

Abstract
Wernicke encephalopathy (WE), a neurological disorder caused by thiamine deficiency (TD), is characterized by structural damage in brain regions that include the thalamus and cerebral cortex. The basis for these lesions is unclear, but may involve a disturbance of glutamatergic neurotransmission. We have therefore investigated levels of the astrocytic glutamate transporters EAAT1 and EAAT2 in order to evaluate their role in the pathophysiology of this disorder. Histological assessment of the frontal cortex revealed a significant loss of neurons in neuropathologically confirmed cases of WE compared with age-matched controls, concomitant with decreases in α-internexin and synaptophysin protein content of 67 and 52% by immunoblotting. EAAT2 levels were diminished by 71% in WE, with levels of EAAT1 also reduced by 62%. Loss of both transporter sites was confirmed by immunohistochemical methods. Development of TD in rats caused a profound loss of EAAT1 and EAAT2 in the thalamus accompanied by decreases in other astrocyte-specific proteins. Treatment of TD rats with N-acetylcysteine prevented the downregulation of EAAT2 in the medial thalamus, and ameliorated the loss of several other astrocyte proteins, concomitant with increased neuronal survival. Our results suggest that (1) loss of EAAT1 and EAAT2 glutamate transporters is associated with structural damage to the frontal cortex in patients with WE, (2) oxidative stress plays an important role in this process, and (3) TD has a profound effect on the functional integrity of astrocytes. Based on these findings, we recommend that early treatment using a combination of thiamine AND antioxidant approaches should be an important consideration in cases of WE.
Keywords: thiamine deficiency, glutamate, astrocyte, vitamin B1, EAAT, excitotoxicity, oxidative stress
Introduction
Ever since the initial report of Wernicke encephalopathy (WE) describing a disorder in three patients characterized by ataxia, ophthalmoplegia, and mental changes (Wernicke, 1881), attempts have been made to determine the underlying processes that lead to its occurrence. One of two components of the Wernicke–Korsakoff syndrome, a neuropsychiatric disorder, WE represents the consequence of a compromise of vitamin B1 (thiamine) status, occurring in disorders of generalized malnutrition, including chronic alcoholism and in nonalcoholic conditions that include malignant disease (Miyajima et al., 1993; Shah and Wolff, 1973), hyperemesis gravidarum (Ebels, 1978; Ohkoshi et al., 1994), a variety of gastrointestinal disorders (Lindboe and Loberg, 1989), and AIDS (Soffer et al., 1989). Neuropathology due to chronic WE can develop following repeated bouts of “subclinical” thiamine deficiency (TD) (Harper, 1983), suggesting that appropriate intervention during these episodes may delay or prevent the development of major histological lesions.
During life, WE is surprisingly difficult to diagnose, with the classical triad of clinical features often being absent in both alcoholic and nonalcoholic cases, leading to only a 20% success rate (Harper et al., 1986). In addition, evidence from studies in a large Australian population suggest that the incidence of WE is higher than anticipated (1.7%), with 88% of these cases being alcohol-related (Harper, 1979). These findings suggest a higher frequency than that of epilepsy or Parkinson's disease, making WE an important health care issue. Although structures such as the mammillary bodies and thalamus are characterized as selectively vulnerable in WE, the cerebral cortex is normally considered to be less susceptible to damage. However, studies have demonstrated that the cortex also sustains histological damage in this disorder (Parkin et al., 1993; Yamashita and Yamamoto, 1995). At present, the cause of these cortical lesions is unclear.
A major function of astrocytes is the efficient removal of glutamate from the extracellular space (Drejer et al., 1983), a process that is instrumental in maintaining normal interstitial levels of this neurotransmitter (Nicholls and Attwell, 1990). This is effected primarily as a result of the activity of two subtypes of glutamate transporter, EAAT1 and EAAT2 (also known as GLAST and GLT-1, respectively), both localized predominantly in astrocytes (for review, see Robinson, 1998), with EAAT2 being considered the major transporter involved in glutamate clearance from the extracellular space. A third transporter subtype, EAAT3 has a postsynaptic neuronal localization (Rothstein et al., 1994), but appears to play less of a role in regulation of external glutamate levels. Decreased expression and function of astrocyte glutamate transporters have the potential to lead to increased extracellular glutamate levels resulting in excitotoxicity and ultimately neuronal cell death (Rothstein et al., 1996). Indeed, mutant mice with a knockout of the EAAT2 gene show more damage following cortical injury than their wild-type counterparts (Tanaka et al., 1997). In addition, a number of neurological disorders have been shown to be associated with astrocyte glutamate transporter dysfunction including ischemia (Martin et al., 1997; Torp et al., 1995), amyotrophic lateral sclerosis (Lin et al., 1998), hepatic encephalopathy (Knecht et al., 1997), and epilepsy (Mathern et al., 1999; Tanaka et al., 1997).
In previous studies, we have demonstrated that loss of astrocytic glutamate transporters is an important feature of TD (Hazell et al., 2001, 2003). Now, for the first time, we have investigated levels of astrocytic glutamate transporters in the frontal cortex of patients with WE. In addition, we have assessed the possible involvement of oxidative stress in loss of glutamate transporter integrity in rats with TD, a well-characterized animal model of WE, using the well-established antioxidant N-acetylcysteine (NAC).
Materials and Methods
Materials
Pyrithiamine hydrobromide, protease inhibitor cocktail, 3,3′-diaminobenzidine (DAB), mouse antiserum against β-actin, and DAPI (4′,6-diamidino-2-phenylindole) were purchased from Sigma-Aldrich (Oakville, ON, Canada). Goat polyclonal antisera against glial fibrillary acidic protein (GFAP), α-internexin, synaptophysin, δ-aminobutyric acid (GABA) transporter-3 (GAT-3), and anti-mouse glutamine synthetase (GS), biotinylated donkey anti-rabbit, -mouse and -goat IgG secondary anti-bodies, and HRP-coupled anti-rabbit, mouse and -goat IgG secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and PerkinElmer Life And Analytical Sciences (Woodbridge, ON, Canada). Anti-mouse CD68 antibody was purchased from AbDSer-otec (Raleigh, NC). NAC (“Parvolex”) was purchased from Bioniche Pharma (Toronto, ON, Canada). Streptavidin-horseradish peroxidase (HRP) conjugate was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Alexa Fluor 488 (green) secondary antibodies were purchased from Invitrogen Canada, Burlington, ON, Canada). Polyvinylidene difluoride (PVDF) membranes and broad-range protein markers were purchased from Bio-Rad Laboratories (Hercules, CA). Enhanced chemiluminescence (ECL) kits were purchased from New England Nuclear (Boston, MA) and X-OMAT autoradiography film was purchased from Kodak (Ile des Soeurs, QC, Canada). All other materials and chemicals were purchased from Amersham Canada (Oakville, ON, Canada).
