

Abstract
Myofilament length-dependent activation is a universal property of striated muscle, yet the molecular mechanisms that underlie this phenomenon are incompletely understood. Additionally, the rate by which sarcomere length (SL) is sensed and then transduced to form length-dependent activation is unknown. Here, using isolated guinea-pig myocardium, we employed a rapid solution-switch single myofibril technique that allows for the study of contractile action/relaxation dynamics in the virtual absence of diffusion delays. We compared contraction kinetics obtained at submaximal activation at steady-state SL with contractions observed after rapid SL ramps to that same SL just before activation. Neither the activation and relaxation kinetics nor the final submaximal force development differed significantly between the two contraction modes for SL ramps as fast as 5 ms. We conclude that the transduction of the length signal by the cardiac sarcomere to modulate thin filament activation levels occurs virtually instantaneously, possibly resulting from structural rearrangements of the contractile proteins.
The Frank-Starling law of the heart describes an important regulatory system whereby the heart has the ability to alter cardiac output to meet changes in the energy demands of the body. The cellular basis for this property resides in the cardiac sarcomere; that is, a change in sarcomere length (SL) alters the responsiveness of the myofilament to Ca2+. This phenomenon has been termed myofilament length-dependent activation (LDA). Despite intense investigation, the molecular mechanisms that underlie LDA remain largely unknown (1). It has been determined that modulation of steady-state contraction force by SL is due to a proportional variation of strongly attached, force-generating cross-bridges. Studies of sinusoidal SL perturbation in activated permeabilized isolated myocardium have revealed two distinct responses to dynamic SL changes: an immediate quasi-elastic force response, and a slower-developing delayed-force response. The time constant reported for the latter phase correlates with the kinetics of cross-bridge cycling, in addition to other factors, and is ∼100 ms in rabbit myocardium at 25°C. These data suggest that LDA develops relatively slowly, i.e., within the time frame of the first ∼25% of the heartbeat (2). However, the term “LDA” principally refers to the impact of an SL change that occurs before myofilament activation. In contrast, the delayed-force response dynamics include both LDA and all subsequent downstream processes, including attachment and detachment of cross-bridges (2). Hence, the rate at which LDA develops, i.e., the rate by which an SL perturbation induces a change in Ca2+ responsiveness of a relaxed myofilament, is not known.
The single-myofibril technique allows for the measurement of activation and relaxation kinetics after a stepwise rapid change in the concentration of activating Ca2+ due to the virtual absence of a diffusion delay in the small preparations required for this approach (3). Therefore, rapid-solution switching results in rapid (∼1 ms) and homogeneous alteration of the ionic milieu surrounding the myofilaments throughout the preparation. Here, we rapidly activated single, isolated guinea-pig myofibrils under submaximal (∼50%) activation conditions at either steady-state SL or after rapid ramp changes to that SL just before activation. We reasoned that a delay in the development of LDA would manifest as delayed-force development dynamics. We chose to use guinea-pig myocardium, a generally slow and predominantly β-myosin-containing species (4), as well as a low temperature (5), to maximize our ability to assess the dynamic rate of LDA development. Both rapid ramp SL stretch (n = 29) and ramp SL release (n = 5) perturbations were examined.
All experiments were performed according to institutional guidelines concerning the care and use of experimental animals. Guinea pigs (Dunkin-Harley, 4–6 months old) were anesthetized with pentobarbital (50 mg/kg) before their hearts were rapidly excised. The hearts were perfused with Krebs-Henseleit solution, and the left ventricle was isolated and cut into ∼100-mg segments that were flash-frozen and subsequently stored in liquid nitrogen for further use. The frozen myocardium was thawed in a high-potassium, calcium-free extracellular solution at 4°C, cut into small strips, and held at near-physiological muscle length while it was permeabilized with 1% Triton-X100 in relaxing solution for 3 h at 4°C (6). Single myofibrils (or small bundles of two to three myofibrils), 1–5 μm in diameter and ∼35–50 μm long (see Fig. 1 A), were obtained by mechanical homogenization (6). The apparatus has been described previously (5,6). An aliquot of myofibril suspension in relaxing solution was injected into a chamber that was filled with ∼2.5 mL bath relaxing solution and mounted on the stage of an inverted microscope (Olympus IX-70). Myofibrils selected for use were attached horizontally between two glass tools: a custom-made, black-ink-coated cantilever of known stiffness (acting as a force probe) and a rigid pipette attached to a piezo motor (Nano-driveOP30 Mad City Labs, Madison, WI) used to impose rapid SL changes. We determined the force by measuring the displacement of the force probe via edge detection, and measured SL by conducting a fast Fourier transform analysis of the myofibril striation pattern (IonOptix, Milton, MA). Experiments were performed at 15°C. Myofibrils were placed in one of the laminar solutions emanating from a double-barreled perfusion pipette (Fig. 1 B). Rapid translation of this pipette allowed for rapid switching between the two solutions. There was an inherent uncertainty regarding the exact time the solution switch took place, due to the inevitable delay between the displacement of the perfusion pipette and the arrival of the new solution at the level of the attached myofibril. To ascertain the proper sequence of rapid muscle length change and subsequent Ca2+ activation, we added a small amount of red dye (ponceau) to the activation solution and measured the transmission of green light in a region just proximal to the attached myofibril. As illustrated in Fig. 1 C, this approach allowed for a protocol in which the muscle length change was completed just before the actual solution switch. Preliminary experiments revealed no impact on force or kinetics in the presence of the small concentration of red dye.
