

Abstract
HIV infection causes loss of CD4+ T cells and type 1 interferon (IFN)–producing and IFN-responsive dendritic cells, resulting in immunodeficiencies and susceptibility to opportunistic infections, such as Pneumocystis. Osteoporosis and bone marrow failure are additional unexplained complications in HIV-positive patients and patients with AIDS, respectively. We recently demonstrated that mice that lack lymphocytes and IFN a/b receptor (IFrag−/−) develop bone marrow failure after Pneumocystis lung infection, whereas lymphocyte-deficient, IFN α/β receptor–competent mice (RAG−/−) had normal hematopoiesis. Interestingly, infected IFrag−/− mice also exhibited bone fragility, suggesting loss of bone mass. We quantified bone changes and evaluated the potential connection between progressing bone fragility and bone marrow failure after Pneumocystis lung infection in IFrag−/− mice. We found that Pneumocystis infection accelerated osteoclastogenesis as bone marrow failure progressed. This finding was consistent with induction of osteoclastogenic factors, including receptor-activated nuclear factor-κB ligand and the proapoptotic factor tumor necrosis factor–related apoptosis-inducing ligand, in conjunction with their shared decoy receptor osteoprotegerin, in the bone marrow of infected IFrag−/− mice. Deregulation of this axis has also been observed in HIV-positive individuals. Biphosphonate treatment of IFrag−/− mice prevented bone loss and protected loss of hematopoietic precursor cells that maintained activity in vitro but did not prevent loss of mature neutrophils. Together, these data show that bone loss and bone marrow failure are partially linked, which suggests that the deregulation of the receptor-activated nuclear factor-κB ligand/osteoprotegerin/tumor necrosis factor–related apoptosis-inducing ligand axis may connect the two phenotypes in our model.
HIV infection results in loss of CD4+ T cells and dendritic cells, which include type 1 interferon (IFN)–producing plasmacytoid dendritic cells.1,2 This process generates a complex immunodeficiency with susceptibilities to opportunistic infections and cancer.3 Autoimmune phenomena,4 bone marrow failure,5,6 and osteoporosis can also occur, but underlying mechanisms are poorly understood. Although the role of CD4+ T cells in the prevention of AIDS progression is undoubted,7,8 the role of type 1 IFNs is controversially discussed. Type 1 IFNs are proinflammatory and antiviral9 but are also important immune modulators.10 There is clear evidence of an independent, protective role of type 1 IFNs in the control of viral replication and protection from opportunistic infection.11–13 However, it is also debated whether type 1 IFN responses during HIV infection are linked to the excessive immune activation seen in patients, causing bystander T-cell depletion or dysfunction.14,15 These data point to the importance of balanced action of mediators to maintain system homeostasis during inflammatory responses.
Although highly active antiretroviral therapy (HAART) has greatly reduced the progression to severe immunodeficiency and thus death due to opportunistic infections, immune recovery can remain incomplete for the CD4+ T-cell repertoire and numbers and type 1 IFN-producing dendritic cells.12,16–18 This incomplete recovery can result in deviated immune responses. Indeed, the epidemiology of disease manifestations in treated HIV-positive individuals has changed. Unexplained chronic pulmonary sequelae, such as chronic obstructive pulmonary disease (COPD)19,20 and accelerated osteoporosis,21–23 now appear at the forefront of disease management.
A close relationship exists between bone metabolism and inflammation as well as hematopoiesis and inflammation.24,25 All blood cells, and therefore immune cells, are generated in the bone marrow, in which hematopoietic stem cells reside in unique niches, interacting with osteoblasts of the bone.26–28 Distinct cytokines relevant in inflammation, such as IL-1, tumor necrosis factor (TNF)-α, and receptor-activated nuclear factor-κB ligand (RANKL), also regulate bone metabolism by activating bone-resorbing osteoclasts, which also mobilize hematopoietic stem cells from their bony niche. In contrast, other mediators, such as osteoprotegerin (OPG), IFN-γ, type 1 IFNs, and IL-4, suppress osteoclast activation.24 Abnormal or chronic immune activation enhances osteoclastogenic cytokine activity and can result in focal bone destruction and accelerated bone loss because it occurs in rheumatoid arthritis and other inflammatory diseases.29,30 In this regard, chronic inflammatory lung diseases, such as COPD, are also consistently associated with accelerated osteoporosis as a systemic manifestation of the disease, although the triggering mechanisms are not clear.31 Interestingly, severe osteoporosis in these chronic lung diseases is also associated with anemia and reduced circulating bone marrow progenitor cells.32 This finding demonstrates that focal diseases can have an unexpected systemic effect on other organ systems, such as the bone and bone marrow.
The prevalence rate of osteoporosis resulting in increased fracture risk is more than three times higher in HIV-positive patients compared with HIV-negative controls.33 The cause of these bone changes is controversial, and treatment with HAART, especially protease inhibitors, appears to contribute to the phenotype.21,22 However, patients who have never received antiretroviral therapy also have an increased incidence of osteoporosis.34 This finding suggests that additional factors, such as deviated systemic immune activation in response to opportunistic infections, might contribute. In this regard, low CD4+ T-cell counts are considered a risk factor for osteoporosis.35 However, the potential contribution of a deviated systemic immune response to low-grade pulmonary opportunistic infections in accelerating osteoporosis in the context of a variable defect of the type 1 IFN system, as can occur in HIV-positive patients, has not been evaluated.
Pneumocystis is a pulmonary fungal pathogen capable of causing a severe interstitial pneumonia in patients with lymphocyte deficiency and/or a defect of the type 1 IFN system.1,11,12 Pneumocystis is the most common opportunistic infection in AIDS, although it also affects patients undergoing chemotherapy, those receiving anti-TNF-α therapy,36–38 or patients with inherited CD4+ T-cell defects.3 In addition, low-grade Pneumocystis colonization exacerbates COPD and other chronic pulmonary disease, not only in HIV-positive patients undergoing HAART39 but also in patients without known immune defects who are likely undergoing glucocorticoid treatments. However, Pneumocystis colonization and COPD are significantly higher in HIV-positive patients undergoing HAART compared with HIV-negative individuals.33,34 Interestingly, anti-inflammatory mechanisms of glucocorticoids also impair the type 1 IFN system.40,41
Given the clear association between chronic inflammatory pulmonary disease and osteoporosis and the implication that Pneumocystis exacerbates such disease, we questioned whether the systemic response to Pneumocystis infection in the context of a defective type 1 IFN system might accelerate osteoporosis.
Using a mouse model of Pneumocystis infection, we recently demonstrated an important role of the type 1 IFN system in regulating the systemic responses to the infection.42,43 Pneumocystis lung infection in mice lacking lymphocytes and type 1 IFN receptor (IFrag−/− mice) results in rapidly progressing bone marrow failure, characterized by severe reduction of bone marrow cellularity affecting all hematopoietic lineages and loss of hematopoietic stem cell function when tested in hematopoietic colony-forming assays, despite minimal pathogen burden within 16 days due to accelerated apoptosis of mature and precursor cells. During progression of bone marrow failure in IFrag−/− mice, we also observed increasingly brittle bone. In contrast, IFN α/β receptor (IFNAR)–competent but lymphocyte-deficient RAG−/− mice have normal hematopoiesis and die of Pneumocystis pneumonia with high pathogen burden after 4 to 5 weeks. Furthermore, no apparent bone changes were observed in RAG−/− mice.
This study aimed to evaluate and quantify the impression of accelerated bone loss in response to Pneumocystis lung infection in our mouse model, elucidate mechanisms associated with this phenotype, and determine whether a causal link exists between bone loss and bone marrow failure. Our data suggest that bone loss in IFrag−/− mice in response to Pneumocystis lung infection is caused by inflammation-induced accelerated osteoclastogenesis and that bone loss and bone marrow failure are partially linked, possibly involving deregulation of the RANKL/OPG/TNF-related apoptosis-inducing ligand (TRAIL) axis.
