

Abstract
All-trans retinoic acid, controlled by cytochrome P450, family 26 (CYP26) enzymes, potentially has beneficial effects in atherosclerosis treatment. This study investigates CYP26 subfamily B, polypeptide 1 (CYP26B1) in atherosclerosis and the effects of a genetic polymorphism in CYP26B1 on retinoid catabolism. We found that CYP26B1 mRNA was induced by retinoic acid in human atherosclerotic arteries, and CYP26B1 and the macrophage marker CD68 were colocalized in human atherosclerotic lesions. In mice, Cyp26B1 mRNA was higher in atherosclerotic arteries than in normal arteries. Databases were queried for nonsynonymous CYP26B1 single nucleotide polymorphisms (SNPs) and rs2241057 selected for further studies. Constructs of the CYP26B1 variants were created and used for production of purified proteins and transfection of macrophagelike cells. The minor variant catabolized retinoic acid with significantly higher efficiency, indicating that rs2241057 is functional and suggesting reduced retinoid availability in tissues with the minor variant. rs2241057 was investigated in a Stockholm Coronary Atherosclerosis Risk Factor (SCARF) subgroup. The minor allele was associated with slightly larger lesions, as determined by angiography. In summary, this study identifies the first CYP26B1 polymorphism that alters CYP26B1 capacity to metabolize retinoic acid. CYP26B1 was expressed in macrophage-rich areas of human atherosclerotic lesions, induced by retinoic acid and increased in murine atherosclerosis. Taken together, the results indicate that CYP26B1 capacity is genetically regulated and suggest that local CYP26B1 activity may influence atherosclerosis.
INTRODUCTION
Atherosclerosis is a chronic inflammatory disease of blood vessels (1). Retinoic acid has powerful biological effects that may treat and prevent atherosclerosis. For example, activation of retinoic acid receptors (RARs) reduces inflammation, vascular cell proliferation and migration, apoptosis, coagulation and matrix remodeling (2–5), and retinoic acid upregulates a set of antiatherogenic genes in macrophages (6). Furthermore, retinoic acid promotes differentiation of regulatory T cells, an immune cell subset that ameliorates inflammation and atherosclerosis (1,7,8). RARs and retinoic receptor ligands are present in atherosclerotic lesions, and retinoic acid is known to regulate macrophage expression of scavenger receptors (9). In line with this, low plasma retinol, the substrate for the active all-trans retinoic acid (atRA), is an independent risk factor for coronary events (10,11). Administration of retinoids also reduces post–balloon injury stenosis in rats (4,12,13) and ameliorates disease in atherosclerosis-prone mice (14).
Increased retinoid levels reduce experimental atherosclerosis, but long-term systemic treatment with retinoids is associated with serious adverse effects (15). Therapeutic targeting of local retinoid turnover to increase local retinoid levels is an alternative strategy (2), and inhibitors of cytochrome P450, family 26 (CYP26) enzymes have been used in clinical studies (16,17). However, knowledge about the metabolism of atRA and other RAR ligands in atherosclerosis has been lacking. In general, synthesis of active retinoids by retinol and retinal dehydrogenases and catabolism by members of the CYP26 subfamily A, B and C, polypeptide 1 (CYP26A1, CYP26B1 and CYP26C1) is tightly controlled (18–21). CYP26A1 was the first member of the CYP26 family to be identified, characterized and cloned (22,23). CYP26B1 has 41% amino acid identity with CYP26A1 but similar functional activity (24,25). In subsets of vascular and immune cells, interference with CYP26 has profound effects on atRA levels (26–28), and increased levels of endogenous atRA result in induction of a number of retinoid-responsive genes in vascular cells (27). To date, little is known about the significance of genetic polymorphisms that occur in the CYP26 enzymes. A CYP26A1 variant reportedly has a significantly reduced activity compared with wild-type (29), but single nucleotide polymorphisms (SNPs) in the CYP26A1 gene have not been linked to disease. The rs707718 polymorphism in CYP26B1 has been associated with oral squamous cell carcinoma in a subgroup of patients (30), but the mechanism behind the link is unclear. It has been unknown if genetic polymorphisms in CYP26B1 influence enzyme efficiency. We reasoned that if CYP26 enzymes would be expressed in atherosclerosis, local influence by CYP26 enzymes might affect retinoid availability, inflammation and disease development. In this study, we investigated CYP26 in atherosclerosis and discovered a CYP26B1 polymorphism that alters the rate of retinoid catabolism.
MATERIALS AND METHODS
These studies were approved by the regional ethical committees for human or animal studies, and human subjects were included after informed consent.
