

Abstract
The histone-like nucleoid structuring protein (H-NS) is a DNA-organizing protein in bacteria. It contains a DNA-binding domain and a dimerization domain, connected by a flexible linker region. Dimerization occurs through the formation of a helical bundle, including a coiled-coil interaction motif. Two conformations have been resolved, for different sequences of Escherichia coli H-NS, resulting in an antiparallel coiled-coil for the shorter wild-type sequence, and a parallel coiled-coil for the longer C21S mutant. Because H-NS functions as a thermo- and osmosensor, these conformations may both be functionally relevant. Molecular simulation can complement experiments by modeling the dynamical time evolution of biomolecular systems in atomistic detail. We performed a molecular-dynamics study of the H-NS dimerization domain, showing that the parallel complex is sensitive to changes in salt conditions: it is unstable in absence of NaCl, but stable at physiological salt concentrations. In contrast, the stability of the antiparallel complex is not salt-dependent. The stability of the parallel complex also appears to be affected by mutation of the critical but nonconserved cysteine residue at position 21, whereas the antiparallel complex is not. Together, our simulations suggest that osmoregulation could be mediated by changes in the ratio of parallel- and antiparallel-oriented H-NS dimers.
Introduction
The bacterial genome is compacted and functionally organized by a family of small, DNA-binding proteins. These proteins are generally referred to as architectural or nucleoid-associated proteins. The action of the individual members as well as their interplay determines how the genome is folded (1–3). In addition, most of these proteins act as global regulators of transcription (4,5). The histone-like nucleoid structuring protein (H-NS) has been identified as an important global regulator (regulating several hundreds of genes) and as a key player in genome organization (6). Its role as a global regulator has been coupled to acting as an environmental sensor (7,8), perceiving changes in the growth conditions of the bacterium, and facilitating physiological changes required for adaptation to the new conditions. It is to date unclear how H-NS would be able to perceive and transmit such signals.
The protein contains a DNA-binding domain (residues 91–137) and a dimerization domain (residues 1–64), connected by a flexible linker region (9). The protein has been shown to bind to DNA as a dimer in two different modes—in trans to bridge two DNA duplexes, or in cis, binding with both DNA binding domains to the same duplex (10–13). Up to now, a complete, high-resolution structure of full-length H-NS, either free or complexed with DNA, has proven elusive. Structural models for the N-terminal dimerization domain have been determined by NMR spectroscopy and x-ray crystallography, showing that dimerization occurs through the formation of a coiled-coil interaction motif. Two different structures have been resolved by NMR spectroscopy, revealing two strikingly different coiled-coil complexes (see Fig. 1) (14,15). The structure comprising residues 2–46 contains an antiparallel coiled-coil complex (15), whereas a longer truncate containing residues 2–58 and Cys21-to-serine mutation results in a parallel orientation of the coiled-coil helices (14).
Figure 1.
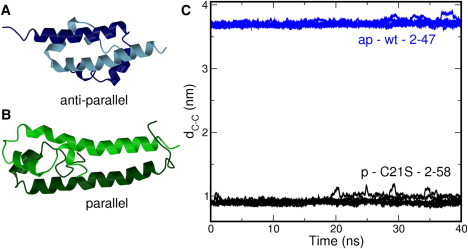
Ribbon representations of the structural data for the H-NS dimerization domain (A) the antiparallel conformation (B) the parallel conformation. The different colors indicate the monomers. (C) Time evolution of distance between C-terminal ends dC-C. For each system, eight simulations were performed initiated from different starting velocities. The labels contain the following information: ap indicates antiparallel conformation and p indicates parallel conformation. The label wt indicates that the wild-type sequence was used, and C21S indicates that cysteine at position 21 was replaced by a serine. The range indicates the residues included in the simulations. The lines show a running average of 100 ps. The actual distances were calculated every 2 ps.