Materials and Methods
Mice
C.B17 SCID mice, as a source for Pneumocystis murina organisms, were bred and maintained at Montana State University (MSU) Animal Resource Center. RAG1−/− mice, herein referred to as RAG−/− mice (C57/BL6 background), were initially purchased from Jackson Laboratories (stock number 002096) and then bred at MSU. IFrag−/− mice were generated by crossing IFNAR knockout mice (129SvEv background and maintained at MSU) with RAG1−/− mice (C57/BL6 background), herein referred to as RAG−/− mice, as previously described42 and have since been twice backcrossed on a C57/BL6 background. Animals were kept in ventilator cages with sterilized food and water. All mouse studies conformed to NIH guidelines and were approved by the Institutional Animal Care and Use Committee MSU. Some mice were treated with risedronate, a bisphosphonate, which inhibits osteoclasts to prevent loss of bone mass. For this, 10 g/kg risedronate (Wako, Tokyo, Japan) was administered intraperitoneally five times per week.44 For in vivo neutralization of TRAIL, mice were injected intraperitoneally with 750 μg of TRAIL-neutralizing antibody (clone N2B2; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) every other day during the infection, starting 1 day before the Pneumocystis infection.
Pneumocystis Lung Infection
Pneumocystis was isolated from heavily infected source mice. Experimental animals were intratracheally infected with 1 × 107 Pneumocystis nuclei in 100 μL of lung homogenate from infected source mice diluted in PBS buffer. Pneumocystis burden of infected animals was assessed microscopically (Zeiss Axiostar microscope; Zeiss, Jena, Germany) by enumeration of trophozoid nuclei count in lung homogenates in 10 to 50 oil immersion fields as previously described. The limit of detection for this technique is log10 4.43.45 A previously established time course of infection was used in the presented studies comparing Pneumocystis-infected IFrag−/− and RAG−/− mice. Bone and bone marrow responses of infected mice were therefore evaluated at day 0, 7, 10, and 16 after Pneumocystis lung infection. These groups are herein referred to as day 0, 7, 10, and 16 IFrag−/− or RAG−/− mice. Each group consisted of four mice per time point, and each experiment was repeated at least twice.
Bone Marrow Flushes, Enumeration, and Differentiation
Bone marrow cells from femur and tibia were collected as previously described by flushing 2 mL of PBS through the bone marrow canal using a 26½-g needle and brought into a single cell suspension.46 Bone marrow cells were diluted 1:10 in PBS, enumerated, spun onto glass slides, and stained with Diff-Quick solution (Dade Behring, Deerfield, IL). Cell differentiation was performed based on morphologic and staining pattern to distinguish myeloid (including myeloblast-myelocyte and metamyelocyte stage), erythroid, neutrophil, and eosinophil cells.46
Osteoclast Activity Assays in Bone Marrow Lysates and on Tissue Sections
Osteoclast formation was assayed in bone marrow cell lysates (preosteoclasts) and on bone sections (mature osteoclasts) from IFrag−/− and RAG−/− mice in response to Pneumocystis lung infection by detecting osteoclast-specific tartrate resistant acidic phosphatase (TRAP) activity. A soluble TRAP assay was performed in bone marrow cell lysates as previously described47,48 using a leukocyte acidic phosphatase kit (386A-1KT; Sigma-Aldrich, St. Louis, MO). Briefly, total bone marrow cells were suspended in PBS and adjusted to 6 × 106 cells/mL. A 50-μL cell suspension was pipetted in duplicate into round bottom 96-well enzyme-linked immunosorbent assay (ELISA) plates (Costar, Milpitas, CA). Cells were pelleted for 30 seconds at 300 × g in a Sorval tabletop centrifuge, the supernatant removed, and cells lysed in 50 μL of cold cell lysis buffer containing 90 mmol/L citrate buffer, pH 5.5, 0.1% Triton X, and 80 mmol/L sodium tartrate. A total of 50 μL of substrate solution [5.4 mmol/L (2 mg/mL) p-nitrophenyl phosphate (Sigma-Aldrich) dissolved in lysis buffer] was added to the lysate, mixed, and incubated for 1 hour at 37°C in the dark. The reaction was stopped by adding 50 μL of 0.5 mol/L NaOH to each well, the assay was read at 405 nm using a plate assay reader, and data were plotted in OD. Positive controls for the assays were bone marrow cells lysed in the same lysis buffer but not containing tartrate to assay for all acidic phosphatases. Negative controls for the assay contained bone marrow lysates pretreated with NaOH, lysates with no substrate added, or assay buffer with no lysate added.
For TRAP staining on formalin-fixed and decalcified bone sections, femur and tibia were placed in 10% ice cold buffered formalin solutions for 24 hours. Bones were decalcified in 5% EDTA in PBS, pH 7.4 to 7.6, at 4°C for approximately 3 to 5 days, rinsed in tap water for 30 minutes, and placed into cold 70% ethanol until further processing. Paraffin embedded bones were cut in 5-μm sections using a Leica RM2255 microtome (Leica Microsystems, Wetzlar, Germany) and deparaffinized. Staining was performed using the leukocyte acidic phosphatase kit from Sigma-Aldrich (St. Louis, MO) (386A-1KT) following the manufacturer's protocol, except the counterstain was performed using hematoxylin 1 (Richard Allen Scientific, Kalamzoo, MI) including a bluing and clarifying step.
Histomorphometric Analysis
Comparative histomorphometric analysis of bones from Pneumocystis-infected IFrag−/− and RAG−/− mice was performed by The University of Alabama at Birmingham Center of Metabolic Bone Diseases Core Laboratory on excised mouse femurs, fixed in 70% ETOH. Longitudinal sections (5 μm thick) were cut at the 50% plane from methyl methacrylate embedded blocks using a Leica 2265 microtome. These were stained with Goldner's trichrome stain for the static measurements. A region of interest was selected that is exactly 250 μm distal to the growth plate and extending 1 mm downward (thereby avoiding the primary spongiosa) through the metaphysis of the femur. Standard bone histomorphometric analysis was performed by the methods of Parfitt et al49,50 using Bioquant Image Analysis software (R & M Biometrics, Nashville, TN). Four types of primary measurements were made—area, length (perimeter), distance, and number. These referents, such as tissue volume, bone volume, bone surface, and osteoid surface, were used to derive other indices, such as trabecular number and trabecular separation.
MicroCT for the Evaluation of Bone Architecture
Microcomputed tomography (microCT) analysis was performed to evaluate structural parameters of cancellous and cortical bone by a nondestructive procedure. Analysis was performed by Scanco USA Inc. (Wayne, PA). The individual specimens were transferred into a Scanco specimen holder and then immersed in 70% ethanol. Specimens were positioned lengthwise in the specimen holder to acquire scans at the highest voxel resolution possible for a specimen holder at that size. All scanning was conducted on a Scanco microCT 40 scanner. X-ray energy of 55 Kvp at a current of 145 μA was used. Images were acquired with a field of view of 12.28 mm and matrix size of 2048 × 2048 (2000 projections at 360°), resulting in a voxel size of 6 μm. Beam-hardening correction (1200 hardening artifact mg/mL) was used (standard on Scanco microCT systems). A single-frame average with an integration time of 300 milliseconds per projection was used for all scans.
Trabecular Bone Imaging
A total of 550 to 650 sections (covering a distance of 3 to 4 mm) were acquired from the metaphyseal region of the proximal femur. The raw data were reconstructed (automatically) to image data using the reconstruction software bundled with the microCT system (Scanco).
Cortical Bone Imaging
A total of 110 to 120 sections (covering 0.6 to 0.7 mm) were acquired from the midshaft of the femur. For trabecular bone analysis a region of interest was manually contoured to include only the trabecular bone compartment for a distance of 1.2 mm (200 sections). Images were filtered using a G filter (σ = 0.8; support = 1.0) and were segmented using a global threshold value of 230. Trabecular bone parameters were calculated using the standard Scanco software suite. A total of 100 sections (covering a distance of 0.6 mm) were analyzed for cortical bone parameters. Images were filtered using a G filter (σ = 0.8; support = 1.0) and were segmented using a global threshold of 350. Cortical bone parameters were calculated using the standard Scanco software suite.
Cytokine Analysis in Bone Marrow Cell Lysates
Bone marrow cells from day 0, 7, 10, and 16 IFrag−/− and Rag−/− mice were isolated, and 5 × 106 total cells were lysed in 100 μL of cell lysis buffer (Biovision Inc, Milipitas, CA). Each group consisted of four animals per time point. Protein assays were performed using bicinchoninic acid assay reagents (Pierce Protein Research Projects, Thermo Fisher Scientific Inc, Rockford, IL) to ensure equal protein concentrations. Twenty-five microliters of lysate from each mouse was assayed in duplicate for an array of inflammatory and bone metabolism–related cytokines using multiplex and single-plex bead ELISA kits from Millipore with subsequent analysis on a Bio-Plex200 system (Bio-Rad Laboratories, Hercules, CA). The following analytes were assessed using reagents from Millipore: macrophage colony-stimulating factor (M-CSF), IL-6, IL-1α, IL-β, TNF-α, RANKL, and OPG. For the detection of the apoptotic factor TRAIL, a sandwich ELISA, was established using a mouse TRAIL capture and detection antibody pair from R&D Systems following the manufacturer's instructions (R&D Systems Duo Set mouse TRAIL; R&D Systems, Minneapolis, MN). A standard curve was generated, the assay was read at 450 nm on a plate reader, and results were analyzed using Softmax Pro software (Molecular Devices, Sunnyvale, CA).