Human Biopsies
Fifteen patients scheduled for carotid endarterectomy were included (31). Nine atherosclerotic lesions were snap-frozen. Six lesions were divided in two and incubated in Dulbecco’s modified Eagle’s medium/F12 medium enriched with 30 mg/mL human albumin (Biovitrum AB, Stockholm, Sweden) for 6 h with 1 μmol/L atRA (Sigma-Aldrich, St. Louis, MO, USA) or vehicle. Ribonucleic acid (RNA) was isolated using the E.Z.N.A. Total RNA kit (Omega Bio-Tek, Doraville, GA, USA). Semiquantitative polymerase chain reaction (PCR) was performed in a 7900HT Fast Real-Time PCR System using Assays-on-Demand™ reagents (Applied Biosystems, Foster City, CA, USA) for human CYP26B1. β-Actin was used as a reference (28). Formalin fixed 10-μm cryosections or arterial biopsies were incubated with mouse anti-CYP26B1 (Abnova, Thaipei City, Taiwan), anti-CD68 or anti-α-actin (Dakopatts, Glostrup, Denmark) monoclonal antibodies or a relevant isotype control overnight at 4°C after treatment with 0.3% H2O2 (Sigma-Aldrich). Sections were rinsed, incubated with Imm-Press (Vector, Burlingame, CA, USA) for 30 min, followed by Nova Red substrate (Vector), and counterstained with Hematoxylin QS (Vector). Samples from the Biobank of Karolinska Carotid Endarterectomies (BiKE) were obtained and analyzed using global transcript analysis with Affymetrix® gene arrays as previously described (31,32).
Murine Biopsies
Female Apoe−/− mice (n = 11, local breeding) and C57BL/6 mice (n = 6, Taconic, Ry, Denmark) were used, fed standard mouse chow and euthanized at 18 wks of age. RNA was extracted from atherosclerotic aortas from Apoe−/− mice and atherosclerosis-free aortas from C57BL/6 mice and analyzed as previously described (31). Premanufactured primers and probe sets (Assays-on-Demand) for Cyp26B1 and TATA box binding protein (TATAbbp) were used for quantitative PCR (qPCR). Relative values were calculated with the 2−ΔΔCT method.
Genotyping and Angiography
The National Center for Biotechnology Information (NCBI) Entrez SNP database (http://www.ncbi.nlm.nih.gov/snp) was used to identify SNPs, and prevalence was determined in a cohort of Swedish individuals (33). Genotyping was performed using a TaqMan SNP assay (34). The Stockholm Coronary Atherosclerosis Risk Factor (SCARF) study has been described previously (35) and includes 387 survivors of a first myocardial infarction and 387 controls. Of the 387 patients, 243 underwent quantitative coronary angiography. The QCA-CMS system (Medis Medical Imaging Systems, Leiden, the Netherlands) was used to outline the vessel lumen (36) and measure the area of atherosclerotic lesions. The inner surface of each segment of the vessel wall was longitudinally outlined, and the plaque area was calculated. To compensate for the individual variation in segment length, plaque area was correlated to the actual segment length (mm2/mm).
Construction of Expression Vectors for CYP26B1
Constructs of full-length CYP26B1 with the major (wild-type) variant of rs2241057 was commercially obtained as an Ultimate ORF Clone ID IOH37861 (Invitrogen, Stockholm, Sweden). The minor (mutated) variant of rs2241057 was obtained from the major variant by site-directed mutagenesis using mutagenic primers (forward: 5′ ACACA GGGCA AGGAC TACTC GGACG CCCTG GAC 3′; reverse: 5′ AGTAG TCCTT GCCCT GTGTG CACTG CAGCT TC 3′) and the GeneTailor Site-Directed Mutagenesis System (Invitrogen). Subsequently, the entire plasmid was sequenced in both directions. The rs2241057 major and minor constructs were cloned into a pcDNA3.2/V5-DEST (Invitrogen) vector using Gateway Technology (Invitrogen). Finally, the sequence was verified (37).
Transient Transfection of COS-1 and THP-1 Cells with CYP26B1 Variants and Measurement of atRA Degradation
COS-1 cells were cultured without antibiotics for 24 h. Cells were transfected with 2 μg of the major or minor CYP26B1 construct or pcDNA3.2/V5-DEST vector without insert and 100 ng pSEAP2 (Invitrogen) control vector using Lipofectamin 2000 (Invitrogen). Transfected cells were incubated for 24 h, medium was changed and 2 μCi [3H]atRA (PerkinElmer Life Sciences, Waltham, MA, USA) was added to each well. After 1 h incubation at 37°C, cells were washed and frozen. Sample contents were separated by reverse-phase high-performance liquid chromatography (HPLC) and [3H]atRA detected as previously described (38). Tritium activity was normalized to protein levels and secreted alkaline phosphatase (SEAP) activity using a reporter gene assay chemiluminescent kit (Roche Applied Science, Indianapolis, IN, USA). Experiments were performed in triplicate and repeated four times. THP-1 cells were treated with 20 nmol/L phorbol myristate acetate for 24 h to differentiate the cells to macro phages. The next day, cells were transfected with 0.9 μg major or minor CYP26B1 construct or pcDNA3.2/V5-DEST vector without an insert in combination with 100 ng pSEAP2 (Invitrogen) control vector using 2 μL TurboFect (Fermentas, Leon-Rot, Germany). Transfected cells were incubated for 24 h, medium was changed and 1 μCi [3H]atRA was added per well. Cells were incubated for 1 h followed by HPLC analysis. Tritium activity was normalized as described above. Experiments were performed in duplicate and repeated three times.