Recently, a crystal structure of H-NS containing residues 2–83 and the C21S mutation has been resolved, revealing a conformation with two regions of interaction (16). One region involves residues 1–40 and is similar to the previously determined antiparallel coiled-coil conformation. The second region comprises residues 67–83, at the C-terminus, and interacts with its crystallographic neighbor. It is unclear whether both the parallel and the antiparallel conformations play a role in the function of H-NS. Based on the notion that H-NS protein reacts to differences in temperature and/or osmotic pressure (17–21) it has been suggested that both structures might be functionally relevant.
Molecular simulation can provide atomistic detail for protein conformational change, complementary to experiments (22). In this work, we used conventional molecular dynamics (MD) simulations to investigate the stability of the parallel and antiparallel conformation of the H-NS protein dimerization domain under various conditions. In this study, we do not aim to sample the transition between the antiparallel and parallel conformations. The tested conditions include different sequence lengths and the C21S mutation on the stability of the H-NS dimerization domain in different conformations.
Furthermore, we studied the effect of adding NaCl on the stability of the complexes. Finally, we also investigated the stability of the isolated coiled-coil regions in the antiparallel and the parallel orientation. As the interaction patterns between the monomers in both conformations are completely different, conversion between the two would involve dissociation and rebinding. This suggests that the conformation of the unbound monomer determines which complex is formed. We present a preliminary investigation of the conformational space of the isolated monomers. Our results provide the groundwork for investigating the H-NS dimerization domain using more advanced molecular simulation methods to tackle the dynamics and mechanism of transition between the different conformations.
Methods
Molecular dynamics
In all simulations, we used the GROMACS software package, Ver. 4.0.4 (23) in combination with the OPLS all-atom force field (24,25). We investigated several systems, listed in Table S1 in the Supporting Material. All systems are derived from two structures with PDB codes 1NI8 (15) and 1LR1 (14), originating originally from Escherichia coli and Salmonella typhimurium, respectively. These structures have been determined by NMR spectroscopy. The sequences of H-NS protein from E. coli and S. typhimurium are identical up to residue 79. 1NI8 contains residues 2–47 in the antiparallel conformation and 1LR1 consists of residues 2–58 in the parallel conformation. In one system, we have extended the H-NS antiparallel conformation up to residue 58 using Modeller, Ver. 9.8 (26), to generate additional coordinates for residues 48–58 in a helical conformation.
We indicated the different systems by their orientation (antiparallel, ap; parallel, p), wild-type or mutation and the length of the sequence. The system ap – C21S – 2-47 contains a complex in the antiparallel conformation of two monomers of length 2–47 with a mutation at position 21 of cysteine to serine. The label m indicates that the system contains a monomer, not a dimer.
All systems were solvated in a periodic dodecahedron-shaped box with a minimum distance of 0.8 nm to the box boundaries, except for the systems containing residues 23–43. These smaller systems were placed in a cubic periodic box with a minimum distance of 1 nm to the box boundaries. All systems were filled with TIP4P water molecules (27), followed by the removal of water molecules that overlap with protein atoms or reside in a hydrophobic location isolated from the bulk. NaCl was added by replacing water molecules by Na+ and Cl− ions at random. The number of added ions depended on the concentration and also aimed to charge-neutralize the system.
All systems, except ap – C21S – 2-47, were energy minimized using the conjugate gradient method. To equilibrate the hydrogen atoms and water molecules the heavy atoms in the protein were position-restrained during 10 ps of molecular dynamics at a temperature of 298 K and a pressure of 1 bar. The van der Waals interaction cutoff was 1.1 nm. Electrostatic interactions beyond a cutoff of 1.1 nm were treated with the particle-mesh Ewald method (28,29) using a grid spacing of 0.12 nm. All bonds were constrained using LINCS (30), allowing for a time step of 2 fs. Temperature was kept constant using the V-rescale thermostat (31), and pressure was kept constant using the Parrinello-Rahman barostat (32,33). For sampling, we performed eight MD runs of either 10 ns or 40 ns, listed in Table 1, each starting with velocities randomly drawn from a Maxwell-Boltzmann distribution at 298 K. All simulations were performed in parallel on a pSeries 575 supercomputer (IBM, North Castle, NY).
