

Abstract
Febrile seizures are the most common seizure type in young children. Whether they induce death of hippocampal and amygdala neurons and consequent limbic (temporal lobe) epilepsy has remained controversial, with conflicting data from prospective and retrospective studies. Using an appropriate-age rat model of febrile seizures, we investigated the acute and chronic effects of hyperthermic seizures on neuronal integrity and survival in the hippocampus and amygdala via molecular and neuroanatomical methods. Hyperthermic seizures–but not hyperthermia alone–resulted in numerous argyrophilic neurons in discrete regions of the limbic system; within 24 hr of seizures, a significant proportion of neurons in the central nucleus of the amygdala and in the hippocampal CA3 and CA1 pyramidal cell layer were affected. These physicochemical alterations of hippocampal and amygdala neurons persisted for at least 2 weeks but were not accompanied by significant DNA fragmentation, as determined by in situend labeling. By 4 weeks after the seizures, no significant neuronal dropout in these regions was evident. In conclusion, in the immature rat model, hyperthermic seizures lead to profound, yet primarily transient alterations in neuronal structure.
Keywords: seizures, animal model, febrile seizures, epilepsy, neuronal death, excitotoxicity, apoptosis, in situ end labeling
Febrile seizures are the most common seizure type in the human infant and young child (Verity et al., 1985;Shinnar, 1990; Hauser, 1994). Prospective epidemiological studies have indicated that febrile seizures do not progress to temporal lobe epilepsy (TLE) (Nelson and Ellenberg, 1976; Shinnar, 1990; Knudsen, 1996). However, retrospective analyses of adults with TLE document a high prevalence (30–50%) of a history of febrile seizures during early childhood, suggesting an etiological role for these seizures in the development of TLE (Gloor, 1991; Cendes et al., 1993). A role for neuronal damage induced by febrile seizures in the pathogenesis of mesial temporal sclerosis, the pathological hallmark of TLE, has been postulated (Falconer et al., 1964; Sagar and Oxbury, 1987; Gloor, 1992). An alternative mechanism for the correlation of febrile seizures and TLE involves pre-existing neuronal injury that triggers both the febrile seizures and the subsequent TLE.
In general, both electrophysiological and behavioral manifestations of seizures in the immature human and in developing experimental animals are more severe than are those resulting from equivalent proconvulsant drugs or insults in the mature brain (Sperber et al., 1992). For example, during the second postnatal week in the rat, kainic acid leads to status epilepticus with dramatic mortality at doses that cause only mild seizures in the adult (Albala et al., 1984; Holmes and Thompson, 1988; Chang and Baram, 1994). Seizure induction by a hypoxic insult is also age-dependent, peaking on the 10th postnatal day (Jensen et al., 1991; Owens et al., 1997). In addition, seizures induced by hyperthermia and fever in the rat and human, respectively, are almost exclusive to the developmental period (Berg et al., 1992; Baram et al., 1997).
Despite the severity of seizures in a number of experimental paradigms, neuronal death, i.e., cell loss as a result of these seizures, does not appear to occur during the first 2 postnatal weeks in the rat (Nitecka et al., 1984; Sperber et al., 1992; Holmes, 1997). Studies using DNA fragmentation or silver-staining methods have failed to reveal acute cell death after severe seizures induced by convulsants such as kainic acid (Sperber et al., 1991), pilocarpine, or hypoxia (Owens et al., 1997), even though all three treatments cause widespread acute cell death in older animals (Ben-Ari et al., 1981; Clifford et al., 1987). Furthermore, long-term studies of kainic acid-induced seizures have revealed neither a decrease in neuronal number in limbic areas that are vulnerable in the adult nor a sprouting response that is observed in adult animals after loss of “target” postsynaptic neurons (Sperber et al., 1992; Baram and Ribak, 1995).
A model of febrile seizures in the immature rat has recently been characterized (Baram et al., 1997). This paradigm relies on rat pups during a brain-development age equivalent to that of the human infant and young child (Dobbing and Sands, 1973). Furthermore, the model is associated with little immediate morbidity, permitting prospective long-term analyses of the effects of hyperthermic seizures on neuronal integrity and survival. The goals of the current study were to (1) investigate potential acute injury induced by hyperthermic seizures, (2) define the topographical distribution of vulnerable neurons, and (3) determine whether acute neuronal injury after hyperthermic seizures produces significant neuronal loss.
MATERIALS AND METHODS
Hyperthermic seizures experimental design
Animals. Rat pups were offspring of time-pregnant Sprague Dawley-derived rats obtained from Zivic-Miller (Zelienople, PA). Mothers were kept on a 12 hr light/dark schedule and given lab chow and water ad libitum (Yi and Baram, 1994; Baram et al., 1997). The time of birth of pups was determined every 12 hr, and the day of birth was considered day 0. Litters were culled to 12 pups on the first postnatal day and kept in quiet, uncrowded American Association for Accreditation of Laboratory Animal Care-approved facilities at a room temperature of 21–22°C. Overall, 89 rat pups participated in the study and were divided into experimental groups as described below.
Hyperthermia induction paradigm. The hyperthermic seizure paradigm has been described previously in detail (Baram et al., 1997). Briefly, on postnatal day 10, the core temperature of pups was raised using a regulated stream of moderately heated air. Pups were placed on the floor of a 3 l glass container, and the air stream was directed ∼50 cm above them. Rectal temperatures were measured at baseline, at 2 min intervals, and at the onset of hyperthermic seizures, which occur in >93% of rats (Baram et al., 1997). Hyperthermia was maintained for 30 min, aiming for a core temperature of 41–42°C, and the presence and duration of seizures for each pup were noted at 2 min intervals. After the hyperthermia period, rats were placed on a cool surface, monitored for 15 min, and then returned to home cages for rehydration by the mothers. Pups who were sedated because of diazepam or pentobarbital pretreatment were hydrated orally and returned to their cages when their behavior normalized (typically <1 hr). The behavioral seizures in this paradigm are stereotyped and easily monitored and have been shown to correlate with EEG rhythmic epileptiform discharges from the hippocampus and amygdala (Baram et al., 1997). These seizures consist of complete arrest of the heat-induced hyperkinesis, unilateral body flexion, and biting of an extremity, occasionally followed by clonic or “swimming” motions.
Experimental groups. The overall strategy was to compare the presence of neuronal injury in three experimental groups. (1) Controls (n = 24) were maintained normothermic for age (rectal temperature, 33–34°C) throughout the experiments. (2) Hyperthermia controls (n = 14) were rendered hyperthermic, but seizures were prevented by pretreatment with intraperitoneal diazepam (5 mg/kg) or the short-acting barbiturate pentobarbital (30 mg/kg) before hyperthermia induction. (3) The third group experienced hyperthermic seizures (n = 51).
For determination of DNA fragmentation, groups (n = 3–4 per group) of animals subjected to hyperthermic seizures were killed at the following time points after the seizures: 1, 4, 8.5, 20, and 48 hr. An additional group (n = 4) was subjected to hyperthermia twice, leading to a total seizure duration of 60 ± 2 min, and was killed 20 hr after the second seizure episode. Normothermic animals (n = 7) and pups in whom hyperthermia-induced seizures were prevented with pentobarbital (see above) were used as controls. For analysis of neuronal injury using the Gallyas “dark”-neuron stain, animals were killed 24 hr, 1 week, or 2 weeks after seizure induction. For cell counting, animals (n = 12; 4 per experimental group) were killed 4 weeks after the hyperthermic seizures.
