

Abstract
The role of apoptosis in the formation and regression of neovascularization is largely hypothesized, although the detailed mechanism remains unclear. Inflammatory cells and endothelial cells both participate and interact during neovascularization. During the early stage, these cells may migrate into an angiogenic site and form a pro-angiogenic microenvironment. Some angiogenic vessels appear to regress, whereas some vessels mature and remain. The control mechanisms of these processes, however, remain unknown. Previously, we reported that the prevention of mitochondrial apoptosis contributed to cellular survival via the prevention of the release of proapoptotic factors, such as apoptosis-inducing factor (AIF) and cytochrome c. In this study, we investigated the regulatory role of cellular apoptosis in angiogenesis using two models of ocular neovascularization: laser injury choroidal neovascularization and VEGF-induced corneal neovascularization in AIF-deficient mice. Averting apoptosis in AIF-deficient mice decreased apoptosis of leukocytes and endothelial cells compared to wild-type mice and resulted in the persistence of these cells at angiogenic sites in vitro and in vivo. Consequently, AIF deficiency expanded neovascularization and diminished vessel regression in these two models. We also observed that peritoneal macrophages from AIF-deficient mice showed anti-apoptotic survival compared to wild-type mice under conditions of starvation. Our data suggest that AIF-related apoptosis plays an important role in neovascularization and that mitochondria-regulated apoptosis could offer a new target for the treatment of pathological angiogenesis.
The mitochondrial apoptosis pathways are important mechanisms of cell death.1 Mitochondria contain proapoptotic factors such as cytochrome c and apoptosis-inducing factor (AIF) in their intermembrane space. Furthermore, mitochondrial outer membrane permeabilization is a critical event during apoptosis, representing the “point of no return” of the lethal process. Cytochrome c is released from mitochondria on mitochondrial outer membrane permeabilization and binds to cytosolic apoptotic protease activating factor-1 to induce its dimerization and a conformational change.2 Apoptotic protease activating factor-1 then oligomerizes into apoptosomes that recruit and activate caspase-9 followed by serial activation of apoptosis-execution molecules.3,4 Mitochondrial outer membrane permeabilization, however, may cause cell death even if caspases are inhibited5 and a broad caspase inhibitor (z-VAD-fmk) fails to block apoptosis in retinal neurons.6 AIF is a caspase-independent apoptogenic factor and is normally confined to the mitochondrial intermembrane space.7 Most cell death in vertebrates proceeds via the mitochondrial pathway of apoptosis, especially in mammalian cells.8,9 During apoptosis, AIF translocates to the cytosol and then to the nucleus where it triggers peripheral chromatin condensation and interacts with cyclophilin A to generate a DNase complex, which is responsible for the so-called “large-scale” DNA degradation to fragments of approximately 50 kbp.10 AIF is strongly conserved among mammalian species (>95% amino acid identity between mouse and human) and bears a highly significant homology with flavoprotein oxidoreductases from all eukaryotic and prokaryotic kingdoms in its C-terminal portion.7 Because AIF, a central player in mitochondrial apoptotic pathways is essential in the developmental process, AIF knockout mice die in utero.11 Based on these findings, it is reasonable to speculate that AIF may be a phylogenetically old major mediator participating in various aspects of the apoptotic process. Because we originally reported AIF translocation in mammalian cells in vivo in retinal cell death,6,9,12–16 the translocation of AIF has been reported in neurodegeneration17,18 and retinal degeneration.19 The contribution of apoptosis, especially phylogenetically old major factors (ie, AIF), however, has remained elusive in the field of neovascularization.
Choroidal neovascularization (CNV) is a pathological process involving the formation of new blood vessels from choroidal vasculature through Bruch's membrane breaks. CNV is associated with a variety of ocular diseases, including age-related macular degeneration (AMD), myopia, histoplasmosis, angioid streaks, tumors, and traumatic and idiopathic conditions, all of which often cause severe visual loss via retinal degeneration. CNV could be induced by focally increased inflammatory and proangiogenic factors, and/or by a decrease of anti-angiogenic factors. Various clinical, as well as experimental, studies have shown that vascular endothelial growth factor (VEGF)-A could be the most important factor for CNV.20 Recent observations in age-related macular degeneration patients with VEGF-A inhibition strongly support the importance in CNV. In CNV, macrophages may be major sources of VEGF-A, which would enhance vascular leakage, as well as angiogenesis via vascular endothelial growth factor receptor (VEGFR)-2.21 Macrophage also expresses VEGFR-1 and VEGF-A that may induce macrophage infiltration. Thus, VEGF is an inflammatory cytokine targeting both leukocytes and endothelial cells. Various studies have shown attachment of mural cells is important for the vascular stability that is dependent on angiopoietin/Tie system and VEGF.22 Tie2 is known to play a direct role in pericyte recruitment and Tie2-knockout blood vessels that lack mural cells.23 The loss of periendothelial cells in the mutants is secondary to endothelial cell apoptosis.24
The efficient clearance of excessive inflammatory cells and neovascular endothelial cells from the pathological sites may be essential for restoration of tissue homeostasis.13 The regulation of apoptosis in angiogenesis-related cells, including leukocytes and endothelial cells, may occur in various disorders. The detailed mechanism, however, remains unclear.25 In this study, we focused on the roles of a major proapoptotic molecule, AIF in the formation and regression of neovascularization.
Materials and Methods
Experimental Animals
All animal procedures were performed in accordance with the statement of the Association for Research in Vision and Ophthalmology and the protocol approved by the Animal Care Committee of Massachusetts Eye and Ear Infirmary. The AIF mutant mice (B6CBACa Aw-J/A-Aifm1Hq/J, stock number 000501; Jackson Laboratory, Bar Harbor, ME) and wild type (WT) from the colony were purchased from the Jackson Laboratory and bred in our laboratory. Adult male mice (8 weeks of age) were used for the following experiments.