Human Tissue Samples
Samples of frontal cortex were obtained at autopsy from a total of 10 patients, including 5 cases with a diagnosis of WE either neuropathologically or during life and 5 control cases (Table 1). Tissue was obtained from the New South Wales Tissue Resource Centre that is supported by the University of Sydney, Neuroscience Institute of Schizophrenia and Allied Disorders, National Institutes of Alcohol Abuse and Alcoholism, and New South Wales Department of Health. Pathological diagnosis of WE was based on classical periventricular changes, notably degenerative changes in the mamillary bodies (Harper, 1979). These cases included a clinical history of consumption of greater than 80 g of absolute alcohol per day. Control cases did not meet any criteria for a neurological disease (Alzheimer's disease, stroke, etc) either clinically or neuropathologically, with a history that included consumption of less than 20 g of absolute alcohol per day. Informed consent from the next of kin was obtained in all cases. Studies were performed in accordance with guidelines set out by the Human Ethics Committee of the University of Montreal.
Table 1. Patient Information.
| Case No. | Age (yr) | PMI (hr) | Sex | ND | COD |
|---|---|---|---|---|---|
| 1 | 46 | 25 | M | Normal | Cardiac arrest |
| 2 | 66 | 22 | M | Normal | Respiratory arrest |
| 3 | 44 | 24 | M | Normal | Acute myocardial infarction |
| 4 | 53 | 26 | M | Normal | Non-Hodgkin's lymphoma |
| 5 | 62 | 9 | M | Normal | Adenocarcinoma |
| 6 | 46 | 28.5 | M | WE | Septicaemia |
| 7 | 47 | 19 | M | WE | Throat cancer |
| 8 | 54 | 12 | M | WE | Septicaemia |
| 9 | 60 | 7 | M | WE | Ischaemic heart disease |
| 10 | 66 | 39 | M | WE | Coronary artery disease |
PMI, postmortem interval; M, male; ND, neuropathological diagnosis; COD, cause of death.
Rat Model of TD
All procedures were undertaken with the approval of the Animal Ethics Committee of Hôpital Saint-Luc and the University of Montreal, and were conducted in accordance with guidelines set out by the Canadian Council on Animal Care. Male Sprague–Dawley rats (225 g) were weighed daily and housed under constant conditions of temperature, humidity, and 12/12 h day/night cycles. Assessment for rotational and backward movements of the animals (Hazell et al., 1998) preceding neurological signs of TD (ataxia, opisthotonus, loss of righting reflexes, convulsions, nystagmus) were made on a daily basis. Experiments were carried out on the following groups of animals: (A) Acute symptomatic group. Rats were made thiamine-deficient by feeding them a diet lacking in thiamine (Ralston Purina, Richmond, VA) supplemented by daily administration of pyrithiamine (0.5 mg/kg body weight, i.p.), and were allowed to progress to a stage of TD characterized by a loss of righting reflexes on Day 14. Animals were studied within 6 h following the appearance of this condition, and none exhibited obvious seizures at this stage. (B) Acute symptomatic group + NAC. Rats were treated as in group (A) plus daily administration of NAC (163 mg/kg body weight, i.p.). (C) Pair-fed control group. Rats were placed on an identical diet to that of groups (A) and (B), but limited in quantity to that consumed by their TD counterparts with daily injections of thiamine (100 μg in 0.2 mL saline, i.p.) that exceed the recommended daily allowance of this vitamin.
Immunoblotting Studies
At the appropriate time, animals (all groups, n = 7) were sacrificed and the brains removed, followed by dissection of the medial thalamus at the level of the medial habenulae and frontal parietal cortex on dry ice. The tissue was stored at −80°C until ready for study. Samples of human and rat brain tissue were homogenized in buffer containing 50 mM Tris, 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 1% NP-40, 0.5% sodium deoxycholate (pH 8.0) and protease inhibitor cocktail, and centrifuged at 10,000g for 10 min, 4°C. Preliminary studies carried out on the pellet and supernatant indicated that both EAAT1 and EAAT2 were completely soluble in this buffer under our conditions. Thus, the supernatant was retained and used for study of both transporters. For extraction of GFAP, buffer containing 2% SDS was used. Protein content of all samples was determined by the method of Lowry et al. (1951) using bovine serum albumin (BSA) as the standard. Sample buffer was added to aliquots of the tissue (30 μg) and the samples boiled for 5 min. Aliquots were subjected to (SDS)-polyacrylamide gel electrophoresis (8% polyacrylamide) and the proteins subsequently transferred to PVDF membranes by wet transfer at 20 V over 24 h. The transfer buffer consisted of 48 mM Tris (pH 8.3), 39 mM glycine, 0.037% SDS, and 20% methanol. Membranes were subsequently incubated in blocking buffer (10 mM Tris, 100 mM NaCl, 5% nonfat dried milk, and 0.1% Tween-20) followed by incubations with rabbit polyclonal antisera directed against EAAT1 {A522 (Ab #314); 0.05 μg/mL}, EAAT2 {B493 (Ab #96); 0.2 μg/mL, and B563 (Ab #355); 0.05 μg/mL}, goat polyclonal antisera against α-internexin (0.4 μg/mL) or GFAP (0.2 μg/mL), or mouse polyclonal antisera directed against synaptophysin (0.2 μg/mL), CD68 (1:1,000) or β-actin (120,000). Details of preparation of rabbit polyclonal antisera to the C-terminal domains of EAAT1 and EAAT2 have previously been described (Beckstrøm et al., 1999; Lehre et al., 1995). Reblocking was followed by incubation with HRP-coupled anti-rabbit IgG (0.01 μig/mL) secondary antiserum. Each incubation step was of 1-h duration following which blots were washed several times with buffer (10 mM Tris, 100 mM NaCl, and 0.1% Tween-20). For the detection of specific antibody binding, the membranes were treated in accordance with the ECL-kit instructions and apposed to photosensitive X-OMAT film for 30–60 s. Signal intensities were subsequently measured by densitometry using a microcomputer-based image display system (Imaging Research, St. Catherines, ON, Canada). Linearity of the relationship between optical density and protein concentration was verified using appropriate standard curves. Blots were reversibly stained with Ponceau-S to assist in determination of uniformity of protein loading and to assess protein transfer efficiency. Negative controls comprised experiments in which transferred proteins were incubated with blocking buffer in which the primary antiserum was replaced with an appropriate amount of BSA protein.