In situ end labeling
In situ end labeling (ISEL) provides a positive label of dying cells, leading to increased sensitivity over methods relying on the “dropout” of neurons. ISEL was modified from Sakhi et al. (1994). Briefly, frozen 20 μm sections were thawed, dried, and fixed in 4% buffered paraformaldehyde, followed by ethanol dehydration. After rehydration and preincubation in a Tris buffer, 50 μl of reaction mix [966 μl of buffer, 4 μl of nucleotide mix (Sakhi et al., 1994), 10 μl of DNA polymerase I (10 U/μl; Promega, Madison, WI), and 20 μl of dUTP–biotin (Sigma, St. Louis, MO) per 1000 μl] were applied for 1 hr. UTP–biotin-tagged cleaved DNA ends were visualized using a commercial avidin–biotin kit (VECTASTAIN; Vector Laboratories, Burlingame, CA). Sections were counterstained with acid fuchsin (Chang and Baram, 1994), and the number of labeled neurons with clumped chromatin in the central nucleus of the amygdala and the hippocampus was determined. Sections from adult rats subjected to kainic acid-induced status epilepticus and allowed a 20 hr survival time were run in parallel as “positive controls” to confirm the validity of the ISEL method at this time point.
Histological methods
For all histological methods, control and experimental groups were processed together, without knowledge of treatment group. Animals were killed and perfused as described elsewhere (Baram and Ribak, 1995;Ribak and Baram, 1996). Briefly, under deep anesthesia, rats were perfused transcardially with saline followed by a 4% paraformaldehyde–2.5% glutaraldehyde–0.1 m phosphate buffer solution. Brains were left in situ overnight and then post-fixed for a week in the same fixative. Brains were sectioned coronally with a vibratome at 100 μm for silver staining and at 20 μm for Nissl staining for neuronal counts. Sections (immersed in fixative) were stored at 4°C.
Silver-staining (dark-neuron) method. This method has been described earlier (Gallyas et al., 1990). Briefly, after dehydration in 50, 75, and 100% propanol, sections were esterified with propanol containing 0.8% sulfuric acid. Sections were rehydrated through 50 and 25% propanol and distilled water, treated with 3% acetic acid for 5 min, and developed for 10 min. The fresh developing solution consisted of equal volumes of 10% Na2CO3 and a solution composed of 0.2% AgNO3, 0.25% NH4NO3, 2% tungstosilicic acid, and 0.4% formaldehyde. When developed, sections were dehydrated and coverslipped.
Semithin sections and electron microscopy methods. Unstained sections adjacent to those containing argyrophilic neurons were post-fixed with 1% osmium tetroxide, dehydrated, and embedded in Epon. Semithin, 2 μm sections were cut and stained with 1% toluidine blue to identify deeply basophilic neurons. For electron microscopy (EM) analysis, ultrathin sections were obtained from the same tissue blocks, subjected to uranyl acetate and lead citrate, and examined with a transmission EM. Electron-dense neuronal profiles and their synapses were further examined in serial sections to confirm their identification.
Semiquantitative neuronal analysis. For counting both silver- and Nissl-stained neurons, sections were analyzed without knowledge of treatment (“blindly”). Based on the limbic phenomenology of hyperthermia-induced seizures (Ben-Ari et al., 1981;Baram et al., 1997), the examination of argyrophilic neurons focused on the amygdala, hippocampus, and cortical regions interconnected with these structures. For determining the fate of most argyrophilic neurons, an estimate of their number was undertaken in the most affected structure, the lateral division (CE-L) of the central nucleus of the amygdala (ACE). For purposes of neuronal counting, the anatomical boundaries of the CE-L were defined using Sherwood and Timiras (1970). Rostral and caudal boundaries were selected to permit an easily identifiable coronal profile of the nucleus. The rostral boundary was at the level of the anterior paraventricular hypothalamic nucleus and the rostral border of the hippocampal CA3 (5.0 mm anterior to bregma in the 39-d-old rat); the caudal boundary was defined at the level of the infundibulum (4.1 mm anterior to bregma). The lateral boundary of the ACE was defined by the clearly visible teardrop-shaped lateral nucleus of the amygdala. The medial border was defined by the cell-poor white matter of the internal capsule. Within the ACE, the CE-L was defined according to Tuunanen et al. (1996). Briefly, the tightly packed capsular region was defined laterally, and the crescent-shaped medial division was distinguished medially (see Figs.3, 5). Because a determination of the absolute, unbiased number of argyrophilic neurons was not needed for the purposes of this study, a modified profile sampling method was used [for a recent discussion, see Popken and Farel (1997); Saper (1997)]. A grid reticule of 100 frames covering an area of 250 × 250 μm was centered over every sixth section of the CE-L, and all frames were subjected to counting for cell bodies throughout the depth of the section (Popken and Farel, 1997). The resulting number was multiplied to yield an estimate of the overall number of argyrophilic neurons in the CE-L as defined above. For Nissl-stained sections, every ninth 20 μm section was subjected to cell counts. To minimize the bias of the “splitting” of large cell bodies resulting in an overestimate of counts, we counted nucleoli (Popken and Farel, 1997). Counts of each section were averaged to yield an estimate of variance, and the overall neuronal numbers in the CE-L, as defined above, were obtained for each experimental animal. The statistical significance of differences among the three treatment groups used ANOVA (PRISM statistical software; GraphPad, San Diego, CA).
Fig. 3.
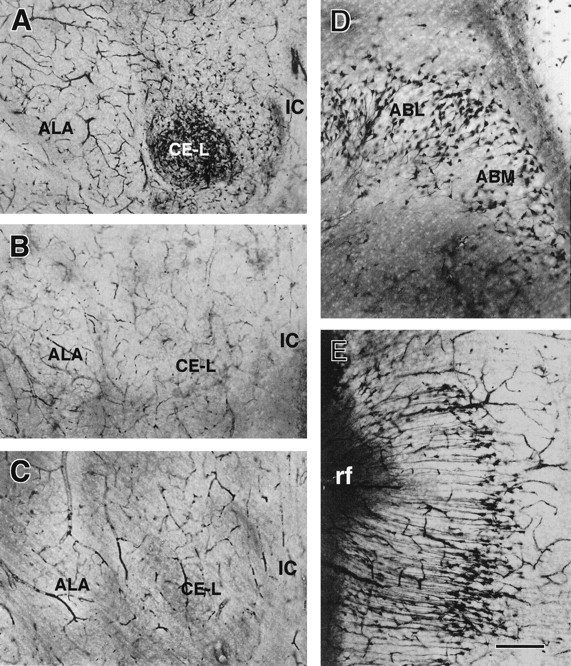
Neuronal injury induced by hyperthermic seizures in the amygdala, demonstrated using the Gallyas dark-neuron method.A, Significant involvement of the lateral division of the central amygdaloid nucleus (CE-L) is manifested by large numbers of densely silver-stained neurons. B,C, Sections from hyperthermic (B) or normothermic (C) controls contain few stained neurons. D, The distribution of silver-stained neurons in other amygdaloid nuclei, the basomedial (ABM) and basolateral (ABL) nuclei, is shown.E, The presence of argyrophilic neurons in the perirhinal cortex is evident. ALA, Lateral amygdala nucleus; IC, internal capsule; rf, rhinal fissure. Scale bars: A–C, 500 μm;D, E, 200 μm.