Purification of CYP26B1 Variants, Measurement of CYP26B1 Activity and Western Blot
CYP26B1 major and minor variants were purified from transfected COS-1 cells using monoclonal antibodies (Abnova) and the Pierce IP Kit (Fisher Scientific, Västra Frölunda, Sweden) and stored in buffer (50 mmol/L KH2PO4, 50 mmol/L K2HPO4, 0.5 mmol/L EDTA [ethylenediaminetetraacetic acid] and 20% of glycerol, pH 7.4) at −80°C. Zero or 5 nmol/L of either major or minor CYP26B1, 10 nmol/L oxidoreductase (Electra-Box Diagnostica, Stockholm, Sweden), 25 nmol/L [3H]atRA and 1 mmol/L reduced nicotinamide adenine dinucleotide (NADPH) in buffer (100 mmol/L KH2PO4 and 100 mmol/L K2HPO4, pH 7.4) were incubated at 37°C for 1–15 min, and the reaction was stopped by the addition of ethanol. Major or minor CYP26B1 without NADPH was used as the control. atRA levels were measured by HPLC (38). The purified protein or total cell lysate from COS-1 cells transfected with CYP26B1 constructs or empty vector were analyzed by Western blot using mouse anti-CYP26B1 monoclonal antibodies (Abnova).
Statistics
SPSS 14.0 (SPSS, Chicago, IL, USA) and Statistica 7.0 (StatSoft, Tulsa, OK, USA) were used for analyses. Comparisons of mRNA levels and of HPLC data, respectively, were performed using a Student t test. Factorial analysis of variance followed by a Tukey honest significance post hoc test was used to analyze enzyme efficiencies. Differences in atherosclerotic lesion size and basic characteristics between genotypes were evaluated by the Mann-Whitney U test. P ≤ 0.05 was considered significant.
All supplementary materials are available online at www.molmed.org.
RESULTS
CYP26B1 mRNA Was Induced by Retinoic Acid in Atherosclerotic Biopsies
Aortas from atherosclerosis-free C57BL/6 mice and atherosclerotic Apoe-deficient mice were analyzed for Cyp26B1 by qPCR. Levels of Cyp26B1 mRNA were significantly higher in atherosclerotic arteries (P = 0.002) (Figure 1A). In line with this, levels of CYP26B1 mRNA were significantly higher in human atherosclerotic carotid endarterectomy samples compared with atherosclerosis-free iliac arteries in BiKE (n = 6 iliac and 107 carotid artery biopsies, P < 0.0001) (Supplementary Figure S1). In vitro treatment of human atherosclerotic tissue with 1 μmol/L atRA for 6 h increased CYP26B1 mRNA levels severalfold (P = 0.02) (Figure 1B).
Figure 1.

CYP26B1 mRNA in atherosclerotic lesions. Murine arterial biopsies from C57Bl/6 (n = 6) and Apoe-deficient (n = 11) mice were analyzed by qPCR for Cyp26B1 mRNA (A). CYP26B1 mRNA levels were measured in human carotid artery biopsies cultured in the absence or presence of atRA (n = 6) (B). Values are mean ± standard error of the mean (SEM) of arbitrary units normalized to TATAbbp (mouse) or β-actin (human). **P < 0.01, *P < 0.05.
CYP26B1 Colocalized with Macrophages in Human Atherosclerotic Lesions
Using immunohistochemistry, CYP26B1 was detected in human atherosclerotic lesions (Figure 2A). To investigate the cellular source of CYP26B1 in atherosclerosis, arterial biopsies were stained with markers for smooth muscle cells (α-actin) and macrophages (CD68). On consecutive serial sections, CYP26B1 and CD68 appeared in the same areas in the vessel wall (Figures 2A, B), whereas there was no apparent colocalization of CYP26B1 and markers for other cell types (data not shown).
Figure 2.

In atherosclerotic lesions, CYP26B1 protein was found in macrophage-rich areas. CYP26B1 protein was detected in human atherosclerotic lesions (A). On consecutive serial sections, CYP26B1 and the macrophage marker CD68 stained similar areas (B). The IgG2a isotype control did not produce any detectable binding (C).
Out of Three Nonsynonymous CYP26B1 SNPs, the Minor Allele Was Detected Only in rs2241057
The (NCBI) Entrez SNP database was searched for nonsynonymous SNPs in CYP26B1. Three described variants were identified: rs2241057, rs2286965 and rs7568553. The suitability of these variants for further studies was probed by approximating the prevalence of the variants in a Swedish cohort (33). The minor allele was detected only in rs2241057, with a C-allele frequency of 0.137, which is similar in the HapMap CEU population (frequency 0.168, ss48402704, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ss.cgi?subsnp_id=48402704). Therefore, this variant, known to cause a Leu-to-Ser substitution at position 264 in CYP26B1, was selected for subsequent studies.