CFC Assay for Mouse Bone Marrow Cells
Hematopoietic precursor cell (HPC) activity in bone marrow from IFrag−/− and RAG−/− mice was assessed by performing colony-forming cell (CFC) assays in methylcellulose media. For these assays, 1 × 105 bone marrow cells were plated in MethCult GF M3534 media (StemCell Technologies, Vancouver, BC), which was formulated to support the optimal growth of granulocyte and macrophage precursor cells. Cells from each sample were plated in duplicate according to the manufacturer's protocol in 35-mm sterile culture dishes (StemCell Technologies) and placed in 100-mm petri dishes in the presence of one 35-mm dish that contained sterile water. Cultures were incubated for 7 days in a water-jacketed incubator maintained with 5% CO2. Colony recognition (granulocyte-macrophage–, granulocyte-, and macrophage-forming colonies) and enumeration were performed according to the StemCell Technologies guidelines.
Statistical Analysis
Statistical analysis was performed using either a one-way or two-way analysis of variance followed by either a Tukey or a Bonferroni posttest.
Results
Association of Bone Marrow Failure in IFrag−/− Mice with Increased Osteoclast Differentiation and Activity along the Trabecular Bone in Response to Pneumocystis Lung Infection
We previously reported that IFrag−/− mice develop progressive bone marrow failure during systemic responses to Pneumocystis lung infection, whereas hematopoiesis was unaffected in RAG−/− mice.42 In a previously established time course (day 0, 7, 10, and 16 after Pneumocystis), we demonstrated that events around day 7 after Pneumocystis lung infection are critical in determining the bone marrow response.43 We also show that during this time course IFN-α serum concentrations in RAG−/− mice peak at day 7 after infection (Figure 1A), clearly demonstrating that a systemic type 1 IFN response is induced during Pneumocystis lung infection to which IFrag−/− mice are unable to respond.
Figure 1.

Induction of bone marrow failure in IFrag−/− mice is associated with increased preosteoclast differentiation. A: Levels of IFN-α in serum of RAG−/− mice were determined over time, from day 0 to day 16. B: Bone marrow cell counts were performed microscopically from bone marrow flushes of both hind legs from day 0, 7, 10, and 16 IFrag−/− and RAG−/− mice during Pneumocystis lung infections, demonstrating progressive bone marrow failure in IFrag−/− mice but not in RAG−/− mice. Each group consisted of four animals per time point. C: TRAP activity was measured in bone marrow cell lysates using 3 × 105 bone marrow cells from each animal and time point. Activity was assayed and plotted in OD. Statistical analysis was performed using a two-way analysis of variance. Values shown are mean ± SEM (n = 4). ***P < 0.001.
During the progression of bone marrow failure, IFrag−/− mice also appear to develop brittle bone, suggesting loss of bone mass possibly due to inflammation-induced accelerated osteoclast activity. We wanted to verify this finding, quantify osteoclast activity, and determine whether the bone loss is causally related to the development of bone marrow failure in our system. Therefore, we determined whether preosteoclast differentiation was enhanced in bone marrow from IFrag−/− but not RAG−/− mice after Pneumocystis lung infection. For this we measured TRAP activity in bone marrow cell lysates from equal cell numbers harvested during the infection as a relative indicator for pre osteoclast numbers. Figure 1B shows typical bone marrow cell counts comparing IFrag−/− versus RAG−/− mice during Pneumocystis lung infection, with bone marrow cell numbers decreasing only in IFrag−/− mice as an indicator of bone marrow failure.43 TRAP activity measured in lysates from these bone marrow cells significantly increased in IFrag−/− compared with RAG−/− mice on day 7 after infection (Figure 1C). However, at day 16 after infection this activity disappeared, suggesting either death of these precursors or further differentiation and attachment to the bone.
To determine whether increased preosteoclast differentiation would also result in enhanced accumulation of mature osteoclasts active along the trabecular bone of IFrag−/− mice, TRAP stains were performed on bone sections of day 0 (uninfected) and day 16 IFrag−/− mice. Figure 2 shows a representative TRAP stain on decalcified and paraffin-embedded femurs from day 0 (Figure 2A) and day 16 IFrag−/− mice (Figure 2B). The osteoclasts stained red and were visible along the trabecular bone, consistently giving the impression that more osteoclasts were lining bone structures of infected compared with uninfected IFrag−/− mice. We also noted that the number and sizes of trabecular bone of infected IFrag−/− mice appeared diminished. To quantify this observation and compare this response to RAG−/− mice, bone histomorphometric studies were performed on calcified and plastic-embedded femurs excised from day 0 and day 16 IFrag−/− and RAG−/− mice. Shown in Figure 3A are von Kossa stains demonstrating mineralized bone structures. Goldner's trichrome stain was used for histomorphometric analysis [Figure 3B; original magnification: ×40 (B); ×200 (insets)], and results are summarized in Figure 3, C-G. For statistical analysis all comparisons were made to day 0 RAG−/− mice. This analysis showed the bone perimeter per tissue area (Figure 3C) and trabecular bone numbers (Figure 3D) significantly reduced only in day 16 IFrag−/− mice in response to Pneumocystis lung infection and no changes in day 16 RAG−/− mice. These data are consistent with loss of bone mass. Also consistent with this finding are an increased number of osteoclasts per bone perimeter (Figure 3E) and increased erosion perimeters per bone perimeter (Figure 3F) only in infected IFrag−/− mice, suggesting accelerated osteoclasts activity. Interestingly, osteoblast numbers and bone perimeter also increased, suggesting an increased, compensatory activity (Figure 3G).
Figure 2.

Osteoclast numbers increase along the trabecular bone of IFrag−/− mice in response to Pneumocystis lung infection. TRAP staining was performed on formalin-fixed and paraffin-embedded bone sections from day 0 (A) and day 16 (B) IFrag−/− mice. TRAP-positive osteoclasts are stained red along the trabecular bone, demonstrating increased osteoclast numbers along the trabecular bone of infected compared with uninfected IFrag−/− mice. Also of note is the decreased bone marrow cellularity in bone marrow space of infected IFrag−/− mice.
Figure 3.

Bone histomorphometric analysis reveals loss of trabecular bone numbers and increased osteoclast activity in IFrag−/− mice compared with RAG−/− mice in response to Pneumocystis lung infection. A: von Kossa staining of calcified, methyl methacrylate embedded bone sections of day 0 and day 16 IFrag−/− and RAG−/− mice demonstrates mineralized bone. B: Adjacent bone sections stained with Goldner's trichrome highlight osteoid seams and were used to perform the bone histomorphometric analysis. Original magnification: ×40 for all sections; ×200 for the overlaid images. Standard bone histomorphometric analysis was performed at the Center of Metabolic Bone Diseases, University of Alabama, by the methods of Parfitt et al49,50 using Bioquant Image Analysis software (R & M Biometrics). Analysis revealed reduced bone perimeter per tissue area (C) and trabecular bone numbers per millimeter (D), associated with increased osteoclast numbers per bone perimeter (E) and increased bone erosion perimeters per millimeter (F) and increased osteoblast numbers per bone perimeter (G) only in IFrag−/− mice in response to Pneumocystis lung infection. Statistical analysis was performed using a one-way analysis of variance. Values were compared to uninfected RAG−/− mice and are shown as mean ± SEM (n = 4). *P < 0.05, **P < 0.01, and ***P < 0.001.