Laser Injury-Induced Choroidal Neovascularization
Mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg). Pupils were dilated with 5.0% phenylephrine and 0.8% tropicamide. CNV was induced with a 532 nm laser (Oculight GLx, Iridex, Mountain View, CA) as previously described.20,26–31 Four laser spots (150 mW, 100 msec, 50 μm settings) were placed in each eye using a slit-lamp delivery system and a cover glass as a contact lens (n = 16 for each time point and strain). Production of a bubble at the time of laser confirmed the rupture of the Bruch's membrane. The vessels were stained with fluorescein isothiocyanate-dextran. The eyes were enucleated at 2, 4, and 12 weeks after laser injury and fixed in 4% paraformaldehyde for 3 hours. The anterior segment and retina were removed from the eyecup. Approximately four to six relaxing radial incisions were made, and the remaining retinal pigment epithelial-choroidal-scleral complex was flat mounted and coverslipped. Pictures of the choroidal flat mounts were taken, and Openlab software version 5 (Improvision, Boston, MA) was used to measure the hyperfluorescent areas corresponding to the CNV lesions. The average size of the CNV lesions was then determined and used for the evaluation.
Bone Marrow Transplantation
To characterize the angiogenic roles of infiltrating macrophages, we produced chimera green fluorescent protein (GFP) mice, by a previously described method.13,32 Briefly, we used the transgenic mouse as a cell source for enhanced GFP (EGFP)-positive bone marrow cells. The host mice took 1,4-butanediol dimethanesulfonate, Busulfan 4 × 25 mg/kg on 4 consecutive days followed by bone marrow transplantation (BMT) to deplete stem cells in the host and consequently allow for high levels of long-term, donor-type engraftment. A successful bone marrow transplantation was confirmed by the identification of GFP-positive cells in the blood at 2 and 4 weeks after BMT. The WT and AIF-deficient mice took BMT from EGFP transgenic mice and laser injury 4 weeks after BMT (n = 10, each group). The eyes were harvested and examined 2 weeks after laser injury, as previously described. To visualize EGFP-positive macrophage in the CNV, the vessels were stained with rhodamine-conjugated concanavalin A.
Corneal Micropocket Assay in Mice
Mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg). Poly 2-hydroxyethyl methacrylate pellets (0.3 μL, P3932; Sigma-Aldrich, St. Louis, MO) containing 200 ng VEGF-A (293-VE; R&D Systems, Minneapolis, MN) were prepared and implanted into the corneas. VEGF-A pellets were positioned at approximately 1 mm distant to the corneal limbus. After implantation, bacitracin ophthalmic ointment (E. Fougera & Co., Melville, NY) was applied to each eye to prevent infection. At 6 days (n = 6, each group), 10 days (n = 8), 14 days (n = 10), 28 days (n = 8), 56 days (n = 10), 84 days (n = 5), and 140 days (n = 3) after implantation, digital images of the corneal vessels were obtained and recorded using OpenLab software version (Improvision Inc., Lexington, MA) with standardized illumination and contrast. The quantitative analysis of neovascularization in the mouse corneas was performed using Scion Image software (version 4.0.2; Scion Corp., Frederick, MD).
TUNEL Assay
TUNEL and quantification of TUNEL+ cells were performed as previously described,6 using the ApopTag Fluorescein in Situ Apoptosis Detection Kit (Chemicon/Millipore, Bedford, MA). The center of the choroidal and corneal neovascularization was photographed, and the number of TUNEL+ cells in the microscopic field was counted in a masked fashion. The results are presented as the mean ± SD.
Immunohistochemistry
The eyes were harvested and snap-frozen in optimal cutting temperature compound (Sakura Finetechnical). Sections (10 μm) were prepared, air dried, and fixed in ice-cold acetone for 10 minutes. The sections were blocked with 3% nonfat dried milk bovine working solution (M7409; Sigma-Aldrich) and stained with a macrophage marker, anti-mouse CD11b (1:50, BD Pharmingen, San Diego, CA) or an endothelial marker, anti-mouse CD31 mAb (1:50; BD Pharmingen), a myeloid lineage marker, CD45 (1:100, BD Pharmingen), VEGF, TNF-α (1:100, Santa Cruz Biotechnology, Santa Cruz, CA), basic fibroblast growth factor, IL-1β (Abcam, Cambridge, MA), and rabbit anti-AIF (1:50, R&D Systems).33 After an overnight incubation, sections were washed and stained for 40 minutes with Alexa Fluor488 goat anti-rat IgG (20 μg/mL, A11006; Invitrogen). The specimens were observed with a fluorescent microscope and confocal microscope (Nikon, Tokyo, Japan).
Transmission Electron Microscopy
The eyes were enucleated and the posterior segments were fixed in 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 mol/L cacodylate buffer with 0.08 mol/L CaCl2 at 4°C. The specimens were postfixed for 1.5 hours in 2% aqueous OsO4, dehydrated in ethanol and water, and embedded in Epon (Electron Microscopy Sciences, Hatfield, PA). Ultrathin sections were cut from blocks and stained with saturated, aqueous uranyl acetate and Sato's lead stain. The specimens were observed with a Hitachi H-7650 electron microscope.