Table 1.
List of order parameters
| dC-C | Distance between CA atoms of residues 43 in the two monomers. |
| dH-O | Distance between hydrogen atom HG in the side chain of residue 21 (Cys/Ser) and oxygen atom O in the backbone of residue Gln17. |
| di-j' | Distance between centers of mass of the side chains of residues at positions i and j in the other monomer, indicated by the prime symbol (′). These distances are calculated for the following pairs: |
| (Antiparallel) Leu26-Val37′, Leu30-Leu33′, Leu33-Leu30′, and Val37-Leu26′, | |
| (Parallel) Leu26-Leu26′, Leu30-Leu30′, Leu33-Leu33′, and Val37-Val37′. | |
| N-H–O=C | Distance between hydrogen atom H in the backbone of residue n and oxygen atom O in the backbone of residue n+4. |
| Σnhc | Sum of distances of native hydrophobic contacts. These distances include d26-37′, d30-33′, d33-30′, and d37-26′ for the antiparallel conformation, and d26-37′, d30-33′, d33-30′, and d37-26′ for the parallel conformation. |
| Σα1 | Sum of distances of N-H – O=C of residues 2–6, 3–7, and 4–8. |
| Σα2 | Sum of distances of N-H – O=C of residues 10–14, 11–15, 12–16, and 13–17. |
| Σ8-14-33 | Sum of distances of centers of mass of side chains of residues Leu8, Leu14, and Leu33. |
Analysis
During the simulations coordinates of the systems were stored every 2 ps. After completion of the simulations, we calculated several distances for these trajectories, listed in Table 1. These distances then served as basis for the calculation of other structural measures (see Table 1).
We displayed the results either as time traces, with a running average of 100 ps, or as log-probability plots, in which we plot −ln[P(Q)], the negative natural log of the probability P(Q) in two-dimensional order parameter representations, with P(Q) the probability distribution of order parameter Q.
Results
Stability of the H-NS dimerization domains
Two differently oriented coiled-coil complexes have been resolved for the dimerization domain of H-NS by NMR spectroscopy (Fig. 1). The structure containing residues 2–47 (Fig. 1 A) has an antiparallel conformation (15) and the structure containing residues 2–58 has a parallel conformation (see Fig. 1 B (14)). The parallel conformation contains a mutation: the cysteine at position 21 is changed into a serine. H-NS is involved in environmental adaptation, as reflected in large changes in global gene expression patterns upon changing external conditions, such as temperature and ionic strength (7,8,34,35). Different orientations for the H-NS dimerization domain could facilitate such regulation, as the orientation of this region will affect the way in which the H-NS dimer interacts with DNA, or with other dimers to yield tetramers or larger multimeric forms (36). We have used molecular dynamics (MD) simulations to probe the stability of the dimerization domain in the two different conformations, the effects of the C21S mutation, differences in sequence length, and finally the influence of NaCl on the conformational space. We have not used more advanced molecular simulation methods, as it was our aim to investigate separately the stability of the parallel and antiparallel conformation under various conditions.
To begin, we performed MD simulations of the two NMR structures of the H-NS dimerization domain. Using eight simulations of 40 ns, each started from different initial random velocities, we calculated the distance between the Cα atoms of residues 43, denoted as dC-C, shown as a red-dotted line in the structures depicted in Fig. 2 B. This distance can distinguish between the parallel and antiparallel conformations, as the first has values for dC-C <1 nm, and the second has values close to 4 nm. Note that we used the distance between the Cα atoms of residues 43 rather than those of 47 (the last residue in the antiparallel structure) because the α-helical hydrogen bonds formed by these C-terminal residues are dissolved; this is also true for the NMR structure (14).
Figure 2.