Fig. 5.
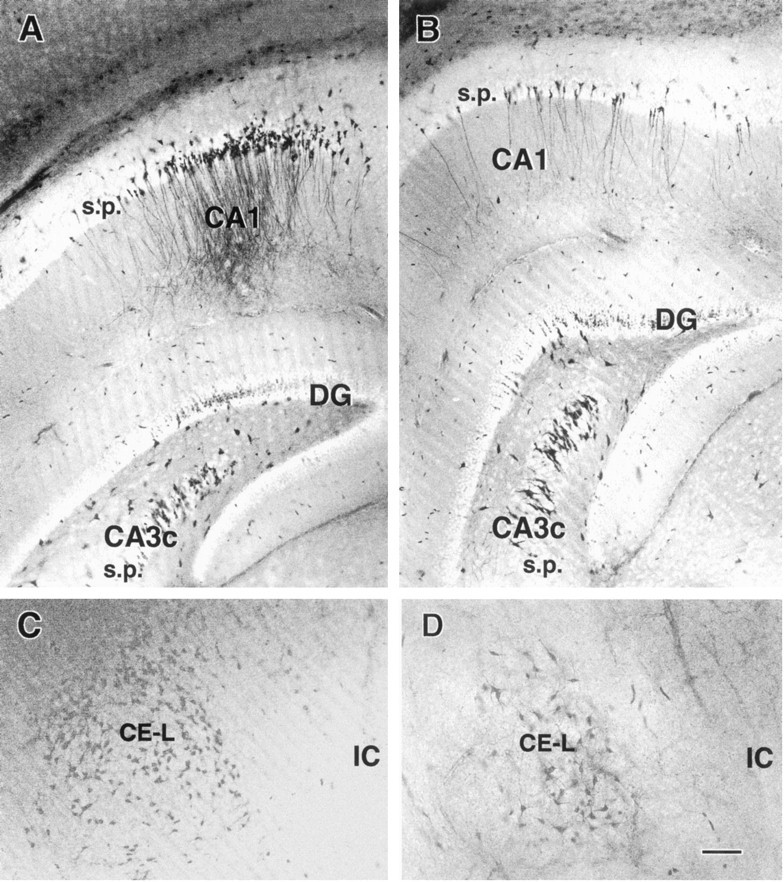
Persistent injury to hippocampal and amygdaloid neurons after hyperthermic seizures. Sections obtained from immature rats killed 1 week (A, C) or 2 weeks (B, D) after hyperthermic seizures are shown. Silver-stained neurons are evident in the CA1 pyramidal cell layer (A, B) and in the lateral division of the central nucleus of the amygdala (CE-L) (C, D) at both time points. Decreased numbers of affected neurons are apparent at the 2-week time point (B, D). DG, Dentate gyrus;IC, internal capsule; s.p., stratum pyramidale. Scale bar, 50 μm.
RESULTS
Using ISEL, hyperthermic seizures lead to little immediate cell death
Direct evidence of DNA fragmentation associated with several forms of neuronal death, including death induced by kainic acid-induced status epilepticus (Pollard et al., 1994), was studied using ISEL. After hyperthermic seizures, only occasional amygdala and hippocampal neurons were labeled in sections derived from rats subjected to hyperthermic seizures and permitted to survive for 1, 4, 8.5, 20, or 48 hr (Fig. 1, Table1). Sections from adult rats subjected to kainic acid-induced status epilepticus and allowed a 20 hr survival time, which were run in parallel, contained abundant labeled neurons in hippocampal CA3 and most amygdala nuclei, confirming the validity of the ISEL method (data not shown).
Fig. 1.
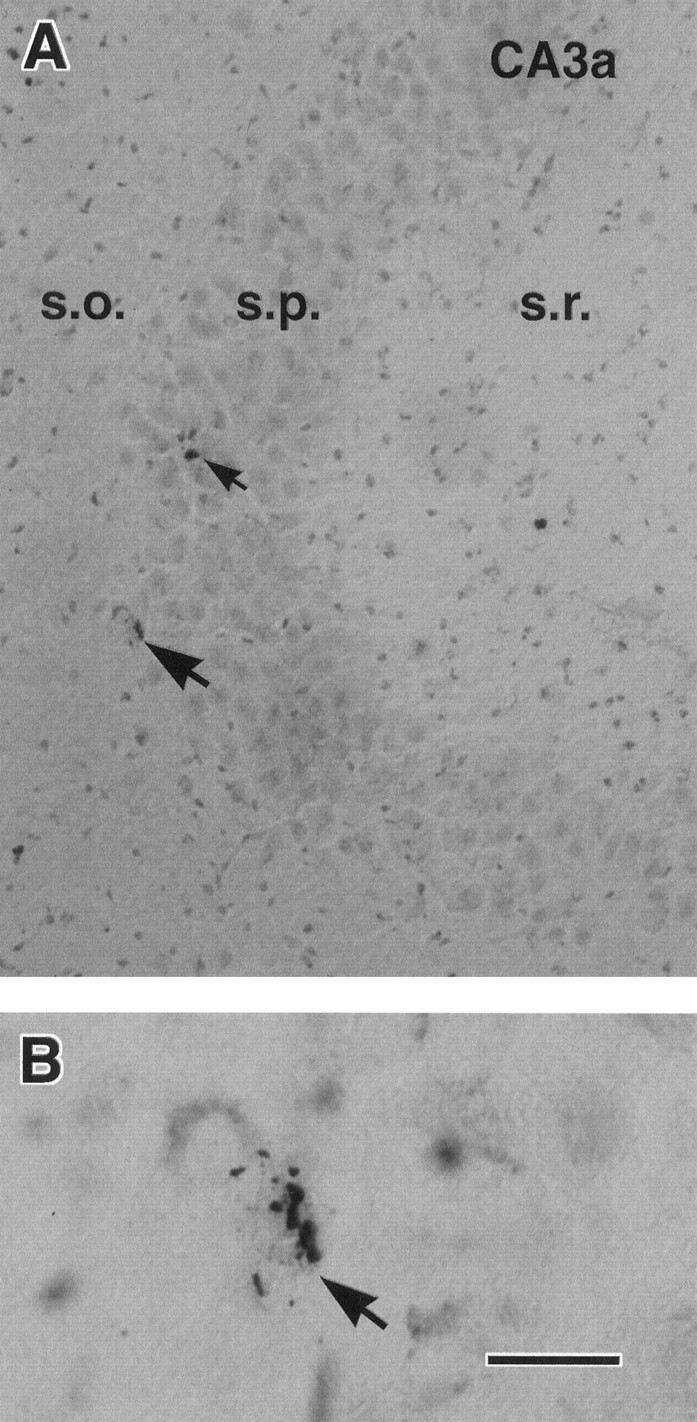
Rare acute neuronal death after hyperthermic seizures in the immature rat. A, In situend labeling reveals two hippocampal CA3 neurons (arrows) undergoing DNA fragmentation from an animal killed 20 hr after hyperthermic seizures. B, Higher magnification shows the typical DNA fragmentation in a pyramidal layer neuron (arrow corresponding to the lower neuron inA). Run in parallel, sections from normothermic controls did not contain labeled neurons, whereas, as expected, sections from kainic acid-treated mature rats revealed numerous dying cells (data not shown). s.o., Stratum oriens; s.p., stratum pyramidale; s.r., stratum radiatum. Scale bars:A, 50 μm; B, 10 μm.