Fluorescein Angiography in CNV Model
Fluorescein angiography was performed by an operator masked to the genetic identity of the animal, with a commercial camera and imaging system (TRC 50 VT camera and IMAGEnet 1.53 system; Topcon, Paramus, NJ), at 2 weeks after laser photocoagulation (WT mice; n = 12, AIF-deficient mice; n = 12). The photographs were captured with a 20-dimensional lens in contact with the fundus camera lens, after intraperitoneal injection of 0.1 mL of 1% fluorescein sodium (Akorn, Decatur, IL). Two masked retina specialists evaluated the fluorescein angiograms at a single sitting. Lesions were graded on an ordinal scale based on the spatial and temporal evolution of fluorescein leakage as follows: grade 0 (nonleaky): no leakage, faint hyperfluorescence, or mottled fluorescence without leakage; grade1 (questionable leakage): hyperfluorescent lesion without progressive increase in size or intensity; grade 2A (leaky): hyperfluorescence increasing in intensity but not in size, no definite leakage; grade 2B (pathologically significant leakage): hyperfluorescence increasing in intensity and in size, definite leakage.34
Macrophage Culture from AIF-Deficient Mice
Briefly, peritoneal macrophages were collected by a previously described method.14,35,36 Peritoneal macrophages were stimulated by injecting Thioglycollate medium into the mouse peritoneal space and collected by washing peritoneal space with 5 mL PBS. Cell density was adjusted to 3.5 × 104 cells each well of an 8-well chamber (Nunc, Thermo Fisher Scientific, Rochester, NY) with Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. We counted the number of TUNEL+ apoptotic macrophages in the culture before and after starvation. For starvation, cells were cultured in Dulbecco's modified Eagle's medium without fetal bovine serum supplement and incubated for 24 hours. The number of TUNEL+ apoptotic macrophages was counted in 10 random fields per well in a masked fashion using ImageJ software version 1.38x (NIH, Bethesda, MD) in Harlequin (Hq) and WT mice. Values are given as the mean ± SEM of 10 replicate wells.
Statistical Analysis
The data from the TUNEL and in vitro survival assays were analyzed with the Scheffé post hoc test using StatView 4.11J software for Macintosh (Abacus Concepts Inc., Berkeley, CA). Significance level was set at P < 0.05 and P < 0.01. The data represent mean ± SD, except for primary culture results.
Results
Nuclear AIF Translocation in TUNEL+ Apoptotic Cells in the Mouse Choroidal Neovascularization
Laser injury-induced neovascular tissue formation from the choroid beneath the retina in 2 weeks in the WT mouse (Figure 1A). TUNEL+ apoptotic cells were observed in the retina, subretinal space, and choroidal neovascularization. Nuclear translocation (accumulation) of AIF was abundantly observed in TUNEL+ apoptotic cells in the subretinal space and the choroid (Figure 1B). In contrast, AIF is diffusely noted in the cytosol of the nonapoptotic normal cells. Confocal microscopy also confirmed co-localization of TUNEL and AIF in high resolution images (Figure 1, C–F). The electron microscopy showed the morphological characteristics of cellular apoptosis, namely chromatin condensation, cellular shrinkage, and apoptotic body formation (Figure 1, G and H).
Figure 1.

A: Apoptotic cell death and mitochondrio-nuclear translocation of AIF in choroidal neovascularization (CNV). The CNV developed from choroidal tissue into the subretinal space in the WT mouse. B: Abundant TUNEL+ cells are observed in the CNV (green), and these cells also showed nuclear accumulation of AIF (red). In contrast, diffuse weak staining of AIF is observed in the cytosol of the nonapoptotic normal cells. C–F: Confocal microscopy also confirms co-localization of TUNEL (C) and AIF (D) in high resolution images. Nuclei are stained blue (E); merge is also shown (F). G and H: The electron microscopy shows the morphological characteristics of cellular apoptosis, namely chromatin condensation, cellular shrinkage, and apoptotic body formation in the CNV. Original magnification: ×200 (A); ×400 (B–F); ×750 (G) and ×2500 (H). ONL, outer nuclear layer.
AIF Deficiency Promotes CNV Expansion and Retards Regression
Hq mice exhibit an X chromosome-linked ataxia due to the progressive degeneration of terminally differentiated cerebellar neurons.37,38 The Hq mutation has been identified as a proviral insertion in the apoptosis-inducing factor (Aif) gene (alias programmed cell death 8, Pdcd8), causing approximately an 80% reduction in AIF expression.38 In contrast to Aif knockout mice, which die in utero,11 Hq mice are born at normal Mendelian ratios and are healthy until the age of 3 months.
Laser-induced CNV in AIF-deficient mice led to lesions 1.7-fold larger than WT 2 weeks after laser (Figure 2, A–C). CNV also continues to grow in size in Hq mice to 4 weeks after laser, in contrast to WT mice (Figure 2C). CNV typically regresses rapidly in WT mice, but persists in Hq mice to 12 weeks. To test the regulatory roles of infiltrating macrophage in the CNV progression, we transplanted bone marrow cells from EGFP transgenic wt mice into WT and AIF-deficient mice 4 weeks before the laser injury (Figure 2, D and E). The EGFP-positive WT macrophages reconstructed from transplanted bone marrow migrated in the CNV (Figure 2D). The CNV size increased in AIF-deficient mice compared to WT mice, whereas the size was decreased in reverse in the AIF-deficient mice with WT bone marrow transplantation (Figure 2E). These experiments support that AIF deficiency resulted in enlarged CNV, which continued to grow and exhibited less regression relative to WT mice.
Figure 2.

Choroidal neovascularization observed by choroidal flat mount. CNV is visualized with fluorescent injection and analyzed with analysis software. AIF-deficient mice (B) induce larger CNV formation than normal WT mice (A). C: Even after 12 weeks AIF-deficient mice (black box) show persistent CNV, whereas WT mice (white box) show regression of formed CNV. D: The EGFP-positive WT macrophages reconstructed from transplanted bone marrow migrate in the CNV stained with rhodamine conjugated concanavalin A. E: The CNV size increased in AIF-deficient mice compared to WT mice; conversely, CNV size was decreased in AIF-deficient mice with WT bone marrow transplantation. *P < 0.05, **P < 0.01. Hq, Harlequin.
AIF Deficiency Decreases Apoptosis of Infiltrating Macrophages and Neovascular Endothelial Cells in CNV
Vertical histological sections also showed a large area of CNV in Hq mice in contrast to WT mice (Figure 3A). A large number of CD45-positive myeloid lineage cells migrated in the laser injury area and accumulated in the CNV. CD11b-positive macrophages were also present in CNV lesions (Figure 3B). In WT mice, TUNEL+ apoptotic cells were abundant among CD11b(+) macrophages and CD31(+) endothelial cells. AIF-deficient mice, however, showed fewer TUNEL+ apoptotic cells in the CNV. To test the roles of CD11b-positive macrophages, immunohistochemistry of CD11b and four major angiogenic or inflammatory factors, namely VEGF, TNF-α, basic fibroblast growth factor, and IL-1β, was examined in the CNV (Figure 3C). These four factors were expressed in CD11b positive macrophages. VEGF was strongly expressed in neovascular tissue, especially in the macrophages. These data suggest that AIF deficiency enhanced macrophage accumulation and angiogenic factor expression in the CNV.