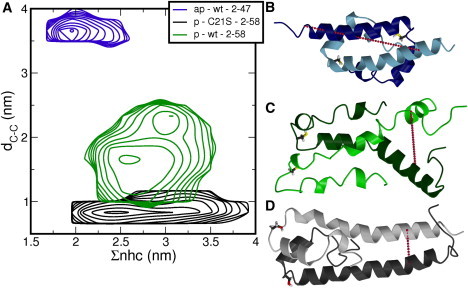
(A) Log-probability plots as a function of the number of hydrophobic contacts Σnhc and the distance between C-terminal ends dC-C. The systems are labeled as described for Fig. 1. Contour lines indicate levels of kBT. (B–D) (Ribbon representations) Structural data for the H-NS dimerization domain (B), the antiparallel conformation (C), the parallel conformation, wild-type, and (D) the parallel conformation, the C21S mutant. (Stick models) Residues at position 21. (Red line) dC-C.
We also used this representation rather than overall root mean square deviation with respect to the experimental structures, as this order parameter gives more insight into the nature of the disruption. Fig. 1 C shows dC-C as a function of the simulation time, and it is clear that both conformations show few fluctuations in dC-C, indicating that the C-terminal ends of the complexes remain at a constant distance. This implies that dissociation and/or large conformational changes do not occur. Further confirmation that the coiled-coil complexes remain intact is shown in Fig. 2 A, showing the log-probability plot as function of the sum of distances between residues in the hydrophobic core of the complex, Σnhc, and dC-C. The antiparallel structure forms a perfect dimeric coiled-coil, as indicated by the small distances between hydrophobic residues (Fig. 2 A, blue lines). In the parallel conformation, hydrophobic residues Ala4, Ile7, Ile11, and Leu14 contribute to the packing of the hydrophobic core, leading to slightly larger distances between leucines 30 and 33 (Fig. 2 A, black lines).
To investigate the effect of the C21S mutation, we changed the serine at position 21 in the parallel conformation to cysteine. This constitutes changing an oxygen atom into a sulfur atom in both monomers. Surprisingly, dC-C increased from <1 nm to values between 1.5 and 2.2 nm, within a few picoseconds, in all eight 40 ns MD runs (see Fig. 2 A, green lines). This increase in dC-C coincides with an increase in Σnhc to 2.5 nm and higher, indicating that the hydrophobic core is disrupted. Fig. 2 C shows a typical snapshot of the distorted parallel complex. On the other hand, changing cysteine into serine in the antiparallel structure does not affect this conformation at all. The main difference between the wild-type and the C21S mutant is that the serine side chain forms a hydrogen bond to the protein backbone, as depicted in Fig. 3 A.
Figure 3.
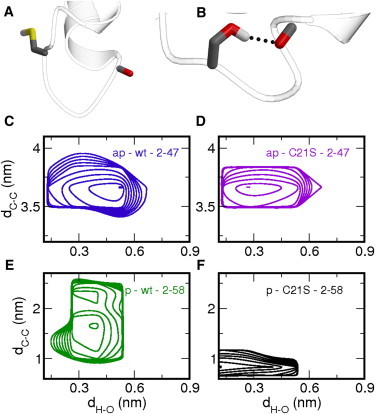
Close-ups of the site of residue 21 with (A) the wild-type with cysteine and (B) the C21S mutant with serine. The C=O group of residue Gln17 is shown as well. (Ribbon) Protein backbone; (stick models) side chains. (Dotted lines) Hydrogen bonds. (C–F) Log-probability plots as a function of the hydrogen bond distance between atom HG in residue 21 and atom O in residue 17 dH-O and the distance between C-terminal ends dC-C for (C) ap – wt – 2-47, (D) p – wt – 2-58, (E) ap – C21S – 2-47, and (F) p – C21S – 2-58. Note that panels C, D, and F represent 8 × 40 ns and panel E represents 8 × 10 ns.