Table 1.
Characteristics of hyperthermia-induced seizures and neuronal in situ end labeling
| Treatment | Hyperthermic seizures (n = 51) | Hyperthermia + AED* (n = 14) | Normothermic controls (n = 24) |
|---|---|---|---|
| Maximal temperature (°C) | 42.22 ± 0.07 | 42.32 ± 0.17 | N/A |
| Seizure duration (min) | 19.13 ± 1.21 | 0 (n = 12); 2; <2 | None |
| Survival time (hr) | 1 | 4 | 8.5 | 20* | 48 |
|---|---|---|---|---|---|
| ISEL-labeled cells: | |||||
| ACE (n) | 0 (9) | 0.36 ± 0.24 (11) | 1.50 ± 0.48 (12) | 0.1 ± 0.1 (10) | 0.37 ± 0.25 (8) |
| Hippocampus (n) | 0 (9) | 0.40 ± 0.21 (11) | 1.15 ± 0.32 (13) | 0 (10) | 0.25 ± 0.25 (8) |
ACE, Central nucleus of the amygdala; AED, antiepileptic drug; ISEL, in situ end labeling; N/A, not applicable; (n) denotes number of sections. Animals (marked with *) had hyperthermic seizures twice totaling 60 ± 2 min (see text). Values are means ± SE.
Hyperthermic seizures lead to neuronal injury in discrete hippocampal and amygdala regions
Sections from animals killed 24 hr after hyperthermic seizures contained significant populations of silver-stained, dark neurons. Figure 2 demonstrates clusters of stained neurons in the hippocampus. All subfields of the CA3 pyramidal cell layer were involved, as well as the entire extent of CA1 and occasional granule cells (Fig. 2A). Affected neurons were interspersed among normal ones, and their stained apical and basal dendrites indicated that they were pyramidal cells. No argyrophilic neurons were apparent in control sections (Fig. 2C) or in sections from animals pretreated with anticonvulsants before hyperthermia induction (Fig. 2B). Higher magnifications of the silver-stained neurons, revealing pronounced physicochemical alterations (shrunken appearance; Gallyas et al., 1992a), are shown in Figure 2, D and E.
Fig. 2.
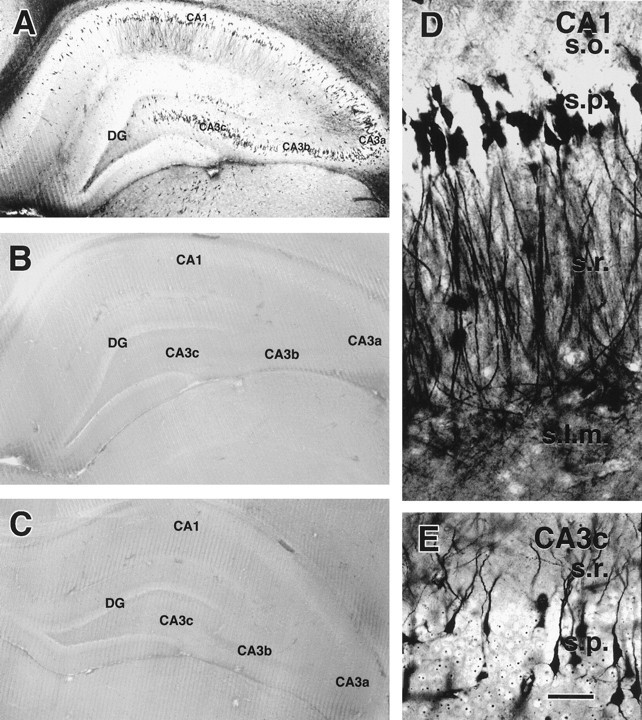
Acute induction of silver-stained neurons in hippocampal sections from immature rats subjected to hyperthermic seizures (A), hyperthermia without seizures (B), or normothermia (C) is shown. Animals were killed 24 hr after treatment, and sections were stained using the Gallyas dark-neuron method. Abundant stained neurons are evident in the CA1 and all three subfields of the CA3 pyramidal cell layers (CA3a–CA3c) of an animal with seizures (A) but not in hyperthermic or normothermic controls (B, C). Higher magnifications (D, E) provide morphological detail of the affected neurons in the CA1 pyramidal layer (D) and in CA3c (E), demonstrating altered physicochemical properties of both the cell body and processes. DG, Dentate gyrus; s.l.m., stratum lacunosum moleculare; s.o., stratum oriens;s.p., stratum pyramidale; s.r., stratum radiatum. Scale bars: A–C, 1 mm;D, E, 50 μm.
The remarkable involvement of the lateral division of the ACE is evident in Figure 3. Sections from rats killed 24 hr after hyperthermia and seizures (Fig. 3A) revealed an abundance of argyrophilic neurons, whereas sections from both of the control groups [hyperthermia without seizures (Fig.3B) and normothermia (Fig. 3C)] did not contain affected cells. Semiquantitative analysis of affected neurons in the CE-L suggested that the number of injured neurons was in the order of 185.8 ± 10.1 per 100-μm-thick section or 1645.5 ± 178 within the defined anatomical boundaries of this nucleus (see Materials and Methods). The distribution of hyperthermic seizures-induced argyrophilic neurons included also the medial and lateral basal amygdala nuclei (Fig. 3D), as well as a delimited portion of the perirhinal cortex (Fig. 3E).
EM was used to show the ultrastructural features of the silver-stained neurons in the hippocampal CA1 and the amygdaloid CE-L. Figure4 demonstrates a hyperelectron-dense shrunken pyramidal cell (Fig. 4B). Intact synapses are apparent along the surfaces of the soma and the shrunken, spiny dendrites of this neuron (Fig. 4C), which are postsynaptic to immature axon terminals. In the CE-L, the electron-dense cells maintained intact cell membranes and contained distinct organelles including Golgi complexes and endoplasmic reticulum (Fig.4D,E).
Fig. 4.