Immunohistochemistry and Histology
Rats (all groups, n = 4) were deeply anesthetized with pentobarbital (60 mg/kg) and perfused transcardially as described previously (Hazell et al., 2001). Brains were removed and postfixed overnight in neutral-buffered formalin containing 4% formaldehyde, 0.5% sodium phosphate buffer, and 1.5% methanol, pH.7.0. Coronal sections (−3.8 to −5.8 mm relative to bregma) of 40-μm thickness were cut using a vibrotome according to the rat brain atlas of Paxinos and Watson (1998). Immunohistochemistry was performed according to Hazell et al. (2001). Frozen sections of human frontal cortex were thawed to room temperature and then fixed for 3 min in acetone. Briefly, both human and rat brain sections were then incubated for 10 min in phosphate-buffered saline (PBS) containing 0.3% hydrogen peroxide to block endogenous peroxidase activity. Tissue sections were washed in PBS (3 × 10 min), blocked for 20 min in 0.5% Triton X-100 and 5% donkey serum, and incubated with or without 0.5% Triton X-100, along with 5% donkey serum and primary rabbit antisera directed against EAAT1 {A522 (Ab #314); 0.25 μg/mL}, EAAT2 {B493 (Ab #96); 0.2 μg/mL, and B563 (Ab #355); 0.5 μg/mL}, goatderived antisera against α-internexin (1:250), the GABA transporter GAT-3 (1:250), GS (1:250), GFAP (1:250), synaptophysin (1:200), or mouse-derived antiserum against CD68 (1:250) at room temperature or 4°C for 1 or 24 h. Sections were then washed (3 × 10 min) and incubated in PBS with 0.5% Triton X-100 containing biotinylated donkey anti-rabbit/goat/mouse IgG (1:100). Sections were then incubated for 1 h in streptavidin-HRP conjugate (1:100) followed by washing (3 × 10 min), and then incubation with DAB (0.05%) in PBS containing in some cases 25 mg/mL nickel ammonium sulfate for signal enhancement and in the presence of H2O2 (0.03%) for 2–10 min. For immunofluorescence experiments, sections were incubated with Alexa Fluor 488 secondary antibodies (1:200). Sections were then mounted on Superfrost Plus slides (Fisher Scientific, Ottawa, ON, Canada), dehydrated in graded alcohols, cleared in xylene, and coverslipped with permount or, in the case of immunofluorescence studies, with Prolong Gold antifade reagent (Invitrogen). Negative controls consisted of omission of primary or secondary antibody, resulting in loss of immunoreactivity. Immunohistochemical assessment of NAC treatment was performed using a 0 (no improvement) to 3+ (marked improvement) grading system. Frozen sections of human frontal cortex (6-μm thickness) and vibratome-cut sections of rat brain were stained with cresyl violet for histological evaluation. DAPI staining was used for detection of cell nuclei. Neuronal cell numbers were counted using Image-Pro Plus (V6.2) software (Media Cybernetics, Bethesda, MD) in four adjacent boxes (0.06 mm2 each) at a magnification of 400× using an Olympus BX51 microscope and attached Spot RT digital camera.
Results
Figure 1 shows bands corresponding to the astrocyte glutamate transporters EAAT1 and EAAT2, the astrocytic cytoskeletal protein GFAP, the neurofilament protein α-internexin, and the nerve terminal protein marker synaptophysin from postmortem frontal cortex of control and WE subjects by immunoblotting. Tissue from WE patients showed a 62 and 71% decrease in EAAT1 and EAAT2 levels compared with controls (Fig. 1A,B). In addition, levels of the astrocytic protein GFAP were dramatically reduced by 70% in WE cases (Fig. 1C). Assessment of synaptophysin in WE revealed a 52% loss in content, while α-internexin levels were decreased by 67% compared with control cases (Fig. 1D,E). Histological assessment of cresyl violet-stained sections of frontal cortex from WE cases showed a decrease in neuronal cell numbers compared with control individuals (57.4% ± 8.4%, P < 0.01) (Fig. 2A–C), with increased numbers of glial cells present in WE brains consistent with a reactive gliosis. Assessment of EAAT1 (Fig. 3A,B) and EAAT2 (Fig. 3C,D) immunoreactivity revealed staining that was localized to astrocytes and their processes, with profound loss of this staining in the cortex of WE cases for both transporters. GFAP immunoreactivity was also considerably decreased in astrocytes, along with reduced synaptophysin and α-internexin immunostaining in WE patients relative to controls, consistent with the immunoblotting findings (not shown).
Fig. 1.
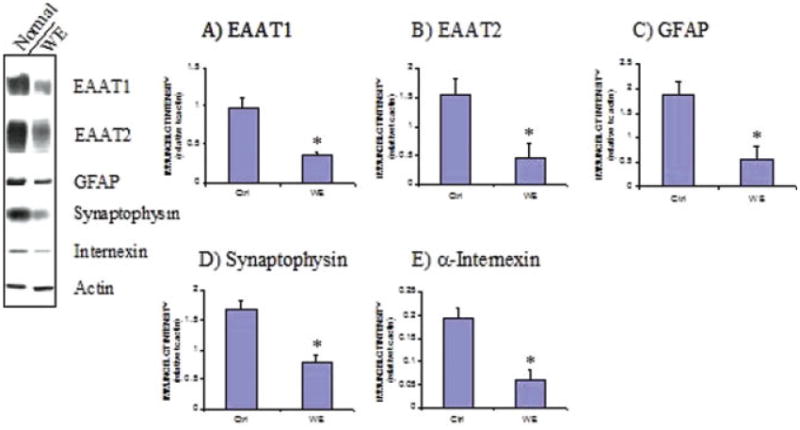
Levels of the astrocytic glutamate transporters EAAT1 and EAAT2 and other astrocytic and neuronal-specific proteins in WE. Representative immunoblots of EAAT1 and EAAT2 are displayed along with the astrocyte-specific cytoskeletal protein GFAP, the synaptic terminal protein synaptophysin, the intermediate neurofilament protein α-internexin, and β-actin in the frontal cortex of control and WE patients. Graphs A–E show densitometric results for EAAT1, EAAT2, GFAP, synaptophysin, and α-internexin proteins following normalization to β-actin. Levels of EAAT1 and EAAT2 along with the other three proteins were decreased in the frontal cortex of WE patients. *P < 0.01 compared with control group (Mann–Whitney U-test). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Fig. 2.
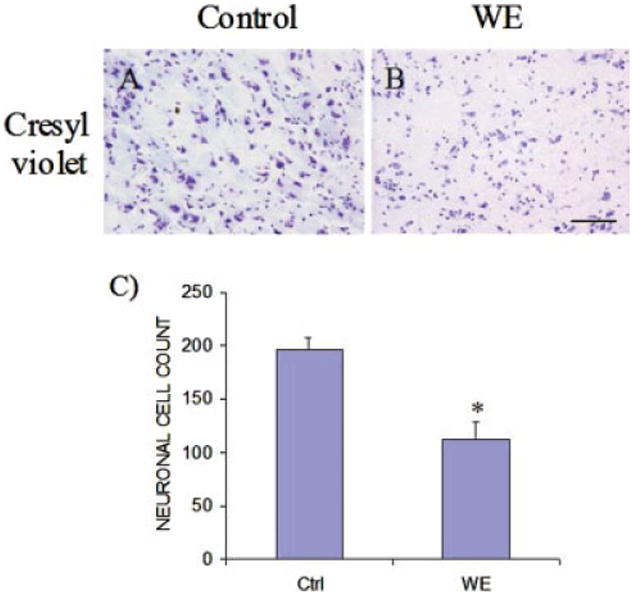
Photomicrographs of representative sections of frontal cortex in Wernicke's encephalopathy (WE). Cresyl violet staining revealed substantial neuronal loss in WE brain (B) compared with controls (A). Graph (C) shows neuronal cell counts for control (Ctrl) and WE samples. *P < 0.01 compared with control group (Mann–Whitney U-test). Bar, 125 lm (A,B). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Fig. 3.