Femoral bones from four uninfected (day 0) and four infected (day 16) IFrag−/− mice were also analyzed by microCT51 to further evaluate structural bone changes following Pneumocystis lung infection. Shown in Figure 4A are three-dimensional images demonstrating trabecular and cortical bone structure of the comparison groups. Images show an increased transition from plate-like to rod-like elements characteristic of deterioration of cancellous bone structure. This transition is defined by a morphometric parameter called Structure Model Index (SMI). The SMI value increases from 0 (ideal plate) to 3 (ideal rod) are reflective of these structural changes. The connectivity density is a measure of the degree to which a structure is multiply connected and reflects the integrity of the microstructure of the trabecular bone. Consistent with our histomorphometric analysis (Figure 3), the SMI values increased in day 16 IFrag−/− mice compared with day 0 mice (Figure 4B), whereas the connectivity density decreased (Figure 4C). In addition and consistent with loss of bone mass, trabecular numbers and bone volume/tissue volume decreased, whereas trabecular separation increased in day 16 IFrag−/− mice (Figure 4, D-F). Although none of these differences were statistically significant, likely because of the variability of disease progression among individual mice (Figure 4A), the trends support our histomorphometric analysis.
Figure 4.
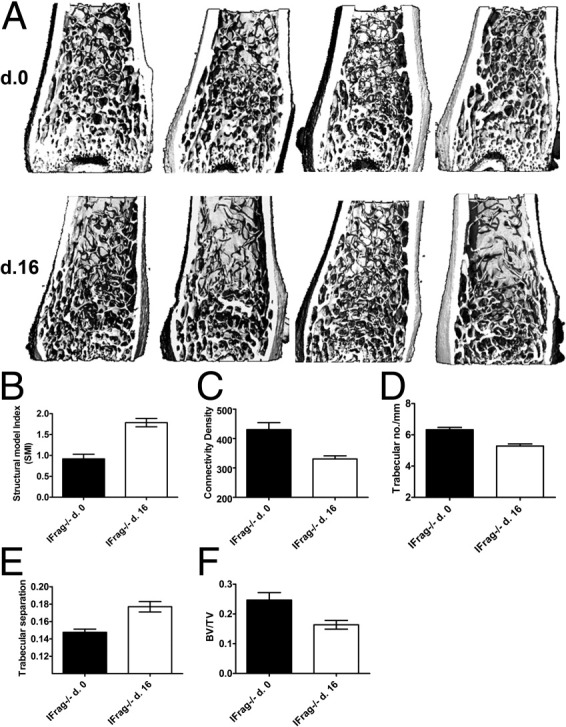
MicroCT analysis of IFrag−/− femoral bones demonstrate structural changes of the trabecular bone in response to Pneumocystis lung infection. A: Three-dimensional images of IFrag−/− femoral bones at days 0 and 16. All scanning was conducted on a Scanco microCT 40 scanner and performed by Scanco. Note the reduction of plate-like elements in the bones from day 16 mice, also reflected in the increase of the SMI (B). Other numerical data from this analysis included connectivity density (C), trabecular bone number per millimeter (D), trabecular separation (E), and BV/TV (F). Statistical analysis was performed using a Mann-Whitney test.
Deviated Bone Marrow Cytokine Profile Caused by Pneumocystis Lung Infection That Supports Osteoclastogenesis in IFrag−/− but Not RAG−/− Mice
Bone loss in IFrag−/− mice is preceded by induction of bone marrow failure after day 7 of Pneumocystis lung infection (Figure 5A),43 although Pneumocystis lung burden (Figure 5B) and the extent of lung inflammation, based on cell numbers in bronchoalveolar lavage, do not significantly differ between the mouse groups (Figure 5C). However, whether and how bone marrow failure and bone loss are interconnected in our model are not clear.
Figure 5.

Bone marrow failure in IFrag−/− mice is not associated with differences in Pneumocystis lung burden and extent of lung inflammation when compared with RAG−/− mice. Tissues were harvested from day 0, 7, 10, and 16 IFrag−/− and RAG−/− mice. A: Bone marrow counts demonstrated stable cell numbers in RAG−/− mice, whereas IFrag−/− mice progressed to bone marrow failure at day 7 after infection. Pneumocystis lung burden (B) and bronchoalveolar lavage cell numbers collected at the same time (C) were not different between the groups. Statistical analysis was performed using a two-way analysis of variance. Values shown are mean ± SEM (n = 4). **P < 0.01, ***P < 0.001.
Hematopoiesis, bone metabolism, and inflammation are linked, and a distinct set of cytokines mediating inflammation is also responsible for promoting osteoclastogenesis.52,53 The cytokines involved are TNF-α, IL-1, IL-6, and the myeloid growth factor M-CSF. However, of specific importance is the osteoclast differentiation factor RANKL. RANKL itself is negatively regulated by its decoy receptor OPG.24,25,52 To determine whether any of these factors were differentially regulated in bone marrow from IFrag−/− versus RAG−/− mice in response to Pneumocystis lung infection, bone marrow cells were harvested at day 0, 7, and 10 after infection, and cytokine analysis of bone marrow cell lysates was performed (Figure 6). Although there were insufficient IFrag−/− bone marrow cell numbers at day 16 after infection to perform extensive cytokine analysis, the data obtained between day 0 and 10 are most relevant because they reflect the induction phase of the condition seen in our system.
Figure 6.

IFrag−/− mice develop a deviated bone marrow cytokine response promoting osteoclastogenesis, suggesting an unbalanced RANKL/OPG/TRAIL axis. Cytokine protein analysis was performed from bone marrow cell lysates at day 0, 7, and 10 after Pneumocystis lung infection using an ELISA-based multiplex technique for M-CSF (A), IL-6 (B), IL-1α (C), IL-1β (D), TNF-α (E), RANKL (F), OPG (G), and TRAIL (H). Results demonstrate a deviated cytokine response in bone marrow from IFrag−/− mice consistent with osteoclastogenesis and bone marrow cell apoptosis. Statistical analysis was performed using a two-way analysis of variance. Values shown are mean ± SEM (n = 4). *P < 0.05, **P < 0.01, and ***P < 0.001.
The findings revealed that virtually all proinflammatory cytokines relevant to bone remodeling and osteoclastogenesis, with the exception of IL-6, were up-regulated in bone marrow from IFrag−/− compared with RAG−/− mice in response to Pneumocystis lung infection. Factors involved in promoting preosteoclast differentiation, such as M-CSF, TNF-α, and IL-1 family members, were already high at day 7 after infection (Figure 6, A-E), whereas RANKL, the key factor for osteoclast maturation and differentiation, was highly induced in day 10 IFrag−/− mice (Figure 6F). Surprisingly, at the same time, its decoy receptor OPG was also significantly induced in bone marrow from day 10 IFrag−/− mice (Figure 6G).
Although capable of neutralizing RANKL actions,54 OPG can also act as a decoy receptor for the proapoptotic cytokine TRAIL.55 Because bone marrow cell apoptosis was a prominent presenting symptom in our infection model,43 we also measured TRAIL levels in the same bone marrow cell lysates. Interestingly, TRAIL expression was already significantly up-regulated in day 7 IFrag−/− mice (Figure 6H) and thus preceded the induction of RANKL and OPG. The cytokine levels presented in Figure 6 are also summarized in Supplemental Table S1 (available at http://ajp.amjpathol.org) as fold change IFrag−/− relative to RAG−/−.
Inhibition of Bone Loss After Pneumocystis Lung Infection and Protection of HPC Viability by Bisphosphonate Treatment of IFrag−/− Mice
To address whether the loss of bone structure in IFrag−/− mice was causally linked to bone marrow failure, we treated Pneumocystis infected IFrag−/− mice with risedronate, a bisphosphonate that inhibits osteoclasts.44 The effects on bone structure, bone marrow cell numbers, and HPC activity were measured at day 16 after infection. As demonstrated in von Kossa stains (Figure 7A), risedronate treatment had a visual effect on the extent of mineralized bone in IFrag−/− mice, and bone histomorphometric analysis revealed that trabecular bone numbers per millimeter increased greatly compared with untreated mice (Figure 7B). Total bone marrow cell numbers of treated and untreated day 16 IFrag−/− mice, however, revealed low counts relative to uninfected mice in both groups (Figure 1A and Figure 5A), indicating progression of bone marrow failure (Figure 7C). Surprisingly, the hematopoietic CFC assay found significantly more colony formation with bone marrow cells from risedronate-treated mice compared with nontreated mice. This finding indicates more viable HPCs in bone marrow from day 16 IFrag−/− mice, in which bone loss was inhibited by risedronate treatment (Figure 7D). These data suggest a partial connection between bone loss and bone marrow failure.
Figure 7.