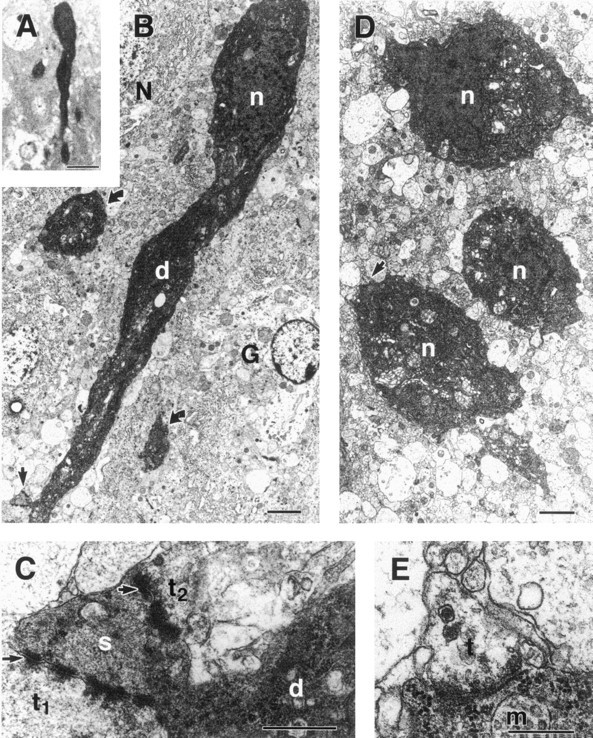
Light and electron micrographs of hyperelectron-dense neurons in the hippocampal CA1 (A–C) and the lateral division of the central amygdaloid nucleus (CE-L) (D,E). A, B, A shrunken, deeply basophilic neuron in A appears electron-dense inB compared with a normal neuron (N). The hyperelectron-dense nucleus (n) and apical dendrite (d) with a spine (arrow) of this neuron, as well as abnormal processes (curved arrows) and a glial cell (G), are shown in B.C, Higher magnification of the dendritic spine (s; arrow in B) reveals two axodendritic synapses (arrows) formed by vesicle-containing axon terminals (t1 andt2). D, Three hyperelectron-dense neurons with intact nuclei (n), Golgi complex, and endoplasmic reticulum are shown. E, Enlargement of an axosomatic synapse (arrow in D) shows synaptic vesicles and a multivesicular body (m) within the electron-dense soma. Scale bars: A, 20 μm;B, D, 2 μm; C,E, 0.1 μm.
Hyperthermic seizures, but not the hyperthermia per se, lead to neuronal injury
Seizure duration in the hyperthermic seizure group averaged 19.13 ± 1.21 min. Animals pretreated with pentobarbital before hyperthermia induction did not have behavioral seizures, although the degree of hyperthermia, as defined by maximal core temperature, and the duration of hyperthermia were comparable with those of non-pretreated pups (Table 1). Sections from anticonvulsant-pretreated animals who did not develop seizures and were killed either 24 hr or a week subsequently did not contain silver-stained neurons (see below). A broad correlation between seizure duration and the extent of neuronal injury was evident. Thus, sections from one of two animals, in whom short seizures (<2 min) developed despite diazepam pretreatment, contained occasional silver-stained neurons (data not shown).
Neuronal changes induced by hyperthermic seizures persist for at least 2 weeks
Altered structural neuronal properties, manifested as silver staining of affected neurons, persisted in the regions in which they were noted within 24 hr after the seizures. Figure5 demonstrates clusters of injured neurons in the hippocampus of an immature rat surviving for 1 week (Fig. 5A) or 2 weeks (Fig. 5B) after hyperthermic seizures. In the CE-L, another region with abundant dark neurons, persistent staining was also observed. Figure 5Cdemonstrates abundant stained neurons in the CE-L of an animal surviving for a week after hyperthermic seizures. A section from a rat surviving for 2 weeks is shown in Figure 5D, suggesting decreasing abundance of stained neurons at the latter time point.
Hyperthermic seizures do not result in the dropout of a significant number of neurons
Sections from 12 animals surviving for 4 weeks after hyperthermic seizures, hyperthermia alone, or no treatment were subjected to neuronal counts by an investigator unaware of treatment (blinded). Because an abundance of argyrophilic, injured neurons occurred in the CE-L, this region was chosen for semiquantitative assessment of neuronal dropout. The three experimental groups did not differ significantly in total neuronal number in the anatomically defined CE-L (4295 ± 97, 4358 ± 139, and 4428 ± 54 cells per CE-L for the hyperthermic seizures, hyperthermia alone, and normothermic groups, respectively). Variance among samples was small, and the maximal difference in CE-L neurons among groups was an order of magnitude lower than was the estimated number of argyrophilic, injured neurons in this structure (see above). These results do not exclude death of some injured neurons; however, they indicate that the majority of hyperthermic seizures-induced argyrophilic neurons do not die and drop out.
DISCUSSION
The principal findings of this study are the following: (1) Hyperthermic seizures alter the structure of select neuronal populations in the hippocampus and the amygdala. (2) These changes are predicated on the presence of the seizures rather than on hyperthermia per se. (3) The distribution of affected neurons provides the first reported topographical “map” of neuronal vulnerability to febrile seizures. (4) In regions where the physicochemical changes induced by hyperthermic seizures are highly abundant, few neurons manifest apoptotic changes, i.e., DNA fragmentation. (5) Although significant numbers of neurons are rendered argyrophilic for at least 2 weeks after the seizures, particularly in the amygdaloid CE-L, no significant neuronal dropout can be demonstrated by 4 weeks after the seizures.
Hyperthermic seizures produce injury of discrete limbic neuronal populations
The current study demonstrates significant and prolonged alterations in the physicochemical properties of neurons in the pyramidal layer of the hippocampal CA1 and all of the CA3 subfields. In addition, a large proportion of pyramidal and nonpyramidal neurons in the CE-L are consistently rendered argyrophilic by this febrile seizures model. The distribution of the argyrophilic neurons is consistent with both the behavioral and the electrophysiological characteristics of hyperthermic seizures (Baram et al., 1997). The behavioral aspects, freezing and oral automatisms, indicate a limbic onset in the amygdala (Gloor, 1992) or the hippocampus (Ben-Ari, 1985). Our electrophysiological studies of the seizures in this model revealed epileptiform discharges in the amygdala and the hippocampus but not the neocortex (Baram et al., 1997). Thus the pattern of distribution of the argyrophilic neurons, taken together with the earlier studies, constitutes a map of the propagation of hyperthermic seizures. At a minimum, the distribution of injured neurons provides the neuroanatomic matrix of neuronal vulnerability to febrile seizures in this model. In this context, it is interesting that electrophysiological and anatomical data regarding the distribution of human febrile seizures are scarce, because the seizures are unpredictable, short, and ethically impossible to induce (Morimoto et al., 1991).
Unique aspects of the distribution of neuronal injury in the febrile seizures model
The distribution of neuronal injury observed in this study possesses features both common to and distinct from injury found with other limbic seizure types. In the hippocampus, major involvement of CA3 and CA1 pyramidal cell layers and relative sparing of the granule cell layer and subiculum are consistent with vulnerability patterns in adult models of kainic acid- (Nadler et al., 1978; Sperk et al., 1983;Pollard et al., 1994) and pilocarpine-induced status epilepticus (Clifford et al., 1987). In the amygdala, however, the almost exclusive involvement of CE-L neurons distinguishes the injury pattern in this model from the adult pilocarpine and kainic acid seizure models, in which neuronal death was found preferentially in the cortical, medial, and lateral nuclei (Clifford et al., 1987) and in the basal nuclear group, respectively (Schwob et al., 1980; Sperk et al., 1983). Involvement of the ACE has been documented (Ben-Ari et al., 1981), and a detailed analysis of amygdala neuropathology after kainic acid seizures suggests that within the ACE, most argyrophilic neurons occur in the CE-L (Tuunanen et al., 1996). Thus, although predominant ACE injury in limbic excitotoxicity models is unusual, the preferential involvement of the CE-L in this febrile seizures model is supported by the argyrophilia pattern documented in a different (adult) model of limbic epilepsy.