The Minor CYP26B1 Variant Catabolized atRA at a Higher Rate
To investigate the biological effects of the Leu-to-Ser substitution in CYP26B1 associated with the minor allele of rs2241057, plasmid constructs of the human CYP26B1 gene variants were created and used for production of purified proteins and transfection of the genetic variants to cell cultures. First, to examine the CYP26B1 variants’ effect on atRA metabolism exclusively, the purified minor and major enzyme variants were added to a cell-free system consisting of a reaction buffer at physiological pH with a known starting concentration of [3H]atRA. [3H]atRA levels were followed over time using HPLC. The minor CYP26B1 variant catabolized atRA at a significantly higher rate than the major variant (Figure 3). Then, to explore if this difference in catabolic rate was significant in actual cell cultures, COS-1 cells were transfected with constructs of CYP26B1 with the major or minor allele of rs2241057 or empty vector. Transfection produced similar cellular levels of the CYP26B1 major and minor protein variants (Figure 4A). After 1 h incubation with [3H]atRA, cellular levels of [3H]atRA as measured by HPLC were significantly lower in cells with mutated CYP26B1 (P = 0.004), supporting a higher enzymatic efficiency of the minor CYP26B1 variant (Figure 4B) and indicating that the rs2241057 polymorphism is functional. Because CYP26B1 appeared in macrophage-rich areas in atherosclerosis, we proceeded to explore CYP26B1 activity in human macrophagelike differentiated THP-1 cells. THP-1 cells were transfected with the different plasmids and [3H]atRA administered and measured as above. Levels of [3H]atRA were lower in cultures transfected with the minor CYP26B1 variant (Figure 4C).
Figure 3.
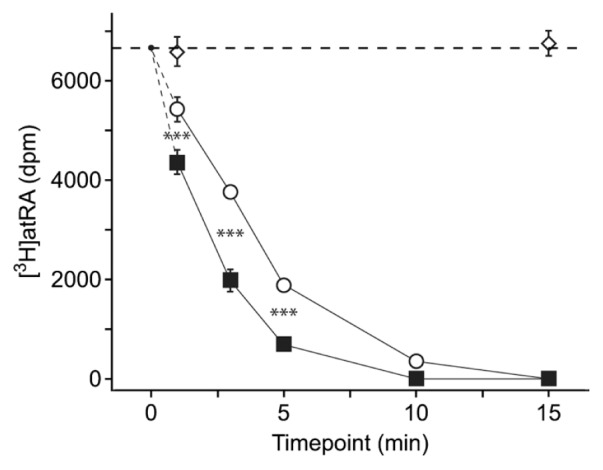
Increased catabolic efficiency of the minor CYP26B1 variant. Purified minor (open circles) and major (filled squares) rs2241057 CYP26B1 variants were introduced in a cell-free system. [3H]atRA was added and its concentration was measured over time using HPLC. Open diamonds show levels in the absence of CYP26B1. The horizontal dashed line indicates baseline concentration. Values are mean ± SEM. dpm, disintegrations per minute. ***P < 0.001.
Figure 4.

Increased efficiency of cellular retinoic acid catabolism by the minor CYP26B1 variant. Plasmid constructs containing the major or minor variant of rs2241057 were transfected into COS-1 cells and THP-1 macrophagelike cells, and levels of the 55-kDa CYP26B1 were determined by Western blot in COS-1 cells (A). Cellular levels of supplemented [3H]atRA were measured by HPLC in COS-1 cells (B) and THP-1 cells (C). Values are relative to protein concentration and SEAP activity, and shown as mean ± SD. **P < 0.01, ***P < 0.001.
The difference in catabolic rate between the CYP26B1 variants suggested that the atRA equilibrium concentration would be reduced in tissues with the minor CYP26B1 enzyme variant. Association of the CYP26B1 genotype with atherosclerosis was, therefore, investigated in the SCARF cohort. There was no significant association between rs2241057 genotype and basic clinical characteristics in the 387 patients and 387 matched controls (Table 1). Association of the rs2241057 polymorphism and local angiography findings on atherosclerotic lesions was investigated in the subgroup of 233 patients with complete CYP26B1 genotyping and angiography results. Carriers of the minor allele had a small increase in atherosclerotic lesions detected by angiography compared with individuals homozygous for the major allele (P = 0.01) (Table 2).
Table 1.