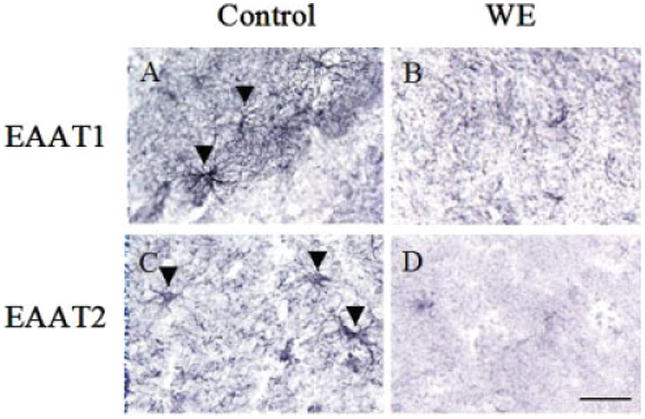
Photomicrographs of representative sections of frontal cortex in Wernicke's encephalopathy (WE). Immunohistochemical assessment revealed a loss of EAAT1 and EAAT2 immunoreactivity in WE brain compared with controls. Arrowheads indicate representative astrocytes staining for EAAT1 or EAAT2. Bar, 60 μm. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Assessment of GFAP immunoreactivity in the posterior medial thalamus of rats treated with TD revealed a profound loss of staining compared with control animals (Fig. 4A,B). EAAT1 and EAAT2 immunostaining were also decreased considerably in this brain region (Fig. 4C–F). GS and GAT-3 immunoreactivity also displayed a pattern indicative of considerable reduction in the levels of these proteins in the medial thalamus (Fig. 4G–J). DAPI staining of brain sections indicated viability of this area of the thalamus, with no evidence of pannecrosis, although neuronal cell numbers were reduced. In support of this, induction of CD68 was detected by immunoblotting and immunohistochemistry in this vulnerable brain region, indicative of microgliosis (see Fig. 5).
Fig. 4.
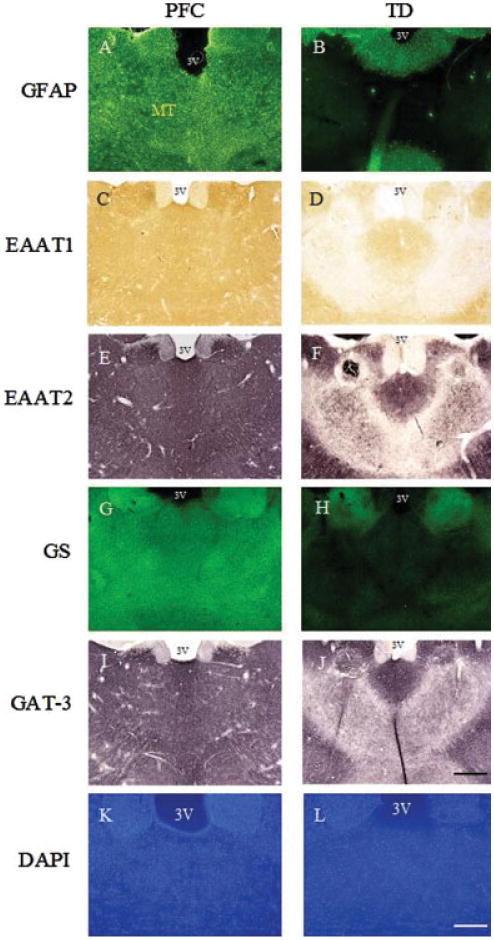
Photomicrographs of representative sections of brain from TD rats at the level of the posterior thalamus. Immunohistochemical assessment revealed a loss of GFAP, EAAT1, EAAT2, GS, and GAT-3 in the area of the medial thalamus of TD rats compared with pair-fed control (PFC) animals. Staining with DAPI indicated the presence of decreased cell nuclei but no pannecrosis in the medial thalamic region. MT, medial thalamus; 3V, third ventricle. Bars: A–J, 600 μm; K and L, 200 μm.
Fig. 5.
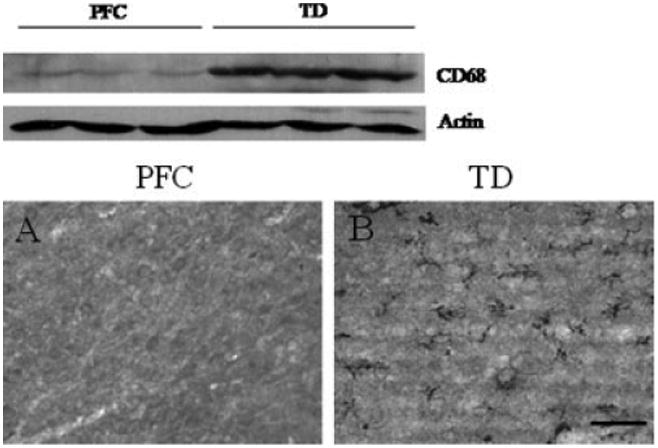
Microgliosis in the medial thalamus of pair-fed controls (PFC) and TD rats. Results show representative immunoblots of CD68 and β-actin at the level of the posterior medial thalamus, and photomicrographs of CD68 immunoreactivity in the same area of brain, in which TD rats (B) display a considerable induction of CD68 compared with PFC animals (A). Bar, 60 μm.
Examination of levels of EAAT2 in the medial thalamus of rats with TD revealed that levels of this transporter were decreased by 68% compared with pair-fed control animals, and which was associated with a 42.5% ± 5.1% loss of neuronal cell numbers (P < 0.01, Mann– Whitney U-test) (see Fig. 6). When similar groups of TD rats were co-treated with NAC, however, downregulation of EAAT2 was prevented in this part of the thalamus, concomitant with increased neuronal survival (see Fig. 6). Immunohistochemical staining for EAAT2 and GS showed marked improvement with NAC treatment (see Fig. 7), while EAAT1 and GAT-3 immunostaining were moderately improved. However, loss of GFAP immunoreactivity due to TD did not respond to NAC treatment (see Fig. 7).
Fig. 6.