Bisphosphonate treatment of Pneumocystis-infected IFrag−/− mice prevents bone loss and maintains HPC activity but does not protect from mature bone marrow cell loss. Two groups of IFrag−/− mice were infected with Pneumocystis, with one group receiving the osteoclast inhibitor risedronate during the infection. Mice were harvested at day 16 after infection. A: von Kossa stains of calcified bone sections from representative IFrag−/− mice of each group. B: Trabecular bone numbers were assessed by bone histomorphometric analysis, which revealed increased numbers in treated day 16 IFrag−/− mice. C: Total bone marrow cell counts at day 16 do not differ between the groups. D: HPC viability, assessed by CFC assays, is significantly higher in bone marrow cells from risedronate-treated compared with untreated day 16 IFrag−/− mice. Statistical analysis was performed using a Mann-Whitney two-tailed test. *P < 0.05, ***P < 0.001.
Amelioration of Progression of Bone Marrow Failure by Treatment of IFrag−/− Mice with Neutralizing Anti-TRAIL Antibody After Pneumocystis Lung Infection
We recently determined that increased reactive oxygen species, possibly due to reduced growth factor production, and increased TNF-α in bone marrow of IFrag−/− mice during Pneumocystis lung infection are partially responsible for the progression of bone marrow failure in our model.43 To determine whether increased TRAIL production in bone marrow of IFrag−/− mice also contributed to the phenotype, we treated IFrag−/− mice with neutralizing TRAIL antibody (750 μg of N2B2 three times weekly) during infection. This approach reduced TRAIL concentrations in the bone marrow by approximately 50% (data not shown). Under this treatment regimen, progression of bone marrow failure was ameliorated at day 10 after infection (Figure 8) but subsequently continued to progress (data not shown). These data support a partial role of TRAIL in the pathogenesis of bone marrow failure in our system.
Figure 8.
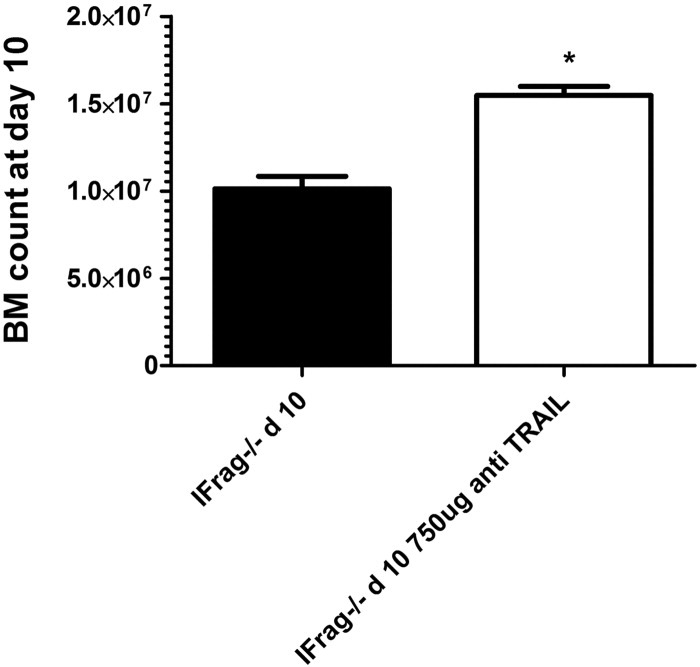
Neutralization of TRAIL ameliorates progression of bone marrow failure in response to Pneumocystis lung infection. Two groups of IFrag−/− mice were infected with Pneumocystis. One group was also intraperitoneally administered anti-TRAIL antibody (clone N2B2) at 750 μg three times per week throughout the infection and starting 1 day before infection. Total bone marrow cell counts at day 10 after infection. *P < 0.05.
Discussion
We found that the absence of type 1 IFN signaling during systemic stress responses to Pneumocystis lung infection not only impairs hematopoiesis but also greatly accelerates osteoclastogenesis and induces significant bone loss.
Osteoclasts are cells of myeloid origin that are uniquely capable of rapidly resorbing bone and are considered an innate immune cell of the bone.56 Soluble TRAP assays from bone marrow cell lysates during Pneumocystis infection indicated increased differentiation of preosteoclasts from myeloid precursor cells after the early systemic response, specifically in IFrag−/− but not RAG−/− mice. Comparative bone histomorphometric studies confirmed increased osteoclast numbers along the trabecular bone and loss of trabecular bone numbers exclusively in IFrag−/− mice in response to Pneumocystis infection. Osteoblast numbers in infected IFrag−/− mice were also increased, possibly as a compensatory effect, and make a defect of these bone-producing cells unlikely the cause of the rapidly progressing bone loss observed in our model.57 Type 1 IFNs are known to negatively regulate osteoclastogenesis,58 although our model also reveals that the systemic response to the focal Pneumocystis lung infection seems to accelerate osteoclastogenesis in the absence of type 1 IFN signaling. This finding suggests a unique role of type 1 IFNs specifically in the modulation of the systemic responses to Pneumocystis lung infection.
Low-grade Pneumocystis infection is common in HIV-positive patients, even those undergoing HAART, in which the type 1 IFN system may still be impaired.39 Pneumocystis colonization has also been detected in lungs from non-AIDS patients with COPD and is linked to exacerbation of the disease.59,60 Interestingly, COPD is often treated with glucocorticoids, which impair the type 1 IFN system by the nature of their action.41 Both HIV infection and COPD are associated with osteoporosis, and patients with either disease may also have blood abnormalities.6,61,62 Although mechanisms are not clearly understood, a deviated systemic immune response is thought to be involved.61 Therefore, the implication that a deviated systemic inflammatory response to Pneumocystis lung infection, possibly due to impaired type 1 IFN signaling, could be causally linked to accelerated osteoporosis is intriguing.
Cytokines are important communicators of inflammation, and osteoclastogenesis and inflammation are tightly linked.53 Further analysis indeed revealed a deviated cytokine profile, supporting osteoclastogenesis uniquely present in the bone marrow of day 7 and 10 IFrag−/− but not RAG−/− mice. M-CSF supports the differentiation of myeloid precursors toward the macrophage lineage and preosteoclasts63 and was also up-regulated by day 7 after infection in bone marrow from IFrag−/− mice. This finding was consistent with increased TRAP activity in bone marrow cell lysates by day 10. TNF-α and IL-1 cytokines were also up-regulated in day 7 and day 10 IFrag−/− mice but not in RAG−/− mice. Both cytokines are known to initiate RANKL production, a key osteoclast maturation factor.64 RANKL was indeed up-regulated in bone marrow from day 10 IFrag−/− mice, which was followed by subsequent maturation and activity of osteoclasts along the trabecular bone in day 16 IFrag−/− mice. The early deviation toward increased IL-1 and TNF-α production in bone marrow from IFrag−/− and not RAG−/− mice is striking. Although type 1 IFNs are primarily considered a proinflammatory cytokine,9 more recent research clearly identified their importance in immune modulation and down-regulation of inflammation. In this regard, type 1 IFNs have been shown to induce IL-10 production via STAT-1–mediated pathways65 and also directly suppress TNF-α gene transcription via the induction of TWIST transcriptional repressors.66 Furthermore, they interfere with IL-1β maturation by repressing the activity of NLRP1 and NLRP3 inflammasomes.67 The profound deviation of the cytokine profile in bone marrow from IFrag−/− compared with RAG−/− mice stresses the importance of type 1 IFNs in the regulation of the systemic response to Pneumocystis lung infection and how it may affect tissue homeostasis at distant organ sites.
Type 1 IFNs are known to negatively regulate osteoclast maturation via direct interference in the RANKL/RANK-signaling pathway by blocking c-FOS protein synthesis and thus disrupting further differentiation signals.68 This interference would be consistent with the phenotype of osteoporosis in our model, except that uninfected IFrag−/− mice did not seem to have significantly accelerated bone loss when compared with uninfected RAG−/− mice, and the differences between the groups evolved in response to Pneumocystis lung infection. Surprisingly, protein levels for the RANKL decoy receptor OPG were also greatly induced in bone marrow from IFrag−/− but not in RAG−/− mice in response to Pneumocystis lung infection. High OPG levels should be able to interfere with RANKL/RANK interaction on preosteoclasts and impede their differentiation, independent of the inhibitory effects of type 1 IFN-mediated signals.48 However, OPG also functions as a decoy receptor for the proapoptotic cytokine TRAIL,55 which is considered a regulator of bone marrow cell homeostasis by also inducing apoptosis in senescent neutrophils.69 Furthermore, sensitivity to TRAIL-mediated cytotoxicity may increase under pathologic conditions.55,69,70
Bone marrow failure and osteoporosis occur concomitantly in our model in response to Pneumocystis lung infection, and an imbalance among RANKL, OPG, and TRAIL may be linked to these phenotypes. Indeed, TRAIL protein concentrations, in conjunction with TNF-α and IL-1, were elevated only in bone marrow from IFrag−/− mice starting at day 7 after infection and thus preceded both bone marrow failure and accelerated bone loss. These data imply that type 1 IFN signaling may negatively regulate not only TNF-α and IL-1 but also TRAIL.