Basic clinical characteristics of rs2241057 genotypes in the SCARF cohort.
| Homozygous for the major allele | Carriers of the minor allele | ||||
|---|---|---|---|---|---|
|
|
|
||||
| n | n | P | |||
| Age (years) | |||||
| Patients | 282 | 53.0 (49.0–57.0) | 90 | 54.0 (48.8–57.0) | 0.45 |
| Controls | 282 | 54.0 (49.8–57.0) | 99 | 54.0 (49.0–57.0) | 0.35 |
| Body mass index (kg/m2) | |||||
| Patients | 281 | 26.8 (24.7–29.6) | 90 | 26.6 (24.3–29.5) | 0.70 |
| Controls | 282 | 25.6 (23.5–27.8) | 98 | 25.7 (24.1–27.5) | 0.79 |
| Current smokers (%) | |||||
| Patients | 282 | 50.7 | 90 | 51.1 | 0.52 |
| Controls | 280 | 24.3 | 98 | 24.5 | 0.53 |
| Serum cholesterol (mmol/L) | |||||
| Patients | 280 | 5.0 (4.2–5.7) | 90 | 5.2 (4.5–5.7) | 0.12 |
| Controls | 282 | 5.4 (4.8–6.1) | 99 | 5.5 (4.7–6.2) | 0.50 |
| Serum low-density lipoprotein (mmol/L) | |||||
| Patients | 272 | 3.2 (2.5–3.9) | 84 | 3.2 (2.6–3.9) | 0.33 |
| Controls | 281 | 3.4 (2.9–4.1) | 98 | 3.7 (3.0–4.3) | 0.18 |
| Serum high-density lipoprotein (mmol/L) | |||||
| Patients | 272 | 1.1 (0.9–1.4) | 84 | 1.1 (0.9–1.4) | 0.36 |
| Controls | 281 | 1.3 (1.1–1.7) | 98 | 1.4 (1.1–1.6) | 0.58 |
| Serum triglycerides (mmol/L) | |||||
| Patients | 280 | 1.6 (1.2–2.2) | 90 | 1.6 (1.2–2.3) | 0.88 |
| Controls | 282 | 1.2 (0.86–1.6) | 99 | 1.2 (0.82–1.5) | 0.86 |
| Blood CRP (mg/L) | |||||
| Patients | 272 | 1.39 (0.72–3.19) | 88 | 2.01 (0.74–4.08) | 0.18 |
| Controls | 275 | 0.93 (0.51–1.78) | 98 | 1.04 (0.56–2.03) | 0.35 |
Values are medians (interquartile range) or percentage (%) as indicated. CRP, C-reactive protein.
Table 2.
Association of the rs2241057 polymorphism with size of atherosclerotic lesions in the SCARF cohort.
| Homozygous for the major allele | Carriers of the minor allele | P | |
|---|---|---|---|
| n | 177 | 56 | |
| Atherosclerotic lesion size (mm2/mm) | 0.21 (0.16–0.26) | 0.23 (0.19–0.28) | 0.013 |
Values are medians (interquartile range).
DISCUSSION
This study identifies the first SNP with significant impact on enzymatic activity in the retinoic acid catabolizing enzyme CYP26B1. Furthermore, it demonstrates an increase in Cyp26B1 mRNA in murine atherosclerosis, expression of CYP26B1 in macrophage-rich areas of human athero-sclerotic lesions and induction of CYP26B1 mRNA by retinoic acid.
Atherosclerosis is an inflammatory disease in which blood vessel infiltration of cholesterol, macrophages and other immune cells play a key role from the onset to clinical manifestations of the disease (1,39). Retinoids are major regulators of both inflammation and cholesterol flux (9), and increased systemic and local retinoid levels promote antiinflammatory immunity, including differentiation of regulatory T cells, and reduce inflammation (8,40). Levels of atRA are tightly regulated by enzymes in the CYP26 family (41), and our finding of CYP26B1 expression inside human atherosclerotic lesions implies that atRA levels are regulated locally in the vessel wall. To compare Cyp26B1 in normal and atherosclerotic arteries, we analyzed Cyp26B1 mRNA in atherosclerosis-free arteries from C57BL/6 mice and atherosclerotic arteries from Apoe-deficient mice. In all investigated specimens, the atherosclerotic arteries had higher levels of Cyp26B1, indicating that Cyp26B1 is increased in atherosclerosis. Because of methodological constraints, it was not possible to directly compare CYP26B1 levels in human atherosclerotic carotid biopsies to a healthy control. To gain knowledge on normal levels of CYP26B1 mRNA in healthy human arteries, we studied atherosclerosis-free iliac arteries. The mean level of CYP26B1 mRNA was almost three times higher in atherosclerotic carotid endarterectomy samples than in atherosclerosis-free iliac arteries, and levels did not overlap between atherosclerotic carotid and healthy iliac specimens, which is in line with the murine results where atherosclerotic arteries showed higher values than healthy controls. The distribution of the atRA-catabolizing enzyme CYP26B1 in atherosclerotic lesions coincided anatomically with the expression of the macrophage marker CD68, suggesting that enzymatic regulation of atRA levels in lesions could affect ligand availability for these inflammatory cells. We and others previously showed that pharmacological inhibition of CYP26, or RNA interference (RNAi)-mediated CYP26 silencing, increases levels of retinoic acid and retinoid signaling (27,28,42). Hence, because our data indicate that the CYP26B1 enzyme was present and upregulated in atherosclerosis, regulation of CYP26B1 activity may influence atherosclerosis development by altering the local availability of retinoid ligands.