Figure 3.

Immunohistochemistry for inflammatory and endothelial cells in CNV, which is observed at the laser injury site. A larger number of myeloid lineage cells (CD45) and macrophages (CD11b) migrate into CNV in apoptosis-inducing factor (AIF)-deficient mice compared to WT mice (A). TUNEL+ inflammatory and endothelial cells are abundant in WT mice compared to AIF-deficient mice (A, B). C: Immunohistochemistry of CD11b and four major angiogenic or inflammatory factors (VEGF, TNF-α, bFGF, and IL-1β) were examined in the CNV. These four factors are expressed in CD11b-positive macrophages. VEGF is strongly expressed in neovascular tissue; however, it is especially expressed in the macrophages. AIF-deficiency enhances macrophage accumulation and angiogenic factor expression in the CNV. bFGF, basic fibroblast growth factor; Hq/Y, harlequin hemizygous male mice.
AIF Deficiency Promotes CNV Leakage on Fluorescein Angiography
CNV leakage can be graded by fluorescein angiography, and in the clinical settting CNV leakage seems to correlate with CNV activity. AIF-deficient mice showed a trend toward a greater number of large, leaky, grade-2B lesions (denoting clinical significant leakage) than WT mice 2 weeks after laser (Figure 4).
Figure 4.
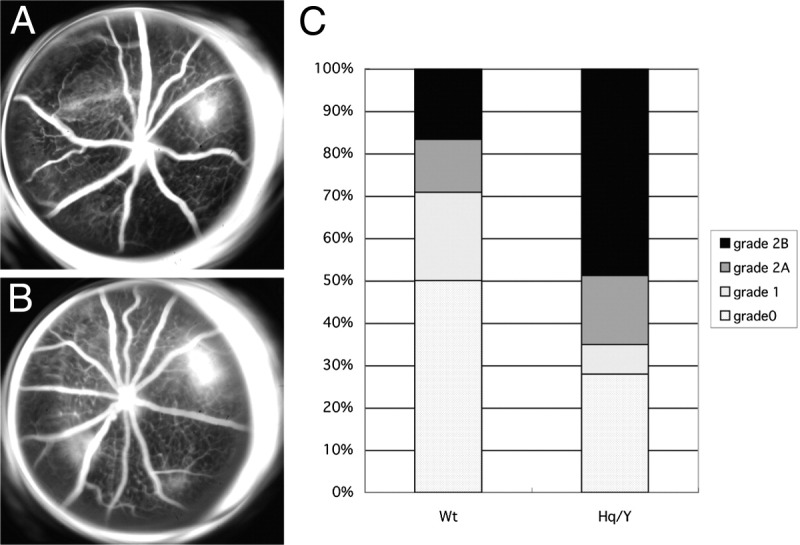
Fluorescein angiography of the laser-induced CNV. Compared to WT mice (A), AIF-deficient mice (B) developed larger CNV formation and more severe leakage. C: Lesions were graded on an ordinal scale based on the spatial and temporal evolution of fluorescein leakage as follows: grade 0 (nonleaky): no leakage, faint hyperfluorescence, or mottled fluorescence without leakage; grade 1 (questionable leakage): hyperfluorescent lesion without progressive increase in size or intensity; grade 2A (leaky): hyperfluorescence increasing in intensity but not in size; no definite leakage; grade 2B (pathologically significant leakage): hyperfluorescence increasing in intensity and in size; definite leakage. Hq/Y, harlequin hemizygous male mice.
AIF Deficiency Promotes Expansion of the Corneal Neovascularization and Retards Regression
Laser injury-induced choroidal neovascularization captures many of the important features of human conditions (ie, age-related macular degeneration), although this model includes various biological responses, such as thermal burn, tissue destruction, apoptosis and necrosis of retinal pigment epithelial cells, choroidal melanocytes, photoreceptors, or disruption of Bruch's membrane, and so forth. To clarify the role of AIF in VEGF-A–dependent angiogenesis, we used a simpler corneal pocket assay to induce angiogenesis in the corneas with AIF-deficient mice and WT mice. VEGF-A was implanted into these mice and the time course of VEGF-A–induced angiogenesis was followed until day 140. On day 6 after VEGF-A stimulation, the angiogenic area in AIF-deficient mice was similar with WT mice, but by day 10 AIF-deficient mice showed a significantly greater area of corneal neovascularization than WT mice (Figure 5). The peak of the corneal neovascularization shifted from day 28 in WT mice to day 56 in AIF-deficient mice. These data suggest that AIF may regulate VEGF-A-dependent angiogenesis at later stages of the process rather than the earlier step of angiogenic sprouting.
Figure 5.

A: VEGF-induced corneal neovascularization by corneal micropocket assay. B: Pellets were implanted in the cornea, and the quantitative analysis of neovascularization in the mouse corneas was performed using image software. AIF-deficient mice (black box) develop larger neovascularization than WT mice (white box). The regression of the formed neovascularization is retarded in AIF-deficient mice compared to WT mice (A, B). Hq/Y, harlequin hemizygous male mice.
AIF Deficiency Decreases Apoptosis of Infiltrating Macrophages and Neovascular Endothelial Cells in VEGF-A-Induced Corneal Neovascularization
To investigate how AIF could regulate VEGF-A-induced angiogenesis, we hypothesized that cellular apoptosis by AIF could regulate the angiogenesis and checked apoptotic cells in VEGF-A-implanted AIF-deficient and WT mice with TUNEL staining. Immunostaining showed abundant infiltrated inflammatory cells (CD11b) and migrated neovascular endothelial cells (CD31) in AIF-deficient mice compared to WT mice (Figure 6A). Less TUNEL+ cells in AIF-deficient mice could be observed than in WT mice at day 6 and at day 14 after VEGF-A implantation (Figure 6B). More TUNEL+ apoptotic cells are noted on day 6 (early) than on day 14 (late) in WT mice. In contrast, more apoptotic cells are observed on day 14 (late) in AIF-deficient mice. AIF deficiency retarded cellular apoptosis in corneal neovascularization. These data suggest AIF could be important for cellular apoptosis during VEGF-A-induced angiogenesis.