Bone marrow failure in IFrag−/− mice is associated with loss of precursor cell activity and numbers.43 The HSC resides in a protected niche along the trabecular bone, and loss of bone structure is associated with loss of HSC numbers.71 This finding raised the question of what extent the evolving bone loss itself may contribute to bone marrow failure in IFrag−/− mice after Pneumocystis lung infection. Bisphosphonate treatment effectively inhibited osteoclast activity and increased trabecular bone numbers and volume of IFrag−/− mice during Pneumocystis lung infection compared with untreated, infected mice. However, the visual effect on bone marrow failure progression was minimal because total bone marrow cell numbers still rapidly decreased. Nevertheless, although band neutrophils and myelocytes disappeared from the marrow space (data not shown), the HPC activity (measured by CFC assays) was significantly improved in treated compared with untreated day 16 IFrag−/− mice, suggesting a protective role of bone with regard to early progenitor cell viability. Interestingly, we previously demonstrated that treatment of IFrag−/− mice with an anti-TNF-α antibody during infection had no effect on neutrophil numbers but improved precursor cell activity.43 Given the supportive role of TNF-α in osteoclast maturation, the mechanisms underlying the partial effect of TNF blockage on improved HPC activity may potentially be due to effects on bone metabolism rather than inhibition of apoptotic effects of TNF-α. The loss of more mature bone marrow cells may be due to an additional lack of growth factors, as previously reported,43 and apoptotic activity of other mediators, such as TRAIL. Experiments aiming to block TRAIL activity in vivo using neutralizing antibody treatment also showed only a partial effect on the progression of bone marrow failure, supporting the notion of a multifactorial pathogenesis. However, assays performed to determine the TRAIL neutralization efficiency within the bone marrow, using ELISA with DR5-TRAIL receptor as a capture reagent, revealed only partial neutralization, with concentrations well above baseline levels (data not shown). Therefore, other means to more efficiently block TRAIL activity (eg, TRAIL receptor blocking antibodies or OPG administration) are needed to further assess the exact role of TRAIL in bone marrow failure and bone loss in our model.
Together these data demonstrate that type 1 IFN signaling regulates multiple aspects of the systemic response to Pneumocystis lung infection, including bone marrow homeostasis and bone metabolism possibly involving direct regulation the RANKL/OPG/TRAIL axis in our mouse model of disease. A defect in type 1 IFN signaling and low-grade Pneumocystis lung infection are common in patients with HIV (undergoing HAART) and those with chronic lung disease undergoing corticosteroid treatment. Whether a deviated systemic immune response to Pneumocystis lung infection or colonization due to impaired type 1 IFN signaling could also accelerate osteoporosis in humans, and thus affect bone marrow functions, is worth investigating. Such a study could have implications for prevention and treatment strategies.
Acknowledgments
We thank Dr. David Pascual for critical reading of the manuscript.
Footnotes
Supported by National Institutes of Health grants RO1HL090488 and COBRE2P20RR020185-06.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at http://dx.doi.org/10.1016/j.ajpath.2012.03.023.
CME Disclosure: The authors of this article and the planning committee members and staff have no relevant financial relationships with commercial interest to disclose.
Supplementary data
References
- 1.Siegal F. Interferon-producing plasmacytoid dendritic cells and the pathogenesis of AIDS. Res Initiat Treat Action. 2003;8:10–13. [PubMed] [Google Scholar]
- 2.Levy J.A. The importance of the innate immune system in controlling HIV infection and disease. Trends Immunol. 2001;22:312–316. doi: 10.1016/s1471-4906(01)01925-1. [DOI] [PubMed] [Google Scholar]
- 3.Sepkowitz K.A. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin Infect Dis. 2002;34:1098–1107. doi: 10.1086/339548. [DOI] [PubMed] [Google Scholar]
- 4.Zandman-Goddard G., Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329–337. doi: 10.1016/s1568-9972(02)00086-1. [DOI] [PubMed] [Google Scholar]
- 5.Isgro A., Mezzaroma I., Aiuti A., De Vita L., Franchi F., Pandolfi F., Alario C., Ficara F., Riva E., Antonelli G., Aiuti F. Recovery of hematopoietic activity in bone marrow from human immunodeficiency virus type 1-infected patients during highly active antiretroviral therapy. AIDS Res Hum Retroviruses. 2000;16:1471–1479. doi: 10.1089/088922200750005994. [DOI] [PubMed] [Google Scholar]
- 6.Moses A., Nelson J., Bagby G.C., Jr The influence of human immunodeficiency virus-1 on hematopoiesis. Blood. 1998;91:1479–1495. [PubMed] [Google Scholar]
- 7.Wolbers M., Babiker A., Sabin C., Young J., Dorrucci M., Chene G., Mussini C., Porter K., Bucher H.C. Pretreatment CD4 cell slope and progression to AIDS or death in HIV-infected patients initiating antiretroviral therapy–the CASCADE collaboration: a collaboration of 23 cohort studies. PLoS Med. 2010;7:e1000239. doi: 10.1371/journal.pmed.1000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolbers M., Battegay M., Hirschel B., Furrer H., Cavassini M., Hasse B., Vernazza P.L., Bernasconi E., Kaufmann G., Bucher H.C. CD4+ T-cell count increase in HIV-1-infected patients with suppressed viral load within 1 year after start of antiretroviral therapy. Antivir Ther. 2007;12:889–897. [PubMed] [Google Scholar]
- 9.Levy D.E., Marie I., Prakash A. Ringing the interferon alarm: differential regulation of gene expression at the interface between innate and adaptive immunity. Curr Opin Immunol. 2003;15:52–58. doi: 10.1016/s0952-7915(02)00011-0. [DOI] [PubMed] [Google Scholar]
- 10.van Boxel-Dezaire A.H., Rani M.R., Stark G.R. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 11.Levy J.A., Scott I., Mackewicz C. Protection from HIV/AIDS: the importance of innate immunity. Clin Immunol. 2003;108:167–174. doi: 10.1016/s1521-6616(03)00178-5. [DOI] [PubMed] [Google Scholar]
- 12.Soumelis V., Scott I., Gheyas F., Bouhour D., Cozon G., Cotte L., Huang L., Levy J.A., Liu Y.J. Depletion of circulating natural type 1 interferon-producing cells in HIV-infected AIDS patients. Blood. 2001;98:906–912. doi: 10.1182/blood.v98.4.906. [DOI] [PubMed] [Google Scholar]
- 13.Hosmalin A., Lebon P. Type I interferon production in HIV-infected patients. J Leukoc Biol. 2006;80:984–993. doi: 10.1189/jlb.0306154. [DOI] [PubMed] [Google Scholar]
- 14.Catalfamo M., Wilhelm C., Tcheung L., Proschan M., Friesen T., Park J.H., Adelsberger J., Baseler M., Maldarelli F., Davey R., Roby G., Rehm C., Lane C. CD4 and CD8 T cell immune activation during chronic HIV infection: roles of homeostasis HIV, type I IFN, and IL-7. J Immunol. 2011;186:2106–2116. doi: 10.4049/jimmunol.1002000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boasso A., Hardy A.W., Anderson S.A., Dolan M.J., Shearer G.M. HIV-induced type I interferon and tryptophan catabolism drive T cell dysfunction despite phenotypic activation. PLoS One. 2008;3:e2961. doi: 10.1371/journal.pone.0002961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chehimi J., Campbell D.E., Azzoni L., Bacheller D., Papasavvas E., Jerandi G., Mounzer K., Kostman J., Trinchieri G., Montaner L.J. Persistent decreases in blood plasmacytoid dendritic cell number and function despite effective highly active antiretroviral therapy and increased blood myeloid dendritic cells in HIV-infected individuals. J Immunol. 2002;168:4796–4801. doi: 10.4049/jimmunol.168.9.4796. [DOI] [PubMed] [Google Scholar]