Figure 1.
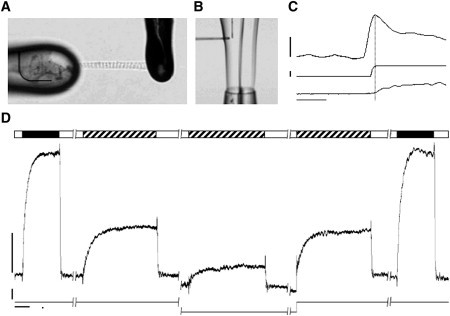
Myofibrils were attached to glass micro tools (A) that were placed in a laminar solution flow emanating from a double-barreled pipette (B). Rapid translation of the pipette allows for rapid solution switching. (C) Force (top), myofibril length (middle), and PMT output (bottom). (D) The five consecutive myofibril activation-relaxation cycles (open bar, pCa = 9; closed bar, pCa = 4.0; hatched bar, pCa = 5.7). Calibration bars: 40 nN for force, 0.15 Lo for length, 2 s (D), and 200 ms (C).
Fig. 1 D shows representative recordings. Initially, the myofibril was maximally activated (pCa = 4) at SL = 2.3 μm to measure Ca2+ saturated force development. This contraction also served as a reference to assess rundown. Next, two submaximal (pCa = 5.7) contractions were initiated at the long and short steady-state SL (2.3 μm and 2.0 μm, respectively). Steady-state SL was attained by holding SL constant for 5 min. Next, the myofibril was rapidly stretched from SL = 2.0 to SL = 2.3 μm and immediately activated with the pCa = 5.7 solution (cf. Fig. 1 C). A final contraction at maximum activation at SL = 2.3 allowed us to assess force rundown. Myofibrils that displayed >20% rundown were not included in the study. The order of activation at either steady-state SL or after a rapid SL change at pCa = 5.7 was randomized. As illustrated in this panel, guinea-pig myocardium displays robust LDA; that is, submaximal force development strongly depends on SL (compare the second and third contractions in Fig. 1 D). Submaximal activation at steady-state SL = 2.0 μm was, on average, 23% ± 2%, whereas submaximal activation at SL = 2.3 resulted in 51% ± 6% force development (normalized to maximum force at SL = 2.3 μm). In addition, the rate of force development qualitatively appeared slower at the short SL; however, the low force levels at this SL prevented quantification of this parameter.
Rapid stretch in the absence of Ca2+ activation resulted in a rapid increase in passive force during the stretch that subsequently decayed to the steady-state passive force seen at SL = 2.3 μm (Fig. 2 A, black trace; see also fourth contraction in Fig. 1 D). Subtraction of the passive force response resulted in the traces shown in Fig. 2 B. On average, the final steady-state level of submaximal force development was 51% ± 6% at steady-state SL, compared with 50% ± 5% for a contraction that initiated ∼5 ms after a rapid stretch from the short SL (n = 22). The rate of submaximal force development, as indexed by the time required to reach 50% of final steady-state force development, was likewise not affected by the mode of contraction (T50 = 0.85 ± 0.04 and 0.84 ± 0.03 s for steady-state and rapid stretch, respectively).
Figure 2.
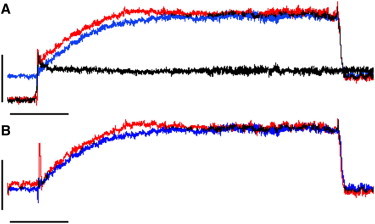
(A) Submaximal force at steady-state SL = 2.3 μm (blue trace) or after a quick stretch from steady-state SL = 2.0 μm (red trace; black trace = passive force). (B) The developed force (total force minus passive force). Calibration bars: 40 nN and 2 s.