Figure 6.

Immunohistochemistry for inflammatory and endothelial cells in the corneal micropocket assay. A: Immunostaining shows abundant infiltrated inflammatory cells (CD11b) and migrated neovascular endothelial cells (CD31) in AIF-deficient mice compared to WT mice. B: More TUNEL+ apoptotic cells are observed in WT mice than in AIF-deficient mice. TUNEL+ apoptotic cells had a trend to decrease on day 14 (late) in WT mice. In contrast, apoptotic cells had a trend to increase on day 14 (late) in AIF-deficient mice. Hq/Y, harlequin hemizygous male mice.
Peritoneal Macrophages Are Resistant to Apoptosis in AIF-Deficient Mice
AIF deficiency promoted choroidal and corneal neovascularization, as well as accumulation of inflammatory cells (ie, CD45, 11b positive cells) at the local sites. To test the hypothesis that macrophages from AIF-deficient mice are resistant to apoptotic stimuli, we examined primary macrophage culture from Hq and WT mice. Peritoneal macrophages were collected and cultured with or without starvation. TUNEL+ apoptotic macrophage increased after starvation, and AIF translocation from the cytosol into the nuclei were noted in these cells undergoing apoptosis (Figure 7). AIF deficiency diminished AIF translocation and protected macrophages from apoptotic cell death.
Figure 7.

Peritoneal macrophage culture under starvation. Macrophages were collected from the peritoneal space and cultured using serum-free medium. A: WT mice show cytosolic (normal) and nuclear (apoptotic) staining for AIF, whereas AIF-deficient mice show no staining. B: TUNEL+ apoptotic macrophages increase after 24 hours of starvation in WT mice. In contrast, macrophages from AIF-deficient mice are resistant to starvation. *P < 0.05. Hq/Y, harlequin hemizygous male mice.
Discussion
AIF deficiency substantially expanded and prolonged formation of neovascularization, and retarded regression of neovascularization in both laser injury CNV- and VEGF-induced corneal neovascularization models. Furthermore, both anatomical and functional metrics of CNV (flat mount analysis and fluorescein angiography) supported the notion that AIF deficiency leads to more robust and larger neovascularization. We have reported that laser photocoagulation incites inflammation, leading to endothelial upregulation of intercellular adhesion molecule-1, which binds to CD18, mediating firm leukocyte-endothelial adhesion and transmigration.34 Laser photocoagulation leads to production of VEGF by retinal pigment epithelial cells,20,39 stimulating proliferation of adjacent choroidal vascular endothelium and upregulation of intercellular adhesion molecule-1 expression on endothelium.40 Circulating leukocytes, which migrate in response to VEGF, also release VEGF, amplifying the locally produced VEGF response as they bind to the endothelium. In addition, leukocyte-derived cytokines can stimulate retinal pigment epithelial cells and fibroblasts to express VEGF,41,42 as well as the chemotaxins IL-8, monocyte chemotactic protein-1,43 metalloproteinases44 and intercellular adhesion molecule-1.45 These observations are consistent with our findings that AIF deficiency protected macrophages and neovascular endothelial cells from apoptosis, result in accumulation and prolongation of inflammatory macrophages and neovascular endothelial cells at the site of the developing neovascularization.
Macrophages have been shown to exhibit distinct functions during injury and repair, especially in neovascularization. We have reported that macrophages are localized to the site of tissue injury before the onset of neovascularization and systemic macrophage depletion resulted in reduced pathological neovascularization in CNV,21 and corneal neovascularization.42 In clinical settings of age-related macular degeneration, it is known that chronic inflammatory responses caused by accumulation of lipids and/or immune complex induce further infiltration of inflammatory cells, and CNV.46 In the current study, cultured peritoneal macrophages from AIF-deficient mice are resistant to apoptotic stimuli, compared to macrophages from WT mice. In line with these in vitro observations, immunohistochemistry showed accumulation and prolongation of macrophages at the site in both laser-induced CNV and VEGF-induced corneal neovascularization in vivo in AIF-deficient mice. Thus, AIF deficiency decreased effective apoptotic clearance of excessive inflammatory cells that induced robust and larger neovascularization.
Pathological angiogenesis is controlled by the balance of proangiogenic factors and anti-angiogenic factors. Several proapoptotic molecules have been identified among anti-angiogenic factors that help regulate angiogenesis, including Fas-ligand, thrombospondin-1, and endostatin. The concept that the balance between pro- and anti-apoptotic factors plays a critical role in the regulation of angiogenesis has been postulated for ocular neovascularization.47–49 The reports, however, mainly focus on the death receptor family (ie, Fas/Fas-Ligand) and subsequent caspase signaling.48,50–52 Cell death in vertebrates, however, usually proceeds via the mitochondrial pathway of apoptosis.1 In the current article, we focused on the phylogenetically old, death receptor-independent, caspase-independent proapoptotic factor, AIF that mainly signals mitochondrial pathway of apoptosis in mammalian cells.7 We took advantage of AIF-deficient mice (Hq), which cause an 80% reduction of functional proapoptotic protein, AIF.14 Blockade of mitochondrial pathway of apoptosis by AIF substantially accumulated and prolonged inflammatory macrophages and neovascular endothelial cells, resulting in expanding and prolonging destructive inflammation and leakage at the site. Furthermore, recent data has revealed that AIF modulate active caspase-independent necrotic pathways defined as necroptosis (programmed necrosis) additionally to apoptosis.53 AIF and the key executioner, such as the kinase, the receptor-interacting protein 1 have become potential therapeutic targets in apoptosis/necrosis.14,54 In the current article, we hypothesized that decreased apoptotic cell death of inflammatory/endothelial cells may modulate CNV. The bone marrow transplantation experiments also support the hypothesis that reconstructed WT macrophages partially reversed CNV modulation in AIF-deficient mice. Further study is needed to clarify the direct molecular mechanism underlying the phenomena.