- 17.Corbeau P., Reynes J. Immune reconstitution under antiretroviral therapy: the new challenge in HIV-1 infection. Blood. 2011;117:5582–5590. doi: 10.1182/blood-2010-12-322453. [DOI] [PubMed] [Google Scholar]
- 18.Siegal F.P., Fitzgerald-Bocarsly P., Holland B.K., Shodell M. Interferon-alpha generation and immune reconstitution during antiretroviral therapy for human immunodeficiency virus infection. AIDS. 2001;15:1603–1612. doi: 10.1097/00002030-200109070-00002. [DOI] [PubMed] [Google Scholar]
- 19.Hull M.W., Phillips P., Montaner J.S. Changing global epidemiology of pulmonary manifestations of HIV/AIDS. Chest. 2008;134:1287–1298. doi: 10.1378/chest.08-0364. [DOI] [PubMed] [Google Scholar]
- 20.Grubb J.R., Moorman A.C., Baker R.K., Masur H. The changing spectrum of pulmonary disease in patients with HIV infection on antiretroviral therapy. AIDS. 2006;20:1095–1107. doi: 10.1097/01.aids.0000226949.64600.f9. [DOI] [PubMed] [Google Scholar]
- 21.Chew N.S., Doran P.P., Powderly W.G. Osteopenia and osteoporosis in HIV: pathogenesis and treatment. Curr Opin HIV AIDS. 2007;2:318–323. doi: 10.1097/COH.0b013e3281a3c092. [DOI] [PubMed] [Google Scholar]
- 22.Barkhordarian A., Ajaj R., Ramchandani M.H., Demerjian G., Cayabyab R., Danaie S., Ghodousi N., Iyer N., Mahanian N., Phi L., Giroux A., Manfrini E., Neagos N., Siddiqui M., Cajulis O.S., Brant X.M., Shapshak P., Chiappelli F. Osteoimmunopathology in HIV/AIDS: a translational evidence-based perspective. Patholog Res Int. 2011;2011:359242. doi: 10.4061/2011/359242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bongiovanni M., Tincati C. Bone diseases associated with human immunodeficiency virus infection: pathogenesis, risk factors and clinical management. Curr Mol Med. 2006;6:395–400. doi: 10.2174/156652406777435435. [DOI] [PubMed] [Google Scholar]
- 24.Walsh M.C., Kim N., Kadono Y., Rho J., Lee S.Y., Lorenzo J., Choi Y. Osteoimmunology: interplay between the immune system and bone metabolism. Annu Rev Immunol. 2006;24:33–63. doi: 10.1146/annurev.immunol.24.021605.090646. [DOI] [PubMed] [Google Scholar]
- 25.Takayanagi H. Osteoimmunology and the effects of the immune system on bone. Nat Rev Rheumatol. 2009;5:667–676. doi: 10.1038/nrrheum.2009.217. [DOI] [PubMed] [Google Scholar]
- 26.Wilson A., Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6:93–106. doi: 10.1038/nri1779. [DOI] [PubMed] [Google Scholar]
- 27.Adams G.B., Scadden D.T. The hematopoietic stem cell in its place. Nat Immunol. 2006;7:333–337. doi: 10.1038/ni1331. [DOI] [PubMed] [Google Scholar]
- 28.Yin T., Li L. The stem cell niches in bone. J Clin Invest. 2006;116:1195–1201. doi: 10.1172/JCI28568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mundy G.R. Osteoporosis and inflammation. Nutr Rev. 2007;65:S147–S151. doi: 10.1111/j.1753-4887.2007.tb00353.x. [DOI] [PubMed] [Google Scholar]
- 30.Kamen D.L., Alele J.D. Skeletal manifestations of systemic autoimmune diseases. Curr Opin Endocrinol Diabetes Obes. 2010;17:540–545. doi: 10.1097/MED.0b013e328340533d. [DOI] [PubMed] [Google Scholar]
- 31.Huertas A., Palange P. COPD: a multifactorial systemic disease. Ther Adv Respir Dis. 2011;5:217–224. doi: 10.1177/1753465811400490. [DOI] [PubMed] [Google Scholar]
- 32.Palange P., Testa U., Huertas A., Calabro L., Antonucci R., Petrucci E., Pelosi E., Pasquini L., Satta A., Morici G., Vignola M.A., Bonsignore M.R. Circulating haemopoietic and endothelial progenitor cells are decreased in COPD. Eur Respir J. 2006;27:529–541. doi: 10.1183/09031936.06.00120604. [DOI] [PubMed] [Google Scholar]
- 33.Brown T.T., Qaqish R.B. Antiretroviral therapy and the prevalence of osteopenia and osteoporosis: a meta-analytic review. AIDS. 2006;20:2165–2174. doi: 10.1097/QAD.0b013e32801022eb. [DOI] [PubMed] [Google Scholar]
- 34.Gibellini D., Borderi M., De Crignis E., Cicola R., Vescini F., Caudarella R., Chiodo F., Re M.C. RANKL/OPG/TRAIL plasma levels and bone mass loss evaluation in antiretroviral naive HIV-1-positive men. J Med Virol. 2007;79:1446–1454. doi: 10.1002/jmv.20938. [DOI] [PubMed] [Google Scholar]
- 35.Yong M.K., Elliott J.H., Woolley I.J., Hoy J.F. Low CD4 count is associated with an increased risk of fragility fracture in HIV-infected patients. J Acquir Immune Defic Syndr. 2011;57:205–210. doi: 10.1097/QAI.0b013e31821ecf4c. [DOI] [PubMed] [Google Scholar]
- 36.Kaur N., Mahl T.C. Pneumocystis jiroveci (carinii) pneumonia after infliximab therapy: a review of 84 cases. Dig Dis Sci. 2007;52:1481–1484. doi: 10.1007/s10620-006-9250-x. [DOI] [PubMed] [Google Scholar]
- 37.Kaur N., Mahl T.C. Pneumocystis carinii pneumonia with oral candidiasis after infliximab therapy for Crohn's disease. Dig Dis Sci. 2004;49:1458–1460. doi: 10.1023/b:ddas.0000042246.58984.98. [DOI] [PubMed] [Google Scholar]
- 38.Komano Y., Harigai M., Koike R., Sugiyama H., Ogawa J., Saito K., Sekiguchi N., Inoo M., Onishi I., Ohashi H., Amamoto F., Miyata M., Ohtsubo H., Hiramatsu K., Iwamoto M., Minota S., Matsuoka N., Kageyama G., Imaizumi K., Tokuda H., Okochi Y., Kudo K., Tanaka Y., Takeuchi T., Miyasaka N. Pneumocystis jiroveci pneumonia in patients with rheumatoid arthritis treated with infliximab: a retrospective review and case-control study of 21 patients. Arthritis Rheum. 2009;61:305–312. doi: 10.1002/art.24283. [DOI] [PubMed] [Google Scholar]
- 39.Morris A., Alexander T., Radhi S., Lucht L., Sciurba F.C., Kolls J.K., Srivastava R., Steele C., Norris K.A. Airway obstruction is increased in pneumocystis-colonized human immunodeficiency virus-infected outpatients. J Clin Microbiol. 2009;47:3773–3776. doi: 10.1128/JCM.01712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flammer JR, Dobrovolna J, Kennedy MA, Chinenov Y, Glass CK, Ivashkiv LB, Rogatsky I. The type I interferon signaling pathway is a target for glucocorticoid inhibition. Mol Cell Biol 30:4564–4574 [DOI] [PMC free article] [PubMed]
- 41.Bhattacharyya S., Zhao Y., Kay T.W., Muglia L.J. Glucocorticoids target suppressor of cytokine signaling 1 (SOCS1) and type 1 interferons to regulate Toll-like receptor-induced STAT1 activation. Proc Natl Acad Sci U S A. 2011;108:9554–9559. doi: 10.1073/pnas.1017296108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meissner N., Rutkowski M., Harmsen A.L., Han S., Harmsen A.G. Type I interferon signaling and B cells maintain hemopoiesis during Pneumocystis infection of the lung. J Immunol. 2007;178:6604–6615. doi: 10.4049/jimmunol.178.10.6604. [DOI] [PubMed] [Google Scholar]
- 43.Taylor D., Wilkison M., Voyich J., Meissner N. Prevention of bone marrow cell apoptosis and regulation of hematopoiesis by type I IFNs during systemic responses to pneumocystis lung infection. J Immunol. 2011;186:5956–5967. doi: 10.4049/jimmunol.1003558. [DOI] [PubMed] [Google Scholar]