This study shows that a Leu-to-Ser substitution in position 264 in CYP26B1 increases the catabolic activity of CYP26B1 substantially, both in a cell free system and in cell culture. The minor CYP26B1 variant catabolized atRA at a substantially higher rate than the major CYP26B1 variant in all three investigated models. The actual catabolic rates in vivo will depend on several factors, including the local tissue concentration of atRA, and cannot be accurately predicted from the data in this study. However, it is plausible that the considerable difference in capacity between the enzyme variants shown here alters the equilibrium of retinoid synthesis and catabolism (43,44) and translates into alterations of atRA levels in vivo. In this way, carriers of the minor CYP26B1 variant may ultimately have reduced availability of atRA in inflamed tissues.
The notion that retinoid availability regulated by CYP26B1 may influence local atRA levels and inflammation in atherosclerosis implies that carriers of the minor CYP26B1 variant have reduced levels of atRA in atherosclerotic lesions. Hence, carriers would potentially have more local inflammation and atherosclerosis. The strongest expression of CYP26B1 was found in macrophage-rich, inflammatory areas of the lesions, where it is likely to have the strongest influence on inflammation. Interestingly, the different enzyme variants metabolized retinoids at different rates also when expressed in human macrophagelike differentiated THP-1 cells, suggesting that the CYP26B1 genotype may play a role for retinoid availability in human plaque-resident macrophages. Our investigation of this possible link between CYP26B1 variants and size of atherosclerotic lesions in patients showed that carriers of the minor allele indeed had larger atherosclerotic lesions as determined by angiography, albeit the differences were small. Importantly, we were unable to directly measure CYP26B1 activity and atRA levels in these atherosclerotic lesions, and investigate lesion size using the more precise intravascular ultrasound technique. Hence, it was not feasible to show causality, and the limited size of the cohort together with the lack of a replication cohort prevented a robust disease-association case-control analysis. This finding should, therefore, be interpreted with caution. It is also possible that the effect associated with the CYP26B1 variants was linked to the systemic availability of atRA. The lack of association between the CYP26B1 variants and known systemic atherosclerosis-associated factors, however, speaks against a systemic mechanism.
CONCLUSION
In conclusion, this study identifies the first SNP with significant impact on the enzymatic activity in the retinoic acid catabolizing enzyme CYP26B1. CYP26B1 mRNA was increased in murine atherosclerosis and expressed in macrophage-rich areas of human atherosclerotic lesions. Furthermore, retinoic acid induced CYP26B1 in human atherosclerotic lesions. Taken together, the findings suggest that genotype may affect CYP26B1-regulated levels of retinoids and possibly atherosclerosis development. Further studies are warranted to determine the detailed mechanisms of retinoid action in atherosclerotic lesions and the potential role of pharmacological CYP26B1 inhibitors in the treatment of atherosclerosis and its complications.
Supplemental Data
ACKNOWLEDGMENTS
We thank Göran K Hansson and Jesse Roth for helpful comments and for critically reviewing the manuscript.
This study was supported by the Swedish Heart-Lung Foundation, the Swedish Health Care Sciences Postgraduate School (NFVO) at Karolinska Institutet, the Swedish Medical Research Council (grants 521-2009-4203 and 349-2007-8703 [Center of Excellence for Research on Inflammation and Cardiovascular disease, Linnaeus support]), Bergwall’s Foundation and the Wenner-Gren Foundation in Stockholm.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–12. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 2.Gidlof AC, Ocaya P, Krivospitskaya O, Sirsjo A. Vitamin A: a drug for prevention of restenosis/reocclusion after percutaneous coronary intervention? Clin Sci (Lond) 2008;114:19–25. doi: 10.1042/CS20070090. [DOI] [PubMed] [Google Scholar]
- 3.Pino-Lagos K, Benson MJ, Noelle RJ. Retinoic acid in the immune system. Ann N Y Acad Sci. 2008;1143:170–87. doi: 10.1196/annals.1443.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Streb JW, Miano JM. Retinoids: pleiotropic agents of therapy for vascular diseases? Curr Drug Targets Cardiovasc Haematol Disord. 2003;3:31–57. doi: 10.2174/1568006033337393. [DOI] [PubMed] [Google Scholar]