The mechanism for the unique vulnerability of ACE neurons in this model of febrile seizures may involve the preferential activation of this nucleus during the hyperthermic stress (Gray, 1993; Tkacs et al., 1997). Stress leads to rapid immediate-early-gene induction in the ACE (Clark et al., 1991; Honkaniemi et al., 1992), followed by activation of a cluster of neurons producing corticotropin-releasing hormone (CRH) (Clark et al., 1991; Gray, 1993). CRH is capable of producing neuronal death in the amygdala of the immature rat (Baram and Ribak, 1995; Ribak and Baram, 1996), so that a local release of CRH in the ACE may account for the observed neuronal injury.
Neuronal injury in the immature, as compared with the adult, brain
The spatiotemporal evolution of neuronal changes observed in the current studies provides a useful perspective into issues of seizure-induced cell death in the immature brain. Although prolonged and severe seizures typically occur when convulsants such as kainic acid or pilocarpine are administered to immature rats (younger than 20 d), it is generally considered that they are not followed by neuropathological changes (Nitecka et al., 1984; Sperber et al., 1992;Chang and Baram, 1994; but see Wasterlain, 1997). The present study, revealing profound but transient alterations of neuronal integrity in regions known to be affected by other limbic seizure paradigms, may provide a mechanism to reconcile conflicting reports regarding the effects of developmental limbic seizures on neuronal survival. Specifically, our findings suggest that similar neuronal populations share vulnerability to limbic seizures in both the immature and mature rat, but immature neurons may undergo injury followed by recovery, whereas mature neurons progress from injury to death (Chang and Baram, 1994; Owens et al., 1997). In addition, as has been shown for eosinophilic acid fuchsin staining (Chang and Baram, 1994), neuronal argyrophilia may carry a different significance regarding neuronal fate in the immature, as compared with the adult, CNS. Along the same lines, ISEL may underestimate neuronal death particularly in the immature brain, leading to the limited cell death found in the current studies. The ISEL time course study and the lack of significant neuronal dropout using an independent method at the 4 week time point, however, render this possibility less likely (and see below).
An intriguing feature of the distribution of affected neurons after hyperthermic seizures in this study is their concordance with the neuropathological features of human temporal lobe (limbic) epilepsy known as mesial temporal sclerosis. Within the hippocampus, the most common sites of neuronal loss are the CA3 and CA4 (of which the hilus is the rodent counterpart) (Sagar and Oxbury, 1987; Bruton, 1988). Less is known regarding the precise topography of neuronal loss in the TLE amygdala (Gloor, 1992), and most studies refer to gliosis in the “basal” nuclear group (Cavanagh and Meyer, 1956; Bruton, 1988;Gloor, 1992). The amygdala is preferentially affected, however, when TLE is caused by lesions outside the hippocampus (Cavanagh and Meyer, 1956; Gloor, 1992).
Hyperthermic seizures lead to neuronal injury without evidence of significant neuronal dropout
The hyperthermic seizures-induced neuronal injury demonstrated in this study was not associated with significant DNA fragmentation, as determined by ISEL. DNA cleavage is generally considered a marker of cell death (Wyllie, 1993), although the type of cell death (apoptosis vs necrosis) may be unsettled (Kure et al., 1991; Pollard et al., 1994). The advantage of ISEL and similar techniques derives from the resulting positive stain, which renders even single cells highly visible, in contrast to cell counts, which determine a decrease in overall cell numbers. A significant disadvantage of methods using DNA fragmentation markers stems from the short temporal “window” (within the process of neuronal death) during which such DNA fragmentation may be detectable. Thus, these methods tend to underestimate neuronal death, particularly when cells do not die synchronously (Voyvodic, 1996). However, the current study included several time points spanning 1–48 hr after seizure induction, so that temporal underestimation of the number of dying neurons is less likely. Therefore, the discrepancy between the large numbers of argyrophilic neurons and the rare ISEL-labeled cells indicates that argyrophilia in the current study does not constitute evidence of “irreversible injury” or death (Gallyas et al., 1992b; Toth et al., 1997) in the majority of neurons.
A major advantage of the silver-staining method used in the current study lies in its ability to demonstrate substantial populations of injured hippocampal and ACE neurons. The precise alterations in the physicochemical properties of injured neurons that lead to their enhanced affinity to the silver stain has not been resolved (Gallyas et al., 1992b). However, aside from the ISEL data, two additional lines of evidence point against equating argyrophilia with neuronal death. First, Toth et al. (1997) demonstrated that fluid percussion trauma can lead to the formation of argyrophilic neurons even when applied to a dead animal. Second, the number of argyrophilic neurons observed in the current study, at least in the CE-L (∼1700), far exceeded any potential cell loss determined by cell counts 4 weeks later (∼150 neurons, maximal intergroup difference). Counts were performed without knowledge of treatment, minimizing a systematic bias, but inherent estimation errors cannot be excluded (Popken and Farel, 1997). However, in the context of the current study, even a 100% error in counting (Popken and Farel, 1997; Saper, 1997) would not alter the conclusion that the majority of argyrophilic, injured neurons induced by hyperthermic seizures do not drop out.
Implications for human febrile seizures
The studies described provide a prospective analysis of the spatiotemporal profile of neuronal changes induced by hyperthermic seizures, in an appropriate-age model of human febrile seizures. Febrile seizures, the most common human seizures, affect at least 500,000 individuals per year (Hauser, 1994). These seizures are essentially restricted to the developmental period of infancy and early childhood (Knudsen, 1996). The contribution of febrile seizures to the development of TLE has remained controversial (Cendes et al., 1993;Knudsen, 1996) and has centered on the issue of induction of limbic neuronal death by these seizures (Sagar and Oxbury, 1987; Gloor, 1991). These fundamental issues of developmental epilepsy are difficult to study in the human. The current study relied on an appropriate-age model in which seizures can be induced in >93% of animals, thus eliminating any concerns of genetic predisposition and pre-existing lesions. Using this model, we have demonstrated the pattern of vulnerability of hippocampal and amygdala neuronal populations to hyperthermic seizures. The absence of neuronal injury (argyrophilia) in hyperthermic rats in whom the seizures were prevented confirms the specificity of these neuronal changes to the seizures themselves. In addition, the results of this study suggest that the majority of injured neurons do not progress to death. Thus, these data do not support the supposition that early-life febrile seizures result directly in hippocampal cell death, the neuropathological lesion found in human TLE. However, the widespread and prolonged neuronal injury demonstrated in this study may lead to significant alterations of the function and electrophysiology of the affected neuronal circuits (Kapur and Coulter, 1995), which could lead eventually to an epileptic state.