- 44.Kanzaki S., Takada Y., Ogawa K., Matsuo K. Bisphosphonate therapy ameliorates hearing loss in mice lacking osteoprotegerin. J Bone Miner Res. 2009;24:43–49. doi: 10.1359/jbmr.080812. [DOI] [PubMed] [Google Scholar]
- 45.Harmsen A.G., Stankiewicz M. Requirement for CD4+ cells in resistance to Pneumocystis carinii pneumonia in mice. J Exp Med. 1990;172:937–945. doi: 10.1084/jem.172.3.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bolliger A.P. Cytologic evaluation of bone marrow in rats: indications, methods, and normal morphology. Vet Clin Pathol. 2004;33:58–67. doi: 10.1111/j.1939-165x.2004.tb00351.x. [DOI] [PubMed] [Google Scholar]
- 47.Tintut Y., Parhami F., Tsingotjidou A., Tetradis S., Territo M., Demer L.L. 8-Isoprostaglandin E2 enhances receptor-activated NFkappa B ligand (RANKL)-dependent osteoclastic potential of marrow hematopoietic precursors via the cAMP pathway. J Biol Chem. 2002;277:14221–14226. doi: 10.1074/jbc.M111551200. [DOI] [PubMed] [Google Scholar]
- 48.Simonet W.S., Lacey D.L., Dunstan C.R., Kelley M., Chang M.S., Luthy R., Nguyen H.Q., Wooden S., Bennett L., Boone T., Shimamoto G., DeRose M., Elliott R., Colombero A., Tan H.L., Trail G., Sullivan J., Davy E., Bucay N., Renshaw-Gegg L., Hughes T.M., Hill D., Pattison W., Campbell P., Sander S., Van G., Tarpley J., Derby P., Lee R., Boyle W.J. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 49.Parfitt A.M. Bone histomorphometry: proposed system for standardization of nomenclature, symbols, and units. Calcif Tissue Int. 1988;42:284–286. doi: 10.1007/BF02556360. [DOI] [PubMed] [Google Scholar]
- 50.Parfitt A.M., Drezner M.K., Glorieux F.H., Kanis J.A., Malluche H., Meunier P.J., Ott S.M., Recker R.R. Bone histomorphometry: standardization of nomenclature, symbols, and units: report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 51.Bagi C.M., Hanson N., Andresen C., Pero R., Lariviere R., Turner C.H., Laib A. The use of micro-CT to evaluate cortical bone geometry and strength in nude rats: correlation with mechanical testing, pQCT and DXA. Bone. 2006;38:136–144. doi: 10.1016/j.bone.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 52.Taichman R.S. Blood and bone: two tissues whose fates are intertwined to create the hematopoietic stem-cell niche. Blood. 2005;105:2631–2639. doi: 10.1182/blood-2004-06-2480. [DOI] [PubMed] [Google Scholar]
- 53.Kollet O., Dar A., Lapidot T. The multiple roles of osteoclasts in host defense: bone remodeling and hematopoietic stem cell mobilization. Annu Rev Immunol. 2007;25:51–69. doi: 10.1146/annurev.immunol.25.022106.141631. [DOI] [PubMed] [Google Scholar]
- 54.Min H., Morony S., Sarosi I., Dunstan C.R., Capparelli C., Scully S., Van G., Kaufman S., Kostenuik P.J., Lacey D.L., Boyle W.J., Simonet W.S. Osteoprotegerin reverses osteoporosis by inhibiting endosteal osteoclasts and prevents vascular calcification by blocking a process resembling osteoclastogenesis. J Exp Med. 2000;192:463–474. doi: 10.1084/jem.192.4.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Secchiero P., Zauli G. Tumor-necrosis-factor-related apoptosis-inducing ligand and the regulation of hematopoiesis. Curr Opin Hematol. 2008;15:42–48. doi: 10.1097/MOH.0b013e3282f15fa6. [DOI] [PubMed] [Google Scholar]
- 56.Wu Y., Humphrey M.B., Nakamura M.C. Osteoclasts: the innate immune cells of the bone. Autoimmunity. 2008;41:183–194. doi: 10.1080/08916930701693180. [DOI] [PubMed] [Google Scholar]
- 57.Harada S., Rodan G.A. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–355. doi: 10.1038/nature01660. [DOI] [PubMed] [Google Scholar]
- 58.Takayanagi H., Kim S., Taniguchi T. Signaling crosstalk between RANKL and interferons in osteoclast differentiation. Arthritis Res. 2002;4(Suppl 3):S227–S232. doi: 10.1186/ar581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morris A., Sciurba F.C., Lebedeva I.P., Githaiga A., Elliott W.M., Hogg J.C., Huang L., Norris K.A. Association of chronic obstructive pulmonary disease severity and pneumocystis colonization. Am J Respir Crit Care Med. 2004;170:408–413. doi: 10.1164/rccm.200401-094OC. [DOI] [PubMed] [Google Scholar]
- 60.Morris A., Sciurba F.C., Norris K.A. Pneumocystis: a novel pathogen in chronic obstructive pulmonary disease. COPD. 2008;5:43–51. doi: 10.1080/1541255070181756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Isgro A., Aiuti A., Leti W., Gramiccioni C., Esposito A., Mezzaroma I., Aiuti F. Immunodysregulation of HIV disease at bone marrow level. Autoimmun Rev. 2005;4:486–490. doi: 10.1016/j.autrev.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 62.Isgro A., Aiuti A., Mezzaroma I., Ruco L., Pinti M., Cossarizza A., Aiuti F. HIV type 1 protease inhibitors enhance bone marrow progenitor cell activity in normal subjects and in HIV type 1-infected patients. AIDS Res Hum Retroviruses. 2005;21:51–57. doi: 10.1089/aid.2005.21.51. [DOI] [PubMed] [Google Scholar]
- 63.Suda T., Tanaka S., Takahashi N. Macrophage colon-stimulating factor (M-CSF) is essential for differentiation rather than proliferation of osteoclast progenitors. Osteoporos Int. 1993;3(Suppl 1):111–113. doi: 10.1007/BF01621881. [DOI] [PubMed] [Google Scholar]
- 64.Tanaka S., Nakamura K., Takahasi N., Suda T. Role of RANKL in physiological and pathological bone resorption and therapeutics targeting the RANKL-RANK signaling system. Immunol Rev. 2005;208:30–49. doi: 10.1111/j.0105-2896.2005.00327.x. [DOI] [PubMed] [Google Scholar]
- 65.Chang E.Y., Guo B., Doyle S.E., Cheng G. Cutting edge: involvement of the type I IFN production and signaling pathway in lipopolysaccharide-induced IL-10 production. J Immunol. 2007;178:6705–6709. doi: 10.4049/jimmunol.178.11.6705. [DOI] [PubMed] [Google Scholar]
- 66.Sharif M.N., Sosic D., Rothlin C.V., Kelly E., Lemke G., Olson E.N., Ivashkiv L.B. Twist mediates suppression of inflammation by type I IFNs and Axl. J Exp Med. 2006;203:1891–1901. doi: 10.1084/jem.20051725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guarda G., Braun M., Staehli F., Tardivel A., Mattmann C., Forster I., Farlik M., Decker T., Du Pasquier R.A., Romero P., Tschopp J. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–223. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 68.Takayanagi H., Kim S., Matsuo K., Suzuki H., Suzuki T., Sato K., Yokochi T., Oda H., Nakamura K., Ida N., Wagner E.F., Taniguchi T. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature. 2002;416:744–749. doi: 10.1038/416744a. [DOI] [PubMed] [Google Scholar]
- 69.Lum J.J., Bren G., McClure R., Badley A.D. Elimination of senescent neutrophils by TNF-related apoptosis-inducing [corrected] ligand. J Immunol. 2005;175:1232–1238. doi: 10.4049/jimmunol.175.2.1232. [DOI] [PubMed] [Google Scholar]
- 70.Kakagianni T., Giannakoulas N.C., Thanopoulou E., Galani A., Michalopoulou S., Kouraklis-Symeonidis A., Zoumbos N.C. A probable role for trail-induced apoptosis in the pathogenesis of marrow failure: Implications from an in vitro model and from marrow of aplastic anemia patients. Leuk Res. 2006;30:713–721. doi: 10.1016/j.leukres.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 71.Calvi L.M., Adams G.B., Weibrecht K.W., Weber J.M., Olson D.P., Knight M.C., Martin R.P., Schipani E., Divieti P., Bringhurst F.R., Milner L.A., Kronenberg H.M., Scadden D.T. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.