The data shown in Figs. 1 and 2 were obtained in the presence of 2 mM inorganic phosphate, a maneuver that improves the stability of the preparation, presumably due to a reduction in overall force development. However, inclusion of phosphate in the solution also increases the contractile dynamics (3), thereby precluding accurate assessment of the relaxation kinetics. Accordingly, we performed both rapid stretch and rapid release experiments in the absence of inorganic phosphate by employing an enzymatic phosphate mop (3). Fig. 3 shows representative traces obtained in the absence of phosphate for rapid SL stretches (A) and rapid SL releases (B). Removal of inorganic phosphate resulted in ∼3-fold slower contractile dynamics, consistent with previous reports (3). On average, neither Ca2+ activation nor biphasic relaxation kinetics were affected by the contraction mode (n = 5–7).
Figure 3.
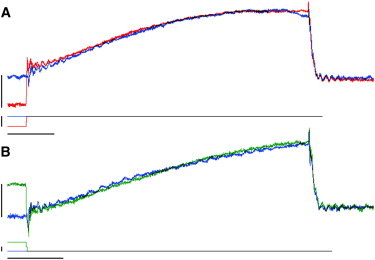
(A) Submaximal force at steady-state SL = 2.3 μm (blue trace) or after a quick stretch from steady-state SL = 2.0 μm (red trace). (B) Submaximal activation at steady SL = 2.3 μm (blue trace) or after a quick release from steady-state SL = 2.5 μm (green trace). Inorganic phosphate was removed by an enzymatic phosphate mop. Calibration bars: 40 nN for force, 0.15 Lo for muscle length, and 2 s for time.
The results from this study indicate that the molecular processes that lead to myofilament LDA occur virtually instantaneously (within ∼5 ms). It is unlikely that we were not able to detect a significant delay in LDA. First, we used guinea-pig myocardium, which contains almost exclusively the slower β-isoform of myosin, which is similar to that found in rabbit myocardium, where a time constant of ∼100 ms for the recruitment phase of the length response has been recorded at 10°C higher temperature (2). Second, the time course of force development after Ca2+ activation was virtually independent of SL history just before activation. Any significant delay in LDA would be expected to alter force development measurably in the early phases of Ca2+ activation, and we detected no such delay in our experiments, for either rapid stretches or rapid releases. The absence of such a delay excludes slow chemical reactions, such as phosphorylation events, as a prominent mechanism in LDA. A more likely mechanism consistent with recently published results is that increasing SL alters the spatial orientation of myosin with respect to binding sites on actin, possibly via a mechanical strain mechanism involving titin (7,8). Overall, our results suggest that LDA results from instantaneous length-induced structural rearrangements of either thin-filament regulatory proteins or thick-filament proteins, or a combination of these structures.
Acknowledgments
This work was supported by the National Institutes of Health (HL62426, HL07692, and HL075494).
References and Footnotes
- 1.de Tombe P.P., Mateja R.D., Irving T.C. Myofilament length dependent activation. J. Mol. Cell. Cardiol. 2010;48:851–858. doi: 10.1016/j.yjmcc.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell K.B., Taheri H., Hunter W.C. Similarities between dynamic elastance of left ventricular chamber and papillary muscle of rabbit heart. Am. J. Physiol. 1993;264:H1926–H1941. doi: 10.1152/ajpheart.1993.264.6.H1926. [DOI] [PubMed] [Google Scholar]
- 3.Tesi C., Colomo F., Poggesi C. The effect of inorganic phosphate on force generation in single myofibrils from rabbit skeletal muscle. Biophys. J. 2000;78:3081–3092. doi: 10.1016/S0006-3495(00)76845-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stehle R., Krüger M., Pfitzer G. Force kinetics and individual sarcomere dynamics in cardiac myofibrils after rapid ca(2+) changes. Biophys. J. 2002;83:2152–2161. doi: 10.1016/S0006-3495(02)73975-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Tombe P.P., Belus A., Poggesi C. Myofilament calcium sensitivity does not affect cross-bridge activation-relaxation kinetics. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;292:R1129–R1136. doi: 10.1152/ajpregu.00630.2006. [DOI] [PubMed] [Google Scholar]
- 6.Colomo F., Piroddi N., Tesi C. Active and passive forces of isolated myofibrils from cardiac and fast skeletal muscle of the frog. J. Physiol. 1997;500:535–548. doi: 10.1113/jphysiol.1997.sp022039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu H., Ait-Mou Y., de Tombe P.P. Structural changes in both the troponin complex and the thick filament may underlie myofilament length dependent activation. Biophys. J. 2012;102:17a. [Google Scholar]
- 8.Farman G.P., Gore D., de Tombe P.P. Myosin head orientation: a structural determinant for the Frank-Starling relationship. Am. J. Physiol. Heart Circ. Physiol. 2011;300:H2155–H2160. doi: 10.1152/ajpheart.01221.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]