To illustrate this, we plotted the two-dimensional log-probability plots of the four simulations as a function of dC-C and dH-O—the distance between the side-chain hydrogen atom, connected to either the sulfur in cysteine or the oxygen atom in serine, to the backbone carbonyl oxygen of residue Gln17. These profiles show that the serine side chain forms a hydrogen bond to the protein backbone, as the minima in Fig. 3, C and E, have values of 0.3 nm for dH-O, which is within the range for a hydrogen-bond interaction. Conversely, the systems containing a cysteine have the minima located at higher values of dH-O, implying that the cysteine does not form a hydrogen bond with the protein backbone. Instead, the cysteine side chain is oriented toward the bulk solvent. Representative snapshots of both situations are shown in Fig. 3 A. These observations imply that the cysteine-to-serine mutation induces a preference to form a hydrogen-bond interaction between the side chain of residue 21 and the protein backbone.
To further investigate the effect of the sequence length on the stability of the complexes, we performed 10-ns MD simulations of three more systems: a longer sequence of 2–58 in an antiparallel conformation, a shorter sequence of 2–47 in a parallel conformation, and the shorter sequence with C21S mutation in a parallel conformation. The results are displayed as log-probability plots of dC-C and Σnhc in Fig. 4, A–C. With a longer sequence, the antiparallel conformation remains intact, as indicated by the single minimum at a low value for Σnhc (Fig. 4 A). This observation is not surprising, as the number of hydrophobic contacts in the coiled-coil complex is the same as for a shorter sequence. The two simulations of shorter sequences in a parallel conformation exhibit several minima. One minimum, representing the intact conformation at Σnhc = 2.5 nm and dC-C = 1 nm occurs for both systems. This minimum extends to higher values in the wt system, which has an additional minimum at Σnhc = 2.7 nm and dC-C = 1.9 nm. The simulation of the C21S mutant has two additional minima at dC-C = 1.6 nm, at Σnhc = 2.3 nm, and at Σnhc > 3 nm.
Figure 4.

Log-probability plots as a function of the number of hydrophobic contacts Σnhc and the distance between C-terminal ends dC-C for systems of different sequence length and different orientation: (A) ap – wt – 2-58, (B) p – wt – 2-47, and (C) p – C21S – 2-47.
Note that the log-probability plots do not represent converged simulations, so no conclusions can be drawn on the barrier heights separating the minima. Nevertheless, these observations suggest that the C21S mutation has a much smaller effect on the shorter sequence. More importantly, these results clearly show that the shorter sequence has a stabilizing effect on the parallel conformation. Finally, the antiparallel conformation remains intact, regardless of sequence length or the presence of the C21S mutation.
The effect of ions on the dimeric conformation
So far, our simulations seem to suggest that the parallel structure occurs only under very specific conditions, i.e., when the protein contains the C21S mutation and has a specific sequence length. The antiparallel conformation is not affected by this mutation or by sequence length. A recent crystal structure of the H-NS dimerization domain, comprising residues 2–83 and the C21S mutation (16), as well as a crystal structure of the H-NS homolog, VicH (37) reveal an antiparallel conformation. It is noteworthy that the 2–83 H-NS protein crystals could only be obtained in absence of inorganic salt (38). This prompted us to perform MD simulations of the dimerization domain at two concentrations of NaCl, as our previous simulations did not include ions. The antiparallel conformation is not affected by the presence of 0.01 M NaCl, as the value of dC-C remains at ∼3.7 nm (Fig. 5 A, 0.01 M). Adding 0.5 M NaCl, however, resulted in the C-terminal helices losing helical structure after ∼30 ns, as indicated by the larger fluctuations in dC-C (Fig. 5 A, 0.5 M).
Figure 5.