Therapeutic strategies directed against pathological neovascularization via appropriate apoptotic clearance of excessive inflammatory cells or endothelial cells may have potential as a novel clinically relevant approach in addition to conventional therapies.
Acknowledgments
We thank Mari Imamura and Fumiyo Morikawa (Kyushu University) and Kennard Thomas, Sreedevi Mallemadugula, Norman Michaud, and Miin Roh (Massachusetts Eye and Ear Infirmary) for their technical assistance. We also thank the Massachusetts Lions Research Fund for generous funds provided for laboratory equipment used in this project and Research to Prevent Blindness for unrestricted funds awarded to the Department of Ophthalmology at Harvard Medical School.
Footnotes
Supported by an Alcon Research Award (J.W.M.), Japan Eye Bank Association (T.H.), a Bausch & Lomb Vitreoretinal Fellowship (T.N.), and National Eye Institute Grant EY014104 (Massachusetts Eye and Ear Infirmary Core Grant).
T.H., S.N., and Y.M. contributed equally to this work.
Contributor Information
Tatsuro Ishibashi, Email: ishi@eye.med.kyushu-u.ac.jp.
Joan W. Miller, Email: Joan_Miller@meei.harvard.edu.
References
- 1.Green D.R., Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 2.Bao Q., Riedl S.J., Shi Y. Structure of Apaf-1 in the auto-inhibited form: a critical role for ADP. Cell Cycle. 2005;4:1001–1003. doi: 10.4161/cc.4.8.1849. [DOI] [PubMed] [Google Scholar]
- 3.Bao Q., Shi Y. Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ. 2007;14:56–65. doi: 10.1038/sj.cdd.4402028. [DOI] [PubMed] [Google Scholar]
- 4.Acehan D., Jiang X., Morgan D.G., Heuser J.E., Wang X., Akey C.W. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell. 2002;9:423–432. doi: 10.1016/s1097-2765(02)00442-2. [DOI] [PubMed] [Google Scholar]
- 5.Bouchier-Hayes L., Lartigue L., Newmeyer D.D. Mitochondria: pharmacological manipulation of cell death. J Clin Invest. 2005;115:2640–2647. doi: 10.1172/JCI26274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hisatomi T., Sakamoto T., Murata T., Yamanaka I., Oshima Y., Hata Y., Ishibashi T., Inomata H., Susin S.A., Kroemer G. Relocalization of apoptosis-inducing factor in photoreceptor apoptosis induced by retinal detachment in vivo. Am J Pathol. 2001;158:1271–1278. doi: 10.1016/S0002-9440(10)64078-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Snow B.E., Brothers G.M., Mangion J., Jacotot E., Costantini P., Loeffler M. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 8.Kroemer G., Martin S.J. Caspase-independent cell death. Nat Med. 2005;11:725–730. doi: 10.1038/nm1263. [DOI] [PubMed] [Google Scholar]
- 9.Hisatomi T., Ishibashi T., Miller J.W., Kroemer G. Pharmacological inhibition of mitochondrial membrane permeabilization for neuroprotection. Exp Neurol. 2009;218:347–352. doi: 10.1016/j.expneurol.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 10.Cande C., Vahsen N., Kouranti I., Schmitt E., Daugas E., Spahr C., Luban J., Kroemer R.T., Giordanetto F., Garrido C. AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene. 2004;23:1514–1521. doi: 10.1038/sj.onc.1207279. [DOI] [PubMed] [Google Scholar]
- 11.Joza N., Oudit G.Y., Brown D., Benit P., Kassiri Z., Vahsen N., Benoit L., Patel M.M., Nowikovsky K., Vassault A. Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol Cell Biol. 2005;25:10261–10272. doi: 10.1128/MCB.25.23.10261-10272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hisatomi T., Sakamoto T., Goto Y., Yamanaka I., Oshima Y., Hata Y., Ishibashi T., Inomata H., Susin S.A., Kroemer G. Critical role of photoreceptor apoptosis in functional damage after retinal detachment. Curr Eye Res. 2002;24:161–172. doi: 10.1076/ceyr.24.3.161.8305. [DOI] [PubMed] [Google Scholar]
- 13.Hisatomi T., Sakamoto T., Sonoda K.H., Tsutsumi C., Qiao H., Enaida H., Yamanaka I., Kubota T., Ishibashi T., Kura S. Clearance of apoptotic photoreceptors: elimination of apoptotic debris into the subretinal space and macrophage-mediated phagocytosis via phosphatidylserine receptor and integrin alphavbeta3. Am J Pathol. 2003;162:1869–1879. doi: 10.1016/s0002-9440(10)64321-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hisatomi T., Nakazawa T., Noda K., Almulki L., Miyahara S., Nakao S., Ito Y., She H., Kohno R., Michaud N. HIV protease inhibitors provide neuroprotection through inhibition of mitochondrial apoptosis in mice. J Clin Invest. 2008;118:2025–2038. doi: 10.1172/JCI34267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murakami Y., Ikeda Y., Yonemitsu Y., Onimaru M., Nakagawa K., Kohno R., Miyazaki M., Hisatomi T., Nakamura M., Yabe T. Inhibition of nuclear translocation of apoptosis-inducing factor is an essential mechanism of the neuroprotective activity of pigment epithelium-derived factor in a rat model of retinal degeneration. Am J Pathol. 2008;173:1326–1338. doi: 10.2353/ajpath.2008.080466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Notomi S., Hisatomi T., Kanemaru T., Takeda A., Ikeda Y., Enaida H., Kroemer G., Ishibashi T. Critical involvement of extracellular ATP acting on P2RX7 purinergic receptors in photoreceptor cell death. Am J Pathol. 2011;179:2798–2809. doi: 10.1016/j.ajpath.2011.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H., Shimoji M., Yu S.W., Dawson T.M., Dawson V.L. Apoptosis inducing factor and PARP-mediated injury in the MPTP mouse model of Parkinson's disease. Ann NY Acad Sci. 2003;991:132–139. doi: 10.1111/j.1749-6632.2003.tb07471.x. [DOI] [PubMed] [Google Scholar]
