 10.1523/JNEUROSCI.20-17-06561.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Fujiki M, Guth L, Steward O. Genetic influences on cellular reactions to spinal cord injury: A wound healing response present in normal mice is impaired in mice carrying a mutation that causes delayed Wallerian degeneration. J Comp Neurol. 1996;371:469–484. doi: 10.1002/(SICI)1096-9861(19960729)371:3<485::AID-CNE10>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Zochodne DW. The challenges and beauty of peripheral nerve regrowth. Journal of the peripheral nervous system, JPNS. 2012;17:1–18. doi: 10.1111/j.1529-8027.2012.00378.x. [DOI] [PubMed] [Google Scholar]