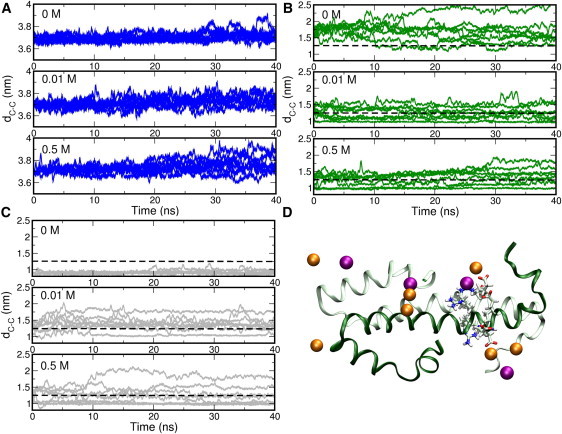
Time evolution of distance between C-terminal ends dC-C. For each system, eight simulations were performed initiated from different starting velocities. (A) The antiparallel complex at 0, 0.01, and 0.5 M NaCl. (B) The parallel complex at 0, 0.01, and 0.5 M NaCl and (C) the parallel complex with C21S mutation at 0, 0.01, and 0.5 M NaCl. (Dotted line) Values of dC-C indicate intact complexes. The range indicates the residues included in the simulations. (Solid lines) Running average of 100 ps. The actual distances were calculated every 2 ps. (D) Snapshot from the p – wt – 2–58 simulations showing the protein backbone (green ribbons), region 39–43 (gray sticks, carbon; blue sticks, nitrogen; red sticks, oxygen; and white sticks, hydrogen), and ions within 0.45 nm of the protein (orange spheres, Na+, purple spheres, Cl−).
More surprising is the effect of NaCl on the parallel conformation. Fig. 5 B shows the time evolution of dC-C of the parallel conformation at different salt concentrations. In absence of salt, it is clear that all simulations have values for dC-C >1.25 nm. When adding salt, several simulations sample dC-C values <1.25 nm, indicating that the complex remains intact. Even though not all simulations resulted in an intact complex, the addition of salt still leads to a stabilization of the parallel complex. The addition of salt diminishes the stabilizing effect of the C21S mutation, as dC-C increases in the presence of salt (Fig. 5 C). At a concentration of 0.01 M NaCl, there are no simulations that sample values of dC-C < 1 nm, whereas at a higher concentration of 0.5 M, half of the runs stay <1 nm for dC-C.
Visual inspection of the trajectories showed that Na+ ions have a preference to occupy specific positions around the complex. These preferred locations center around residues 39–44, exhibiting the highly charged sequence ERREEE (Fig. 5 D). This sequence is located at the end of the coiled-coil region of H-NS, which can be recognized at sequence level via the occurrence of heptad repeats (39). In a perfect heptad repeat, hydrophobic residues occupy the a and d positions, whereas region 39–44 contains a positively charged Arg39 at the d position and negatively charged Glu44 at the a position (Fig. 6 B). In the parallel conformation, these two charged residues are directly in the interface region of the coiled-coil.
Figure 6.
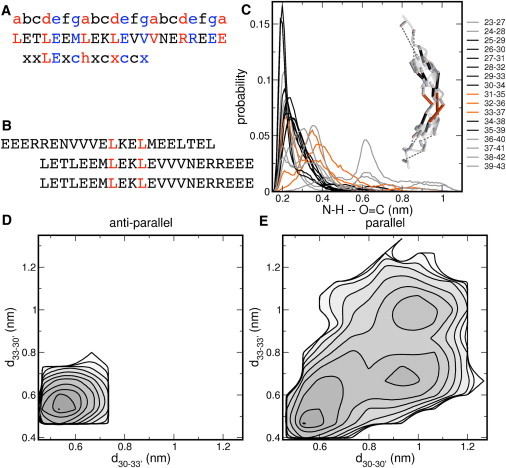
Coiled-coil core of the H-NS dimerization domain. (A) Coiled-coil core sequence of H-NS protein, including residues 23–42, including the heptad-repeat labeling and the location of the trigger sequence. (B) Sequence of the antiparallel and parallel coiled-coil core. (C) Probability distribution of distances of helical hydrogen bonds between atom H of residue n and atom O of residue n+4, n ranges from 23 to 43. (Black lines) Distributions that stay <0.25 nm; (gray lines) distances at the helical ends; and (orange lines) distances that range >0.25 nm and which are not at the termini. (D and E) Log-probability plots of the coiled-coil complexes as a function of the distances between (D) residues Leu30 and Leu33′d30-33′ and Leu30′ and Leu33d30′-33 and (E) residues Leu30 and Leu30′d30-30′ and Leu33 and Leu33d33-33′.