- 5.Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res. 2002;43:1773–808. doi: 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- 6.Langmann T, et al. Gene expression profiling identifies retinoids as potent inducers of macrophage lipid efflux. Biochim Biophys Acta. 2005;1740:155–61. doi: 10.1016/j.bbadis.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 7.Zhou X, et al. Cutting edge: all-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. J Immunol. 2010;185:2675–9. doi: 10.4049/jimmunol.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mucida D, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–60. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 9.Wuttge DM, et al. Induction of CD36 by all-trans retinoic acid: retinoic acid receptor signaling in the pathogenesis of atherosclerosis. FASEB J. 2001;15:1221–3. doi: 10.1096/fj.00-0488fje. [DOI] [PubMed] [Google Scholar]
- 10.Gey KF, et al. Low plasma retinol predicts coronary events in healthy middle-aged men: the PRIME Study. Atherosclerosis. 2010;208:270–4. doi: 10.1016/j.atherosclerosis.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 11.Brazionis L, Walker KZ, Itsiopoulos C, O’Dea K. Plasma retinol: a novel marker for cardiovascular disease mortality in Australian adults. Nutr Metab Cardiovasc Dis. 2011 2011 Nov 25; doi: 10.1016/j.numecd.2011.08.009. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 12.Neuville P, et al. Retinoic acid regulates arterial smooth muscle cell proliferation and phenotypic features in vivo and in vitro through an RARalpha-dependent signaling pathway. Arterioscler Thromb Vasc Biol. 1999;19:1430–6. doi: 10.1161/01.atv.19.6.1430. [DOI] [PubMed] [Google Scholar]
- 13.Miano JM, et al. All-Trans-retinoic acid reduces neointimal formation and promotes favorable geometric remodeling of the rat carotid artery after balloon withdrawal injury. Circulation. 1998;98:1219–27. doi: 10.1161/01.cir.98.12.1219. [DOI] [PubMed] [Google Scholar]
- 14.Takeda N, et al. Synthetic retinoid Am80 reduces scavenger receptor expression and atherosclerosis in mice by inhibiting IL-6. Arterioscler Thromb Vasc Biol. 2006;26:1177–83. doi: 10.1161/01.ATV.0000214296.94849.1c. [DOI] [PubMed] [Google Scholar]
- 15.Brelsford M, Beute TC. Preventing and managing the side effects of isotretinoin. Semin Cutan Med Surg. 2008;27:197–206. doi: 10.1016/j.sder.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 16.Verfaille CJ, et al. Oral R115866 in the treatment of moderate to severe plaque-type psoriasis. J Eur Acad Dermatol Venereol. 2007;21:1038–46. doi: 10.1111/j.1468-3083.2007.02158.x. [DOI] [PubMed] [Google Scholar]
- 17.Baert B, De Spiegeleer B. Local skin pharmacokinetics of talarozole, a new retinoic acid metabolism-blocking agent. Skin Pharmacol Physiol. 2011;24:151–9. doi: 10.1159/000323012. [DOI] [PubMed] [Google Scholar]
- 18.Ray WJ, Bain G, Yao M, Gottlieb DI. CYP26, a novel mammalian cytochrome P450, is induced by retinoic acid and defines a new family. J Biol Chem. 1997;272:18702–8. doi: 10.1074/jbc.272.30.18702. [DOI] [PubMed] [Google Scholar]
- 19.White JA, et al. cDNA cloning of human retinoic acid-metabolizing enzyme (hP450RAI) identifies a novel family of cytochromes P450. J Biol Chem. 1997;272:18538–41. doi: 10.1074/jbc.272.30.18538. [DOI] [PubMed] [Google Scholar]
- 20.White JA, et al. Identification of the human cytochrome P450, P450RAI-2, which is predominantly expressed in the adult cerebellum and is responsible for all-trans-retinoic acid metabolism. Proc Natl Acad Sci U S A. 2000;97:6403–8. doi: 10.1073/pnas.120161397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taimi M, et al. A novel human cytochrome P450, CYP26C1, involved in metabolism of 9-cis and all-trans isomers of retinoic acid. J Biol Chem. 2004;279:77–85. doi: 10.1074/jbc.M308337200. [DOI] [PubMed] [Google Scholar]
- 22.White JA, et al. Identification of the retinoic acid-inducible all-trans-retinoic acid 4-hydroxylase. J Biol Chem. 1996;271:29922–7. doi: 10.1074/jbc.271.47.29922. [DOI] [PubMed] [Google Scholar]
- 23.White JA, et al. Identification of the human cytochrome P450, P450RAI-2, which is predominantly expressed in the adult cerebellum and is responsible for all-trans-retinoic acid metabolism. Proc Natl Acad Sci U S A. 2000;97:6403–8. doi: 10.1073/pnas.120161397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helvig C, Taimi M, Cameron D, Jones G, Petkovich M. Functional properties and substrate characterization of human CYP26A1, CYP26B1, and CYP26C1 expressed by recombinant baculovirus in insect cells. J Pharmacol Toxicol Methods. 2011;64:258–63. doi: 10.1016/j.vascn.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 25.Topletz AR, et al. Comparison of the function and expression of CYP26A1 and CYP26B1, the two retinoic acid hydroxylases. Biochem Pharmacol. 2012;83:149–63. doi: 10.1016/j.bcp.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takeuchi H, Yokota A, Ohoka Y, Iwata M. Cyp26b1 regulates retinoic acid-dependent signals in T cells and its expression is inhibited by transforming growth factor-beta. PLoS One. 2011;6:e16089. doi: 10.1371/journal.pone.0016089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ocaya P, Gidlof AC, Olofsson PS, Torma H, Sirsjo A. CYP26 inhibitor R115866 increases retinoid signaling in intimal smooth muscle cells. Arterioscler Thromb Vasc Biol. 2007;27:1542–8. doi: 10.1161/ATVBAHA.106.138602. [DOI] [PubMed] [Google Scholar]