Footnotes
This work was supported by National Institutes of Health Grants NS28912 and NS35439 to T.Z.B. and NS15669 to C.E.R. The technical assistance of L. Schultz and M. Shiba-Noz is appreciated. We thank Drs. G. Popken and I. Soltesz for helpful discussions, and A. Owens for her help with this manuscript.
Correspondence should be addressed to Dr. Tallie Z. Baram, Departments of Anatomy and Neurobiology and Pediatrics, ZOT 4475, University of California, Irvine, Irvine, CA 92697-4475.
REFERENCES
- 1.Albala BJ, Moshe SL, Okada R. Kainic acid-induced seizures: a developmental study. Brain Res. 1984;315:139–148. doi: 10.1016/0165-3806(84)90085-3. [DOI] [PubMed] [Google Scholar]
- 2.Baram TZ, Ribak CE. Peptide-induced status epilepticus causes neuronal death and synaptic reorganization. NeuroReport. 1995;6:277–280. doi: 10.1097/00001756-199501000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baram TZ, Gerth A, Schultz L. Febrile seizures: an age appropriate model. Brain Res Dev Brain Res. 1997;246:134–143. doi: 10.1016/s0165-3806(96)00190-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- 5.Ben-Ari Y, Tremblay E, Riche D, Ghilini G, Naquet R. Electrographic, clinical and pathological alterations following systemic administration of kainic acid, bicuculline or pentetrazole: metabolic mapping using the deoxyglucose method with special reference to the pathology of epilepsy. Neuroscience. 1981;6:1361–1391. doi: 10.1016/0306-4522(81)90193-7. [DOI] [PubMed] [Google Scholar]
- 6.Berg AT, Shinnar S, Hauser WA, Alemany M, Shapiro ED, Salomon ME, Crain EF. A prospective study of recurrent febrile seizures. N Engl J Med. 1992;327:1122–1127. doi: 10.1056/NEJM199210153271603. [DOI] [PubMed] [Google Scholar]
- 7.Bruton CJ. The neuropathology of temporal lobe epilepsy. Oxford UP; New York: 1988. [Google Scholar]
- 8.Cavanagh JB, Meyer A. Aetiological aspects of Ammon’s horn sclerosis associated with temporal lobe epilepsy. Br Med J. 1956;2:1403–1407. doi: 10.1136/bmj.2.5006.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cendes F, Andermann F, Dubeau F, Gloor P, Evans A, Jones-Gotman M, Olivier A, Andermann E, Robitaille Y, Lopes-Cendes I. Early childhood prolonged febrile convulsions, atrophy and sclerosis of mesial structures, and temporal lobe epilepsy: an MRI volumetric study. Neurology. 1993;43:1083–1087. doi: 10.1212/wnl.43.6.1083. [DOI] [PubMed] [Google Scholar]
- 10.Chang D, Baram TZ. Status epilepticus results in reversible neuronal injury in infant rat hippocampus: novel use of a marker. Brain Res Dev Brain Res. 1994;77:133–136. doi: 10.1016/0165-3806(94)90220-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark M, Weiss SR, Post RM. Expression of c-fos mRNA in rat brain after intracerebroventricular administration of corticotropin-releasing hormone. Neurosci Lett. 1991;132:235–238. doi: 10.1016/0304-3940(91)90309-h. [DOI] [PubMed] [Google Scholar]
- 12.Clifford DB, Olney JW, Maniotis A, Collins RC, Zorumski CF. The functional anatomy and pathology of lithium-pilocarpine and high-dose pilocarpine seizures. Neuroscience. 1987;23:953–968. doi: 10.1016/0306-4522(87)90171-0. [DOI] [PubMed] [Google Scholar]
- 13.Dobbing J, Sands J. Quantitative growth and development of human brain. Arch Dis Child. 1973;48:757–767. doi: 10.1136/adc.48.10.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falconer MA, Serafetinides EA, Corsellis JAN. Etiology and pathogenesis of temporal lobe epilepsy. Arch Neurol. 1964;10:233–248. doi: 10.1001/archneur.1964.00460150003001. [DOI] [PubMed] [Google Scholar]
- 15.Gallyas F, Guldner FH, Zoltay G, Wolff JR. Golgi-like demonstration of “dark” neurons with an argyrophil III method for experimental neuropathology. Acta Neuropathol (Berl) 1990;79:620–628. doi: 10.1007/BF00294239. [DOI] [PubMed] [Google Scholar]
- 16.Gallyas F, Zoltay G, Balas I. An immediate light microscopic response of neuronal somata, dendrites and axons to contusing concussive head injury in the rat. Acta Neuropathol (Berl) 1992a;83:394–401. doi: 10.1007/BF00713531. [DOI] [PubMed] [Google Scholar]
- 17.Gallyas F, Zoltay G, Dames W. Formation of “dark” (argyrophilic) neurons of various origins proceeds with a common mechanism of biophysical nature (a novel hypothesis). Acta Neuropathol (Berl) 1992b;83:504–509. doi: 10.1007/BF00310027. [DOI] [PubMed] [Google Scholar]
- 18.Gloor P. Mesial temporal sclerosis: historical background and an overview from a modern perspective. In: Luders HO, editor. Epilepsy surgery. Raven; New York: 1991. pp. 689–703. [Google Scholar]
- 19.Gloor P. Role of the amygdala in temporal lobe epilepsy. In: Aggleton JP, editor. The amygdala: neurobiological aspects of emotion, memory, and mental dysfunction. Wiley-Liss; New York: 1992. pp. 505–538. [Google Scholar]
- 20.Gray TS. Amygdaloid CRF pathways. Role in autonomic, neuroendocrine, and behavioral responses to stress. Ann NY Acad Sci. 1993;697:53–60. doi: 10.1111/j.1749-6632.1993.tb49922.x. [DOI] [PubMed] [Google Scholar]
- 21.Hauser WA (1994) The prevalence and incidence of convulsive disorders in children. Epilepsia 35 [Suppl] 2:S1–S6. [DOI] [PubMed]
- 22.Holmes GL. Epilepsy in the developing brain: lessons from the laboratory and clinic. Epilepsia. 1997;38:12–30. doi: 10.1111/j.1528-1157.1997.tb01074.x. [DOI] [PubMed] [Google Scholar]
- 23.Holmes GL, Thompson JL. Effects of kainic acid on seizure susceptibility in the developing brain. Brain Res. 1988;467:51–59. doi: 10.1016/0165-3806(88)90066-1. [DOI] [PubMed] [Google Scholar]
- 24.Honkaniemi J, Pelto-Huikko M, Rechardt L, Isola J, Lammi A, Fuxe K, Gustafsson JA, Wikstrom AC, Hokfelt T. Colocalization of peptide and glucocorticoid receptor immunoreactivities in rat central amygdaloid nucleus. Neuroendocrinology. 1992;55:451–459. doi: 10.1159/000126156. [DOI] [PubMed] [Google Scholar]
- 25.Jensen FE, Applegate CD, Holtzman D, Belin TR, Burchfiel JL. Epileptogenic effect of hypoxia in the immature rodent brain. Ann Neurol. 1991;29:629–637. doi: 10.1002/ana.410290610. [DOI] [PubMed] [Google Scholar]