In absence of salt, these charged residues repel each other, therewith causing the disruption of the parallel complex. The presence of ions screen these charges, reducing the repulsive interaction and thus providing stability for the parallel complex. Monitoring the distance between the charged oxygen atoms of Glu44 and the Na+ ion closest to them, and dC-C, illustrates the stabilizing effect for the simulations at 0.01 M NaCl as a log-probability plot, exhibiting two minima (see Fig. S1 A in the Supporting Material). One minimum represents the intact complex at (Na+-Glu44 = 0.25 nm, dC-C = 1 nm), with a sodium ion very close to the glutamate. Another minimum represents disrupted conformations at dC-C ranging from 1.2 to 1.4 nm, and a large distance between Glu44 and the closest Na+ ion. Note that there is no stable state at low dC-C when the ions are far from Glu44. The C21S mutant does not display similar behavior, as the location of an Na+ ion is not related to the value of dC-C (see Fig. S1 B).
The coiled-coil core of H-NS
As both the parallel and the antiparallel complex are coiled-coil complexes, we now focus on the coiled-coil properties of H-NS. H-NS exhibits two heptad repeats with deviations at the hydrophilic g position, a methionine at g in the first heptad repeat, and a valine in the second repeat (see Fig. 6 A). A third heptad repeat contains a more extreme deviation, as elucidated in the previous section. Close inspection of the H-NS coiled-coil region revealed that it exhibits similarity to the consensus trigger sequence, a region that is crucial to coiled-coil formation (40,41). One hypothesis regarding coiled-coil formation requires part of the coiled-coil to retain its helical structure even as a monomer. To test whether the coiled-coil region remains helical in isolation, we cut out residues 23–43 from the antiparallel complex, solvated it, and performed eight 10-ns MD simulations initiated from different starting velocities.
As a measure for the helicity of this fragment, we calculated the distances of the helical hydrogen bonds, between N-H and C=O of residues n and n+4, respectively, and plotted the probability distribution in Fig. 6 B. Distances < 0.3 nm indicate that a hydrogen bond exists for a particular pair and these distances are colored black. Larger distances are colored gray, when these occur at the ends of the peptide, and orange for all other. This visualization facilitates locating helical hydrogen bonds that break during the simulation. The loss of helical structure at the peptide ends is caused by solvation of the hydrogen bonds by bulk water. The increasing hydrogen bond lengths for pairs 31–35, 32–36, and 33–37 display the inherent flexibility of the peptide. In isolation, the coiled-coil fragment does not lose its helical structure entirely, but instead bends in the middle of the second heptad repeat.
From both the parallel and the antiparallel complex, we cut out the coiled-coil region, comprising residues 23–43 (see Fig. 6 B) and performed eight times 10 ns MD simulations. To investigate the stability of the two complexes we used the distances between hydrophobic residues Leu30 and Leu33 in the coiled-coil core. For the antiparallel complex, hydrophobic interactions occur between Leu30 and Leu33′, and Leu33 and Leu30′, indicated by d30-33′ and d33-30′ respectively. For the parallel complex, the interaction pattern consists of Leu30 and Leu30′, and Leu33 and Leu33′, indicated by d30-30′ and d33-33′, respectively. The log-probability of the coiled-coil complexes are displayed in Fig. 6, C and D. They exhibit distinct differences. Both profiles have a minimum at values for the hydrophobic side-chain distances <0.6 nm, indicating that the complex is intact. For the antiparallel conformation, this is the only minimum occurring in the simulations, from which we conclude that the antiparallel complex remains intact in all eight MD runs. For the parallel complex, we sampled two additional minima, at larger values for d30-30′ and d33-33′. Visual inspection indeed showed disruption and dissociation of the complex (see Fig. S2). In absence of the N-terminal helices, we have thus shown that the coiled-coil complex has a strong preference for the antiparallel orientation.