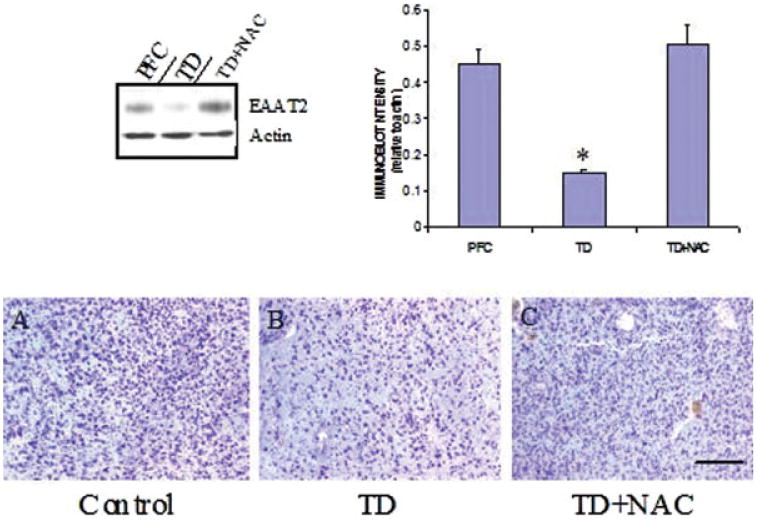
Comparison of EAAT2 levels in the medial thalamus in pair-fed controls (PFC), TD rats, and in TD rats co-treated with NAC. Results show representative blots of EAAT2 and β-actin along with quantitative analysis in which NAC prevented downregulation of the transporter protein. *P < 0.05 compared with PFC group (one-way ANOVA with posthoc Dunnett's test for multiple comparisons). Photomicrographs show representative cresyl violet staining of medial thalamus from a PFC (A), TD (B), and TD rat co-treated with NAC (C). Note neuronal loss in TD and prevention of this effect following NAC treatment. Bar, 300 lm. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Fig. 7.
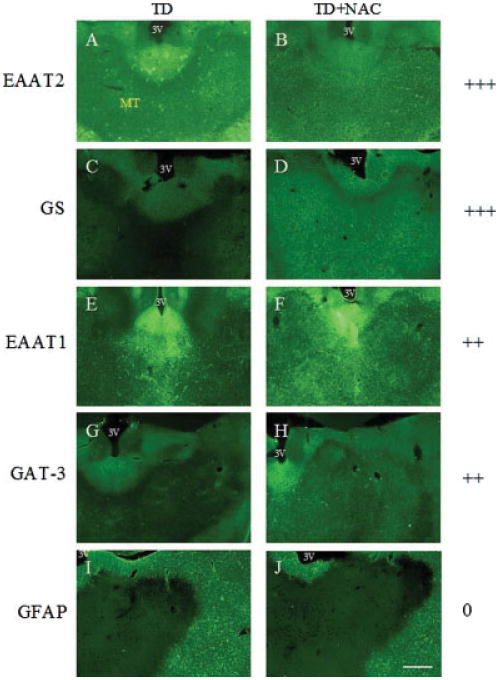
Photomicrographs of representative sections of brain from TD and TD + NAC treated rats at the level of the posterior thalamus. Panels show EAAT2 (A,B), GS (C,D), EAAT1 (E,F), GAT-3 (G,H), and GFAP (I,J) immunostaining in the medial thalamus of animals treated with TD only (A,C,E,G,I) and TD + NAC (B,D,F,H,J). Treatment with NAC prevented loss of EAAT2, GS, EAAT1, and GAT-3 immunoreactivity. No NAC-mediated protection was afforded against the loss of GFAP. MT, medial thalamus; 3V, third ventricle. Grading key: no protection (0), mild protection (+), moderate protection (++), marked protection (+++). Bar: 600 μm. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Discussion
Results of the present study demonstrate a downregulation of the astrocytic glutamate transporters EAAT1 and EAAT2 in the frontal cortex of patients with WE. In recent years, a growing body of evidence has suggested that the pathophysiology of TD involves a disturbance of glutamatergic neurotransmission. Previous reports indicate that loss of the astrocytic glutamate transporters EAAT1 and EAAT2 in the thalamus is a major feature of this disorder (Hazell et al., 2001). These findings are consistent with evidence of increased extracellular glutamate levels in this brain region (Hazell et al., 1993; Langlais and Zhang, 1993), and add credence to the possibility that glutamate-mediated excitotoxicity is an important feature of TD. Such reports are also consistent with earlier studies in which the appearance and development of the central thalamic lesion was found to be similar to that observed following intrathalamic administration of excitatory amino acids (Armstrong-James et al., 1988). In addition, treatment with the noncompetitive NMDA glutamate receptor antagonist MK-801 reduced the extent of cell death in brain regions affected in TD (Langlais and Mair, 1990), providing further evidence of an involvement of glutamate in structural damage in this disorder.
Although the cerebral cortex is normally considered to be less susceptible to damage in TD and WE, several studies have demonstrated that this area of the brain also sustains histological damage under these circumstances in which there is atrophy and white matter regions of the cortex are affected (Kril et al., 1997; Langlais and Zhang, 1997; Yamashita and Yamamoto, 1995). The cause of these lesions is unclear, but may involve an excitotoxic-related process similar to that in the thalamus. Indeed, recent studies have reported evidence for glutamate-mediated excitotoxicity involving white matter injury following stroke and spinal cord injury (Agrawal and Fehlings, 1997; Li and Stys, 2000; Tekkök et al., 2007).
Although the brains of alcoholic patients with WE show evidence of neuronal loss (Kril et al., 1997), studies indicate that TD in the absence of alcohol exposure decreases glutamate uptake in the prefrontal cortex (Carvalho et al., 2006), indicating that a deficit in glutamate transport function is a feature of the cerebral cortex under TD conditions. Thus, the neuronal loss in cases of WE with a history of alcoholism is likely to be due at least partly to the accompanying TD. Since TD results in a loss of EAAT1 and EAAT2 in vulnerable brain regions, it is probable that loss of these glutamate transporters in the frontal cortex of alcoholic patients with WE is also a major contributor to the neuronal cell death occurring in this brain region. Although the Carvalho study described a reduction in glutamate uptake in synaptosomes, recent evidence has been provided for the presence of EAAT2 in the plasma membranes of synaptic terminals (Furness et al., 2008), suggesting that loss of EAAT2 in synaptosomes in TD could at least partly account for their findings. Our results showing a loss of both EAAT1 and EAAT2 in alcoholics with WE suggest that the cortex also experiences a dysfunction of glutamatergic neurotransmission in these cases, possibly leading to increased extracellular glutamate concentration, similar to what is observed in the thalamus of the TD rat model.