- 26.Kapur J, Coulter DA. Experimental status epilepticus alters gamma-aminobutyric acid type A receptor function in CA1 pyramidal neurons. Ann Neurol. 1995;38:893–900. doi: 10.1002/ana.410380609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knudsen FU. Febrile seizures–treatment and outcome. Brain Dev. 1996;18:438–449. doi: 10.1016/s0387-7604(96)00059-9. [DOI] [PubMed] [Google Scholar]
- 28.Kure S, Tominaga T, Yoshimoto T, Tada K, Narisawa K. Glutamate triggers internucleosomal DNA cleavage in neuronal cells. Biochem Biophys Res Commun. 1991;179:39–45. doi: 10.1016/0006-291x(91)91330-f. [DOI] [PubMed] [Google Scholar]
- 29.Morimoto T, Nagao H, Sano N, Takahashi M, Matsuda H. Electroencephalographic study of rat hyperthermic seizures. Epilepsia. 1991;32:289–293. doi: 10.1111/j.1528-1157.1991.tb04653.x. [DOI] [PubMed] [Google Scholar]
- 30.Nadler JV, Perry BW, Cotman CW. Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature. 1978;271:676–677. doi: 10.1038/271676a0. [DOI] [PubMed] [Google Scholar]
- 31.Nelson KB, Ellenberg JH. Predictors of epilepsy in children who have experienced febrile seizures. N Engl J Med. 1976;295:1029–1033. doi: 10.1056/NEJM197611042951901. [DOI] [PubMed] [Google Scholar]
- 32.Nitecka L, Tremblay E, Charton G, Bouillot JP, Berger ML, Ben-Ari Y. Maturation of kainic acid seizure-brain damage syndrome in the rat. Neuroscience. 1984;13:1073–1094. doi: 10.1016/0306-4522(84)90289-6. [DOI] [PubMed] [Google Scholar]
- 33.Owens JJ, Robbins CA, Wenzel HJ, Schwartzkroin PA. Acute and chronic effects of hypoxia on the developing hippocampus. Ann Neurol. 1997;41:187–199. doi: 10.1002/ana.410410210. [DOI] [PubMed] [Google Scholar]
- 34.Pollard H, Charriaut-Marlangue C, Cantagrel S, Represa A, Robain O, Moreau J, Ben-Ari Y. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience. 1994;63:7–18. doi: 10.1016/0306-4522(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 35.Popken GJ, Farel PB. Sensory neuron number in neonatal and adult rats estimated by means of stereologic and profile-based methods. J Comp Neurol. 1997;386:8–15. [PubMed] [Google Scholar]
- 36.Ribak CE, Baram TZ. Selective death of hippocampal CA3 pyramidal cells with mossy fiber afferents after CRH-induced status epilepticus in infant rats. Brain Res Dev Brain Res. 1996;91:245–251. doi: 10.1016/0165-3806(95)00183-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sagar HJ, Oxbury JM. Hippocampal neuron loss in temporal lobe epilepsy: correlation with early childhood convulsions. Ann Neurol. 1987;22:334–340. doi: 10.1002/ana.410220309. [DOI] [PubMed] [Google Scholar]
- 38.Sakhi S, Bruce A, Sun N, Tocco G, Baudry M, Schreiber SS. p53 induction is associated with neuronal damage in the central nervous system. Proc Natl Acad Sci USA. 1994;91:7525–7529. doi: 10.1073/pnas.91.16.7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saper CB. Counting on our reviewers to set the standards. J Comp Neurol. 1997;386:1. [Google Scholar]
- 40.Schwob JE, Fuller T, Price JL, Olney JW. Widespread patterns of neuronal damage following systemic or intracerebral injections of kainic acid: a histological study. Neuroscience. 1980;5:991–1014. doi: 10.1016/0306-4522(80)90181-5. [DOI] [PubMed] [Google Scholar]
- 41.Sherwood NM, Timiras PS. A stereotaxic atlas of the developing rat brain. University of California; Berkeley, CA: 1970. [Google Scholar]
- 42.Shinnar S. Febrile seizures. In: Johnson RT, editor. Current therapy in neurological disease. Decker; Philadelphia: 1990. pp. 29–32. [Google Scholar]
- 43.Sperber EF, Haas KZ, Stanton PK, Moshe SL. Resistance of the immature hippocampus to seizure-induced synaptic reorganization. Brain Res Dev Brain Res. 1991;60:88–93. doi: 10.1016/0165-3806(91)90158-f. [DOI] [PubMed] [Google Scholar]
- 44.Sperber EF, Stanton PK, Haas K, Ackerman RF, Moshe SL. Molecular neurobiology of epilepsy, pp 67–81. Elsevier; Amsterdam: 1992. Developmental differences in the neurobiology of epileptic brain damage. [PubMed] [Google Scholar]
- 45.Sperk G, Lassmann H, Baran H, Kish SJ, Seitelberger F, Hornykiewicz O. Kainic acid-induced seizures: neurochemical and histopathological changes. Neuroscience. 1983;10:1301–1315. doi: 10.1016/0306-4522(83)90113-6. [DOI] [PubMed] [Google Scholar]
- 46.Tkacs NC, Li J, Strack AM. Central amygdala Fos expression during hypotensive or febrile, nonhypotensive endotoxemia in conscious rats. J Comp Neurol. 1997;379:592–602. doi: 10.1002/(sici)1096-9861(19970324)379:4<592::aid-cne9>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 47.Toth Z, Hollrigel GS, Gorcs T, Soltesz I. Instantaneous perturbation of dentate interneuronal networks by a pressure wave-transient delivered to the neocortex. J Neurosci. 1997;17:8106–8117. doi: 10.1523/JNEUROSCI.17-21-08106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tuunanen J, Halonen T, Pitkanen A. Status epilepticus causes selective regional damage and loss of GABAergic neurons in the rat amygdaloid complex. Eur J Neurosci. 1996;8:2711–2725. doi: 10.1111/j.1460-9568.1996.tb01566.x. [DOI] [PubMed] [Google Scholar]
- 49.Verity CM, Butler NR, Golding J. Febrile convulsions in a national cohort followed up from birth. Prevalence and recurrence in the first five years of life. Br Med J. 1985;290:1307–1310. doi: 10.1136/bmj.290.6478.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Voyvodic JT. Cell death in cortical development: how much? Why? So what? Neuron. 1996;16:693–696. doi: 10.1016/s0896-6273(00)80089-6. [DOI] [PubMed] [Google Scholar]
- 51.Wasterlain CG. Recurrent seizures in the developing brain are harmful. Epilepsia. 1997;38:728–734. doi: 10.1111/j.1528-1157.1997.tb01244.x. [DOI] [PubMed] [Google Scholar]
- 52.Wyllie AH. Apoptosis. Br J Cancer. 1993;67:205–208. doi: 10.1038/bjc.1993.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yi SJ, Baram TZ. Corticotropin-releasing hormone mediates the response to cold stress in the neonatal rat without compensatory enhancement of the peptide’s gene expression. Endocrinology. 1994;135:2364–2368. doi: 10.1210/endo.135.6.7988418. [DOI] [PMC free article] [PubMed] [Google Scholar]