- 18.Chu C.T., Zhu J.H., Cao G., Signore A., Wang S., Chen J. Apoptosis inducing factor mediates caspase-independent 1-methyl-4-phenylpyridinium toxicity in dopaminergic cells. J Neurochem. 2005;94:1685–1695. doi: 10.1111/j.1471-4159.2005.03329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanges D., Comitato A., Tammaro R., Marigo V. Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc Natl Acad Sci USA. 2006;103:17366–17371. doi: 10.1073/pnas.0606276103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishibashi T., Hata Y., Yoshikawa H., Nakagawa K., Sueishi K., Inomata H. Expression of vascular endothelial growth factor in experimental choroidal neovascularization. Graefes Arch Clin Exp Ophthalmol. 1997;235:159–167. doi: 10.1007/BF00941723. [DOI] [PubMed] [Google Scholar]
- 21.Tsutsumi C., Sonoda K.H., Egashira K., Qiao H., Hisatomi T., Nakao S., Ishibashi M., Charo I.F., Sakamoto T., Murata T. The critical role of ocular-infiltrating macrophages in the development of choroidal neovascularization. J Leukoc Biol. 2003;74:25–32. doi: 10.1189/jlb.0902436. [DOI] [PubMed] [Google Scholar]
- 22.Gaengel K., Genove G., Armulik A., Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:630–638. doi: 10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- 23.Patan S. TIE1 and TIE2 receptor tyrosine kinases inversely regulate embryonic angiogenesis by the mechanism of intussusceptive microvascular growth. Microvasc Res. 1998;56:1–21. doi: 10.1006/mvre.1998.2081. [DOI] [PubMed] [Google Scholar]
- 24.Jones N., Voskas D., Master Z., Sarao R., Jones J., Dumont D.J. Rescue of the early vascular defects in Tek/Tie2 null mice reveals an essential survival function. EMBO Rep. 2001;2:438–445. doi: 10.1093/embo-reports/kve093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dimmeler S., Zeiher A.M. Endothelial cell apoptosis in angiogenesis and vessel regression. Circ Res. 2000;87:434–439. doi: 10.1161/01.res.87.6.434. [DOI] [PubMed] [Google Scholar]
- 26.Miller J.W., Walsh A.W., Kramer M., Hasan T., Michaud N., Flotte T.J., Haimovici R., Gragoudas E.S. Photodynamic therapy of experimental choroidal neovascularization using lipoprotein-delivered benzoporphyrin. Arch Ophthalmol. 1995;113:810–818. doi: 10.1001/archopht.1995.01100060136048. [DOI] [PubMed] [Google Scholar]
- 27.Zacks D.N., Ezra E., Terada Y., Michaud N., Connolly E., Gragoudas E.S., Miller J.W. Verteporfin photodynamic therapy in the rat model of choroidal neovascularization: angiographic and histologic characterization. Invest Ophthalmol Vis Sci. 2002;43:2384–2391. [PubMed] [Google Scholar]
- 28.Hisatomi T., Sakamoto T., Yamanaka I., Sassa Y., Kubota T., Ueno H., Ohnishi Y., Ishibashi T. Photocoagulation-induced retinal gliosis is inhibited by systemically expressed soluble TGF-beta receptor type II via adenovirus mediated gene transfer. Lab Invest. 2002;82:863–870. doi: 10.1097/01.lab.0000018829.49754.dd. [DOI] [PubMed] [Google Scholar]
- 29.She H., Nakazawa T., Matsubara A., Hisatomi T., Young T.A., Michaud N., Connolly E., Hafezi-Moghadam A., Gragoudas E.S., Miller J.W. Reduced photoreceptor damage after photodynamic therapy through blockade of nitric oxide synthase in a model of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2007;48:2268–2277. doi: 10.1167/iovs.06-0979. [DOI] [PubMed] [Google Scholar]
- 30.She H., Nakazawa T., Matsubara A., Connolly E., Hisatomi T., Noda K., Kim I., Gragoudas E.S., Miller J.W. Photoreceptor protection after photodynamic therapy using dexamethasone in a rat model of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2008;49:5008–5014. doi: 10.1167/iovs.07-1154. [DOI] [PubMed] [Google Scholar]
- 31.Noda K., She H., Nakazawa T., Hisatomi T., Nakao S., Almulki L., Zandi S., Miyahara S., Ito Y., Thomas K.L. Vascular adhesion protein-1 blockade suppresses choroidal neovascularization. FASEB J. 2008;22:2928–2935. doi: 10.1096/fj.07-105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westerhof G.R., Ploemacher R.E., Boudewijn A., Blokland I., Dillingh J.H., McGown A.T., Hadfield J.A., Dawson M.J., Down J.D. Comparison of different busulfan analogues for depletion of hematopoietic stem cells and promotion of donor-type chimerism in murine bone marrow transplant recipients. Cancer Res. 2000;60:5470–5478. [PubMed] [Google Scholar]
- 33.Hisatomi T., Sonoda K.H., Ishikawa F., Qiao H., Nakazawa T., Fukata M., Nakamura T., Noda K., Miyahara S., Harada M. Identification of resident and inflammatory bone marrow derived cells in the sclera by bone marrow and haematopoietic stem cell transplantation. Br J Ophthalmol. 2007;91:520–526. doi: 10.1136/bjo.2006.102046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakurai E., Taguchi H., Anand A., Ambati B.K., Gragoudas E.S., Miller J.W., Adamis A.P., Ambati J. Targeted disruption of the CD18 or ICAM-1 gene inhibits choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:2743–2749. doi: 10.1167/iovs.02-1246. [DOI] [PubMed] [Google Scholar]