Along with a downregulation of astrocytic glutamate transporters, histological assessment revealed the presence of neuronal loss in the frontal cortex of WE patients. We investigated this further using α-internexin and synaptophysin as supporting indices of neuronal damage and discovered that levels of both proteins were also decreased in this area of the brain. In addition, we have previously demonstrated that EAAT2 levels in the thalamus of TD rats are correlated with that of α-internexin (Hazell et al., 2001); thus, loss of EAAT2 may be a major contributing factor to the neuronal loss detected in the frontal cortex of these cases of WE. In contrast, it is possible that loss of EAAT2 may be a consequence of the neuronal injury process itself; such effects have been observed previously (Levy et al., 1995). It is also conceivable that the EAAT2 and neuronal cell loss may simply be correlated with no direct cause–effect relationship. Recently, we demonstrated that experimental WE results in a downregulation of complexin I and complexin II in the medial thalamus (Hazell and Wang, 2005). These two nerve terminal proteins, which are intimately associated with the synaptic vesicle release machinery of the cell and are specifically localized in inhibitory and excitatory synapses respectively (Harrison and Eastwood, 1998; Yamada et al., 1999), are thought to play an important role in the modulation of neurotransmitter release and the maintenance of normal synaptic function (Hu et al., 2002; Pabst et al., 2000). Of particular significance, downregulation of complexin II suggests that a dysregulation of the glutamate release process occurs in experimental WE, and may therefore be an important additional contributing factor to increased extracellular levels of glutamate in vulnerable brain regions.
Understanding the basis of the downregulation of astrocytic glutamate transporters is an important objective in studying the pathophysiology of WE. Recently, we reported a downregulation of EAAT1 protein in cultured astrocytes exposed to TD conditions in which phosphorylation of the transporter protein and Group II metabotropic glutamate receptors are contributing factors (Hazell et al., 2003). In the present study, we have examined the role of oxidative stress in the loss of EAAT2 and EAAT1 by co-treating rats with TD and the antioxidant NAC. Previous studies have demonstrated an increase in oxidative stress in the thalamus of TD rats (Calingasan et al., 1999; Langlais et al., 1997), and several reports have indicated an involvement of oxidative stress in glutamate transporter dysfunction (Allen et al., 2001; Volterra et al., 1994). Studies have also demonstrated that NAC can protect against ROS through restoration of intracellular glutathione (Juurlink and Paterson, 1998; Ratan et al., 1994), a naturally occurring antioxidant predominantly located in astrocytes. Thus, NAC may protect against neuronal death by improving antioxidant status of the tissue. Our present findings that NAC treatment markedly blocked downregulation of EAAT2 and moderately prevented loss of EAAT1, concomitant with increased neuronal survival in the thalamus of TD rats, thus indicate a likely role for oxidative stress in the loss of these glutamate transporters, and a contributing factor to the injury process in WE.
Such an involvement of oxidative stress could also underlie the basis of our findings in the TD rat of a reduction in levels of the other astrocyte-specific proteins GFAP and GS, along with GAT-3, which in the thalamus is localized predominantly in these glial cells (De Biasi et al., 1998). Although NAC afforded marked protection of GS against TD, antioxidant treatment resulted in only moderate protection of GAT-3 immunoreactivity. In contrast, loss of GFAP staining was unresponsive to NAC treatment. These findings suggest that impaired astrocyte function in TD likely has a multifactorial basis, in which other mechanisms in addition to oxidative stress (e.g., inflammation) may be involved. Indeed, previous reports have indicated that inflammatory responses are a feature of both WE (Schwenk et al., 1990) and TD (Karuppagounder et al., 2007; Todd and Butterworth, 1999), consistent with the results of the present study, in which induction of CD68 was observed, indicative of the presence of microgliosis in this damaged area of the thalamus. In addition, although the cause of this focal compromise in astrocyte integrity is unclear, it is possible that the impaired oxidative metabolism due to decreased activity of the thiamine-dependent enzyme α-ketoglutarate dehydrogenase may be a major contributor to this effect in the thalamus. If this turns out to be the case, it would suggest that the astrocyte is targeted metabolically in TD. Indeed, it has been known for decades that a major consequence of TD is the accumulation of lactic acid in brain (Holowach et al., 1968; Kinnersley and Peters, 1930), with the major source of cerebral lactate production being the astrocyte (Pellerin et al., 2007). In contrast, since neurons are net consumers of lactate (Lovatt et al., 2007), and focal accumulation of lactate is associated with areas of neuronal loss in TD, it is arguable that lactate accumulation may be due, at least in part, to decreased neuronal consumption. Since neuronal functional integrity (and survival) is dependent on the presence of astrocytes and efficient intercellular trafficking between these two cell types, it is conceivable that impaired astrocytic function could play a major role in the neuronal loss observed in vulnerable brain regions such as the thalamus in TD, and possibly WE.
In conclusion, our findings suggest that astrocytic glutamate transporter downregulation plays a role in damage to the frontal cortex in WE. As in TD, loss of these transporters may lead to elevated interstitial glutamate levels. These changes in glutamate transporter levels may be due to major astrocyte dysfunction, since levels of GFAP were also decreased. Loss of neurons in the frontal cortex was associated with decreased levels of α-internexin and synaptophysin, consistent with a diminished number of axons and synaptic terminals. Loss of EAAT2 transporter in the vulnerable medial thalamus of TD rats, a model of WE, was preventable by co-treatment with NAC, suggesting that oxidative stress may play an important role in the loss of astrocytic glutamate transporters in cases of WE. Together, these results support the concept of excitotoxic damage being a major contributor to the pathophysiology of WE, with oxidative stress having a major role to play in this process. On the basis of these findings, we recommend that antioxidant treatment be an important consideration in conjunction with thiamine administration in cases of acute WE, and particularly in cases of recurrent bouts of the disorder, in which the underlying damaging processes may be having a cumulative effect.
Acknowledgments
The authors are grateful to Dr. Niels C. Danbolt (University of Oslo) for kindly providing the EAAT1 and EAAT2 antibodies used in this investigation.
Grant sponsor: Canadian Institutes of Health Research; Grant number: MOP-84497.