- 28.Ocaya PA, et al. CYP26B1 plays a major role in the regulation of all-trans-retinoic acid metabolism and signaling in human aortic smooth muscle cells. J Vasc Res. 2010;48:23–30. doi: 10.1159/000317397. [DOI] [PubMed] [Google Scholar]
- 29.Lee S-J, et al. The discovery of new coding alleles of human CYP26A1 that are potentially defective in the metabolism of all-trans retinoic acid and their assessment in a recombinant cDNA expression system. Pharmacogenet Genomics. 2007;17:169–80. doi: 10.1097/FPC.0b013e32801152d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen P-H, et al. CYP26B1 is a novel candidate gene for betel quid-related oral squamous cell carcinoma. Oral Oncol. 2011;47:594–600. doi: 10.1016/j.oraloncology.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 31.Olofsson PS, et al. Genetic variants of TNFSF4 and risk for carotid artery disease and stroke. J Mol Med. 2009;87:337–46. doi: 10.1007/s00109-008-0412-5. [DOI] [PubMed] [Google Scholar]
- 32.Agardh HE, et al. Expression of fatty acid-binding protein 4/aP2 is correlated with plaque instability in carotid atherosclerosis. J Intern Med. 2011;269:200–10. doi: 10.1111/j.1365-2796.2010.02304.x. [DOI] [PubMed] [Google Scholar]
- 33.Van Guelpen B, et al. Plasma folate and total homocysteine levels are associated with the risk of myocardial infarction, independently of each other and of renal function. J Intern Med. 2009;266:182–95. doi: 10.1111/j.1365-2796.2009.02077.x. [DOI] [PubMed] [Google Scholar]
- 34.De la Vega FM, Lazaruk KD, Rhodes MD, Wenz MH. Assessment of two flexible and compatible SNP genotyping platforms: TaqMan SNP Genotyping Assays and the SNPlex Genotyping System. Mutat Res. 2005;573:111–35. doi: 10.1016/j.mrfmmm.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 35.Samnegard A, et al. Serum matrix metallo-proteinase-3 concentration is influenced by MMP-3-1612 5A/6A promoter genotype and associated with myocardial infarction. J Intern Med. 2005;258:411–9. doi: 10.1111/j.1365-2796.2005.01561.x. [DOI] [PubMed] [Google Scholar]
- 36.Tuinenburg JC, et al. One core laboratory at two international sites, is that feasible? An inter-core laboratory and intra-observer variability study. Catheter Cardiovasc Interv. 2002;56:333–40. doi: 10.1002/ccd.10189. [DOI] [PubMed] [Google Scholar]
- 37.Heiner CR, Hunkapiller KL, Chen SM, Glass JI, Chen EY. Sequencing multimegabase-template DNA with BigDye terminator chemistry. Genome Res. 1998;8:557–61. doi: 10.1101/gr.8.5.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gidlof AC, Ocaya P, Olofsson PS, Torma H, Sirsjo A. Differences in retinol metabolism and proliferative response between neointimal and medial smooth muscle cells. J Vasc Res. 2006;43:392–8. doi: 10.1159/000094415. [DOI] [PubMed] [Google Scholar]
- 39.Olofsson PS, et al. CD137 is expressed in human atherosclerosis and promotes development of plaque inflammation in hypercholesterolemic mice. Circulation. 2008;117:1292–301. doi: 10.1161/CIRCULATIONAHA.107.699173. [DOI] [PubMed] [Google Scholar]
- 40.Wiedermann U, et al. Vitamin A deficiency increases inflammatory responses. Scand J Immunol. 1996;44:578–84. doi: 10.1046/j.1365-3083.1996.d01-351.x. [DOI] [PubMed] [Google Scholar]
- 41.Petkovich PM. Retinoic acid metabolism. J Am Acad Dermatol. 2001;45:S136–42. doi: 10.1067/mjd.2001.113715. [DOI] [PubMed] [Google Scholar]
- 42.Stoppie P, et al. R115866 inhibits all-trans-retinoic acid metabolism and exerts retinoidal effects in rodents. J Pharmacol Exp Ther. 2000;293:304–12. [PubMed] [Google Scholar]
- 43.Wolf G. Retinoic acid homeostasis: retinoic acid regulates liver retinol esterification as well as its own catabolic oxidation in liver. Nutr Rev. 2001;59:391–4. doi: 10.1111/j.1753-4887.2001.tb06968.x. [DOI] [PubMed] [Google Scholar]
- 44.Ross AC. Retinoid production and catabolism: role of diet in regulating retinol esterification and retinoic acid oxidation. J Nutr. 2003;133:291S–296S. doi: 10.1093/jn/133.1.291S. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.