- 35.Maruyama K., Ii M., Cursiefen C., Jackson D.G., Keino H., Tomita M., Van Rooijen N., Takenaka H., D'Amore P.A., Stein-Streilein J. Inflammation-induced lymphangiogenesis in the cornea arises from CD11b-positive macrophages. J Clin Invest. 2005;115:2363–2372. doi: 10.1172/JCI23874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakazawa T., Hisatomi T., Nakazawa C., Noda K., Maruyama K., She H., Matsubara A., Miyahara S., Nakao S., Yin Y. Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc Natl Acad Sci USA. 2007;104:2425–2430. doi: 10.1073/pnas.0608167104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barber B.R. Research news. Mouse News Lett. 1971;45:34–35. [Google Scholar]
- 38.Klein J.A., Longo-Guess C.M., Rossmann M.P., Seburn K.L., Hurd R.E., Frankel W.N., Bronson R.T., Ackerman S.L. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- 39.Wada M., Ogata N., Otsuji T., Uyama M. Expression of vascular endothelial growth factor and its receptor (KDR/flk-1) mRNA in experimental choroidal neovascularization. Curr Eye Res. 1999;18:203–213. doi: 10.1076/ceyr.18.3.203.5368. [DOI] [PubMed] [Google Scholar]
- 40.Miyamoto K., Khosrof S., Bursell S.E., Moromizato Y., Aiello L.P., Ogura Y., Adamis A.P. Vascular endothelial growth factor (VEGF)-induced retinal vascular permeability is mediated by intercellular adhesion molecule-1 (ICAM-1) Am J Pathol. 2000;156:1733–1739. doi: 10.1016/S0002-9440(10)65044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oh H., Takagi H., Takagi C., Suzuma K., Otani A., Ishida K., Matsumura M., Ogura Y., Honda Y. The potential angiogenic role of macrophages in the formation of choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1999;40:1891–1898. [PubMed] [Google Scholar]
- 42.Nakao S., Kuwano T., Tsutsumi-Miyahara C., Ueda S., Kimura Y.N., Hamano S., Sonoda K.H., Saijo Y., Nukiwa T., Strieter R.M. Infiltration of COX-2-expressing macrophages is a prerequisite for IL-1 beta-induced neovascularization and tumor growth. J Clin Invest. 2005;115:2979–2991. doi: 10.1172/JCI23298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holtkamp G.M., Van Rossem M., de Vos A.F., Willekens B., Peek R., Kijlstra A. Polarized secretion of IL-6 and IL-8 by human retinal pigment epithelial cells. Clin Exp Immunol. 1998;112:34–43. doi: 10.1046/j.1365-2249.1998.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peppin G.J., Weiss S.J. Activation of the endogenous metalloproteinase, gelatinase, by triggered human neutrophils. Proc Natl Acad Sci USA. 1986;83:4322–4326. doi: 10.1073/pnas.83.12.4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elner S.G., Elner V.M., Pavilack M.A., Todd R.F., 3rd, Mayo-Bond L., Franklin W.A., Strieter R.M., Kunkel S.L., Huber A.R. Modulation and function of intercellular adhesion molecule-1 (CD54) on human retinal pigment epithelial cells. Lab Invest. 1992;66:200–211. [PubMed] [Google Scholar]
- 46.Ishibashi T., Patterson R., Ohnishi Y., Inomata H., Ryan S.J. Formation of drusen in the human eye. Am J Ophthalmol. 1986;101:342–353. doi: 10.1016/0002-9394(86)90830-5. [DOI] [PubMed] [Google Scholar]
- 47.Wang S., Sorenson C.M., Sheibani N. Attenuation of retinal vascular development and neovascularization during oxygen-induced ischemic retinopathy in Bcl-2−/− mice. Dev Biol. 2005;279:205–219. doi: 10.1016/j.ydbio.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 48.Davies M.H., Eubanks J.P., Powers M.R. Increased retinal neovascularization in Fas ligand-deficient mice. Invest Ophthalmol Vis Sci. 2003;44:3202–3210. doi: 10.1167/iovs.03-0050. [DOI] [PubMed] [Google Scholar]
- 49.Davies M.H., Stempel A.J., Powers M.R. MCP-1 deficiency delays regression of pathologic retinal neovascularization in a model of ischemic retinopathy. Invest Ophthalmol Vis Sci. 2008;49:4195–4202. doi: 10.1167/iovs.07-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ilg R.C., Davies M.H., Powers M.R. Altered retinal neovascularization in TNF receptor-deficient mice. Curr Eye Res. 2005;30:1003–1013. doi: 10.1080/02713680500330355. [DOI] [PubMed] [Google Scholar]
- 51.Semkova I., Fauser S., Lappas A., Smyth N., Kociok N., Kirchhof B., Paulsson M., Poulaki V., Joussen A.M. Overexpression of FasL in retinal pigment epithelial cells reduces choroidal neovascularization. FASEB J. 2006;20:1689–1691. doi: 10.1096/fj.05-5653fje. [DOI] [PubMed] [Google Scholar]
- 52.Hubert K.E., Davies M.H., Stempel A.J., Griffith T.S., Powers M.R. TRAIL-deficient mice exhibit delayed regression of retinal neovascularization. Am J Pathol. 2009;175:2697–2708. doi: 10.2353/ajpath.2009.090099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delavallee L, Cabon L, Galan-Malo P, Lorenzo HK, Susin SA: AIF-mediated caspase-independent necroptosis: a new chance for targeted therapeutics. IUBMB Life 63:221–232 [DOI] [PubMed]
- 54.Trichonas G., Murakami Y., Thanos A., Morizane Y., Kayama M., Debouck C.M., Hisatomi T., Miller J.W., Vavvas D.G. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci USA. 2010;107:21695–21700. doi: 10.1073/pnas.1009179107. [DOI] [PMC free article] [PubMed] [Google Scholar]