References
- Agrawal SK, Fehlings MG. Role of NMDA and non-NMDA ionotropic glutamate receptors in traumatic spinal cord axonal injury. J Neurosci. 1997;17:1055–1063. doi: 10.1523/JNEUROSCI.17-03-01055.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JW, Mutkus LA, Aschner M. Methylmercury-mediated inhibition of 3H-D-aspartate transport in cultured astrocytes is reversed by the antioxidant catalase. Brain Res. 2001;902:92–100. doi: 10.1016/s0006-8993(01)02375-7. [DOI] [PubMed] [Google Scholar]
- Armstrong-James M, Ross DT, Chen F, Ebner FF. The effect of thiamine deficiency on the structure and physiology of the rat forebrain. Metab Brain Dis. 1988;3:91–124. doi: 10.1007/BF01001012. [DOI] [PubMed] [Google Scholar]
- Beckstrøm H, Julsrud L, Haugeto O, Dewar D, Graham DI, Lehre KP, Storm-Mathisen J, Danbolt NC. Interindividual differences in the levels of the glutamate transporters GLAST and GLT, but no clear correlation with Alzheimer's disease. J Neurosci Res. 1999;55:218–229. doi: 10.1002/(SICI)1097-4547(19990115)55:2<218::AID-JNR9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Calingasan NY, Chun WJ, Park LC, Uchida K, Gibson GE. Oxidative stress is associated with region-specific neuronal death during thiamine deficiency. J Neuropathol Exp Neurol. 1999;58:946–958. doi: 10.1097/00005072-199909000-00005. [DOI] [PubMed] [Google Scholar]
- Carvalho FM, Pereira SR, Pires RG, Ferraz VP, Romano-Silva MA, Oliveira-Silva IF, Ribeiro AM. Thiamine deficiency decreases glutamate uptake in the prefrontal cortex and impairs spatial memory performance in a water maze test. Pharmacol Biochem Behav. 2006;83:481–489. doi: 10.1016/j.pbb.2006.03.004. [DOI] [PubMed] [Google Scholar]
- De Biasi S, Vitellaro-Zuccarello L, Brecha NC. Immunoreactivity for the GABA transporter-1 and GABA transporter-3 is restricted to astrocytes in the rat thalamus. A light and electron-microscopic immunolocalization. Neuroscience. 1998;83:815–828. doi: 10.1016/s0306-4522(97)00414-4. [DOI] [PubMed] [Google Scholar]
- Drejer J, Larsson OM, Schousboe A. Characterization of uptake and release processes for D- and L-aspartate in primary cultures of astrocytes and cerebellar granule cells. Neurochem Res. 1983;8:231–243. doi: 10.1007/BF00963923. [DOI] [PubMed] [Google Scholar]
- Ebels EJ. How common is Wernicke-Korsakoff syndrome? Lancet. 1978;2:781–782. doi: 10.1016/s0140-6736(78)92663-6. [DOI] [PubMed] [Google Scholar]
- Furness DN, Dehnes Y, Akhtar AQ, Rossi DJ, Hamann M, Grutle NJ, Gundersen V, Holmseth S, Lehre KP, Ullensvang K, Wojewodzic M, Zhou Y, Attwell D, Danbolt NC. A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: New insights into a neuronal role for excitatory amino acid transporter 2. Neuroscience. 2008;157:80–94. doi: 10.1016/j.neuroscience.2008.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper C. Wernicke's encephalopathy: A more common disease than realised. A neuropathological study of 51 cases. J Neurol Neurosurg Psychiatry. 1979;42:226–231. doi: 10.1136/jnnp.42.3.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper C. The incidence of Wernicke's encephalopathy in Australia—A neuropathological study of 131 cases. J Neurol Neurosurg Psychiatry. 1983;46:593–598. doi: 10.1136/jnnp.46.7.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper CG, Giles M, Finlay-Jones R. Clinical signs in the Wernicke-Korsakoff complex: A retrospective analysis of 131 cases diagnosed at necropsy. J Neurol Neurosurg Psychiatry. 1986;49:341–345. doi: 10.1136/jnnp.49.4.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PJ, Eastwood SL. Preferential involvement of excitatory neurons in medial temporal lobe in schizophrenia. Lancet. 1998;352:1669–1673. doi: 10.1016/S0140-6736(98)03341-8. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Butterworth RF, Hakim AM. Cerebral vulnerability is associated with selective increase in extracellular glutamate concentration in experimental thiamine deficiency. J Neurochem. 1993;61:1155–1158. doi: 10.1111/j.1471-4159.1993.tb03635.x. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Hakim AM, Senterman MK, Hogan MJ. Regional activation of L-type voltage-sensitive calcium channels in experimental thiamine deficiency. J Neurosci Res. 1998;52:742–749. doi: 10.1002/(SICI)1097-4547(19980615)52:6<742::AID-JNR13>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Pannunzio P, Rama Rao KV, Pow DV, Rambaldi A. Thiamine deficiency results in downregulation of the GLAST glutamate transporter in cultured astrocytes. Glia. 2003;43:175–184. doi: 10.1002/glia.10241. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Rama Rao KV, Danbolt NC, Pow DV, Butterworth RF. Selective down-regulation of the astrocyte glutamate transporters GLT-1 and GLAST within the medial thalamus in experimental Wernicke's encephalopathy. J Neurochem. 2001;78:560–568. doi: 10.1046/j.1471-4159.2001.00436.x. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Wang C. Downregulation of complexin I, complexin II in the medial thalamus is blocked by N-acetylcysteine in experimental Wernicke's encephalopathy. J Neurosci Res. 2005;79:200–207. doi: 10.1002/jnr.20278. [DOI] [PubMed] [Google Scholar]
- Holowach J, Kauffman F, Ikossi MG, Thomas C, McDougal DB., Jr The effects of a thiamine antagonist, pyrithiamine, on levels of selected metabolic intermediates and on activities of thiamine-dependent enzymes in brain and liver. J Neurochem. 1968;15:621–631. doi: 10.1111/j.1471-4159.1968.tb08961.x. [DOI] [PubMed] [Google Scholar]
- Hu K, Carroll J, Rickman C, Davletov B. Action of complexin on SNARE complex. J Biol Chem. 2002;277:41652–41656. doi: 10.1074/jbc.M205044200. [DOI] [PubMed] [Google Scholar]
- Juurlink BH, Paterson PG. Review of oxidative stress in brain and spinal cord injury: Suggestions for pharmacological and nutritional management strategies. J Spinal Cord Med. 1998;21:309–334. doi: 10.1080/10790268.1998.11719540. [DOI] [PubMed] [Google Scholar]
- Karuppagounder SS, Shi Q, Xu H, Gibson GE. Changes in inflammatory processes associated with selective vulnerability following mild impairment of oxidative metabolism. Neurobiol Dis. 2007;26:353–362. doi: 10.1016/j.nbd.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnersley HW, Peters RA. Brain localization of lactic acidosis in avitaminosis B1 and its relation to the origin of symptoms. Biochem J. 1930;24:711–722. doi: 10.1042/bj0240711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knecht K, Michalak A, Rose C, Rothstein JD, Butterworth RF. Decreased glutamate transporter (GLT-1) expression in frontal cortex of rats with acute liver failure. Neurosci Lett. 1997;229:201–203. doi: 10.1016/s0304-3940(97)00444-8. [DOI] [PubMed] [Google Scholar]
- Kril JJ, Halliday GM, Svoboda MD, Cartwright H. The cerebral cortex is damaged in chronic alcoholics. Neuroscience. 1997;79:983–998. doi: 10.1016/s0306-4522(97)00083-3. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Anderson G, Guo SX, Bondy SC. Increased cerebral free radical production during thiamine deficiency. Metab Brain Dis. 1997;12:137–143. [PubMed] [Google Scholar]
- Langlais PJ, Mair RG. Protective effects of the glutamate antagonist MK-801 on pyrithiamine-induced lesions and amino acid changes in rat brain. J Neurosci. 1990;10:1664–1674. doi: 10.1523/JNEUROSCI.10-05-01664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais PJ, Zhang SX. Extracellular glutamate is increased in thalamus during thiamine deficiency-induced lesions and is blocked by MK-801. J Neurochem. 1993;61:2175–2182. doi: 10.1111/j.1471-4159.1993.tb07457.x. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Zhang SX. Cortical and subcortical white matter damage without Wernicke's encephalopathy after recovery from thiamine deficiency in the rat. Alcohol Clin Exp Res. 1997;21:434–443. doi: 10.1111/j.1530-0277.1997.tb03788.x. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glutamate transporters in rat brain; quantitative and immunocytochemical observations. J Neurosci. 1995;15:1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy LM, Lehre KP, Walaas SI, Storm-Mathisen J, Danbolt NC. Down -regulation of glial glutamate transporters after glutamatergic denervation in the rat brain. Eur J Neurosci. 1995;7:2036–2041. doi: 10.1111/j.1460-9568.1995.tb00626.x. [DOI] [PubMed] [Google Scholar]
- Li S, Stys PK. Mechanisms of ionotropic glutamate receptor-mediated excitotoxicity in isolated spinal cord white matter. J Neurosci. 2000;20:1190–1198. doi: 10.1523/JNEUROSCI.20-03-01190.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, Clawson L, Rothstein JD. Aberrant RNA processing in a neurodegenerative disease: The cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron. 1998;20:589–602. doi: 10.1016/s0896-6273(00)80997-6. [DOI] [PubMed] [Google Scholar]
- Lindboe CF, Loberg EM. Wernicke's encephalopathy in non-alcoholics. An autopsy study. J Neurol Sci. 1989;90:125–129. doi: 10.1016/0022-510x(89)90095-6. [DOI] [PubMed] [Google Scholar]
- Lovatt D, Sonnewald U, Waagepetersen HS, Schousboe A, He W, Lin JH, Han X, Takano T, Wang S, Sim FJ, Goldman SA, Nedergaard M. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J Neurosci. 2007;27:12255–12266. doi: 10.1523/JNEUROSCI.3404-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:263–275. [PubMed] [Google Scholar]
- Martin LJ, Brambrink AM, Lehmann C, Portera-Cailliau C, Koehler R, Rothstein J, Traystman RJ. Hypoxia-ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann Neurol. 1997;42:335–348. doi: 10.1002/ana.410420310. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Mendoza D, Lozada A, Pretorius JK, Dehnes Y, Danbolt NC, Nelson N, Leite JP, Chimelli L, Born DE, Sakamoto AC, Assirati JA, Fried I, Peacock WJ, Ojemann GA, Adelson PD. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology. 1999;52:453–472. doi: 10.1212/wnl.52.3.453. [DOI] [PubMed] [Google Scholar]
- Miyajima Y, Fukuda M, Kojima S, Matsuyama T, Shylaja N, Aso K. Wernicke's encephalopathy in a child with acute lymphoblastic leukemia. Am J Pediatr Hematol Oncol. 1993;15:331–334. [PubMed] [Google Scholar]
- Nicholls D, Attwell D. The release and uptake of excitatory amino acids. Trends Pharmacol Sci. 1990;11:462–468. doi: 10.1016/0165-6147(90)90129-v. [DOI] [PubMed] [Google Scholar]
- Ohkoshi N, Ishii A, Shoji S. Wernicke's encephalopathy induced by hyperemesis gravidarum, associated with bilateral caudate lesions on computed tomography and magnetic resonance imaging. Eur Neurol. 1994;34:177–180. doi: 10.1159/000117034. [DOI] [PubMed] [Google Scholar]
- Pabst S, Hazzard JW, Antonin W, Sudhof TC, Jahn R, Rizo J, Fasshauer D. Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J Biol Chem. 2000;275:19808–19818. doi: 10.1074/jbc.M002571200. [DOI] [PubMed] [Google Scholar]
- Parkin AJ, Dunn JC, Lee C, O'Hara PF, Nussbaum L. Neuropsy-chological sequelae of Wernicke's encephalopathy in a 20-year-old woman: Selective impairment of a frontal memory system. Brain Cogn. 1993;21:1–19. doi: 10.1006/brcg.1993.1001. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic Press; 1998. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. Activity-dependent regulation of energy metabolism by astrocytes: An update. Glia. 2007;55:1251–1262. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MB. The family of sodium-dependent glutamate transporters: A focus on the GLT-1/EAAT2 subtype. Neurochem Int. 1998;33:479–491. doi: 10.1016/s0197-0186(98)00055-2. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Schwenk J, Gosztonyi G, Thierauf P, Iglesias J, Langer E. Wernicke's encephalopathy in two patients with acquired immunodeficiency syndrome. J Neurol. 1990;237:445–447. doi: 10.1007/BF00314738. [DOI] [PubMed] [Google Scholar]
- Shah N, Wolff JA. Thiamine deficiency: Probable Wernicke's encephalopathy successfully treated in a child with acute lymphocytic leukemia. Pediatrics. 1973;51:750–751. [PubMed] [Google Scholar]
- Soffer D, Zirkin H, Alkan M, Berginer VM. Wernicke's encephalopathy in acquired immune deficiency syndrome (AIDS): A case report. Clin Neuropathol. 1989;8:192–194. [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tekkök SB, Ye Z, Ransom BR. Excitotoxic mechanisms of ischemic injury in myelinated white matter. J Cereb Blood Flow Metab. 2007;27:1540–1552. doi: 10.1038/sj.jcbfm.9600455. [DOI] [PubMed] [Google Scholar]
- Todd KG, Butterworth RF. Early microglial response in experimental thiamine deficiency: An immunohistochemical analysis. Glia. 1999;25:190–198. doi: 10.1002/(sici)1098-1136(19990115)25:2<190::aid-glia9>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Torp R, Lekieffre D, Levy LM, Haug FM, Danbolt NC, Meldrum BS, Ottersen OP. Reduced postischemic expression of a glial glutamate transporter, GLT1, in the rat hippocampus. Exp Brain Res. 1995;103:51–58. doi: 10.1007/BF00241964. [DOI] [PubMed] [Google Scholar]
- Volterra A, Trotti D, Tromba C, Floridi S, Racagni G. Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J Neurosci. 1994;14:2924–2932. doi: 10.1523/JNEUROSCI.14-05-02924.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernicke C. Lehrbuch der Gehirnkrankheiten fur Aerzte und Studirende. Vol. 2. Kassel: Theodor Fischer; 1881. pp. 229–242. [Google Scholar]
- Yamada M, Saisu H, Ishizuka T, Takahashi H, Abe T. Immunohistochemical distribution of the two isoforms of synaphin/complexin involved in neurotransmitter release: Localization at the distinct central nervous system regions and synaptic types. Neuroscience. 1999;93:7–18. doi: 10.1016/s0306-4522(99)00104-9. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Yamamoto T. Wernicke encephalopathy with symmetric pericentral involvement: MR findings. J Comput Assist Tomogr. 1995;19:306–308. doi: 10.1097/00004728-199503000-00026. [DOI] [PubMed] [Google Scholar]