The isolated monomer
As the two conformations of the H-NS dimerization domain do not have shared contacts, the conversion mechanism necessarily involves dissociation into monomers followed by association into the other conformation. The rebinding can occur in an antiparallel or a parallel fashion, depending on the conformation of the monomer. If both orientations are accessible for the monomer, the conformational spaces of monomers extracted from the antiparallel and parallel complex overlap. We therefore performed 10-ns MD simulations of the dimerization domain monomer extracted from the antiparallel and the parallel conformation. In all simulations, helix α3 does not remain completely helical, but exhibits a kink, as shown in Fig. 7, A and C. Fig. 7, B and D, shows the probability distributions for the helical hydrogen-bond distances N-H–O=C in helix α3. Distances <0.3 nm indicate that a hydrogen bond exists for a particular pair and these distances are colored black. The probability distributions of distances that peak between 0.3 and 0.35 nm are colored blue, indicating weak helical hydrogen bonds. Larger distances are colored gray, when these occur at the ends of the monomer, and orange for all other.
Figure 7.
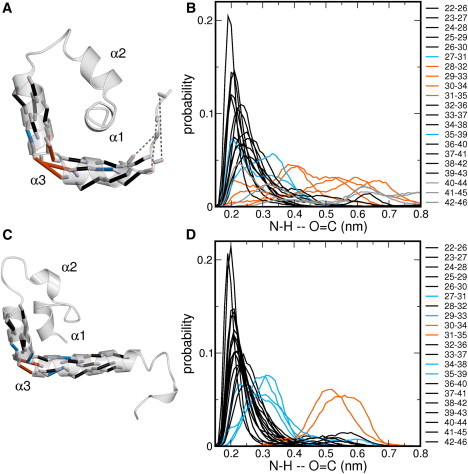
Conformation of the monomer. (A and C) Snapshots (ribbon representation) of monomeric conformations with helical hydrogen bonds (sticks). (A) Monomer extracted from the antiparallel complex and (C) the parallel complex. The labels indicate the three different helices. (B and D) Probability distribution of distances of helical hydrogen bonds between atom H of residue n and atom O of residue n+4 for (B) the antiparallel monomer and (D) the parallel monomer. The value n ranges from 23 to 43. (Black lines) Distributions that stay <0.25 nm; (gray lines) distances at the C-terminal end; (orange lines) distances that range >0.35 nm; and (blue lines) distances that have high probability between 0.25 and 0.35 nm.
This visualization facilitates locating helical hydrogen bonds that break during the simulation, thus pinpointing the location of the kink. The loss of helical structure at the C-terminal end for the antiparallel monomer is caused by solvation of the hydrogen bonds by bulk water. In the monomer simulations originating from the antiparallel complex the kink occurs at hydrogen bonds between 29–33, 30–34, 31–35. and 32–36, and helix α3 is separated into two separate helices. For the longer monomer from the parallel complex, this helix also shows a bend, indicated by increasing distances between residues 31–35 and 32–36. These probabilities have a narrower distribution for the longer sequence. The two monomers extracted from the opposite orientations thus show similar behavior for helix α3, indicating overlap in configuration space.
The snapshots displayed in Fig. 7, A and C, show different conformations for residues 1–20 that include helices α1 and α2. To characterize the differences in orientation and conformation of this region, we calculated log-probability plots using three different order parameters: Σα1, Σα2, and Σ8-14-33. These order parameters indicate the sum of distances of helical hydrogen bonds in helix α1, in helix α2, and the sum of distances among residues Leu8, Leu14, and Leu33, respectively. Fig. 8 shows the resulting log-probability plots. The sampling of the configurational space clearly has not converged. The diamond and the circle indicate the location of the conformations in the antiparallel and parallel complexes, respectively. These locations correlate strongly with the location of the deepest minima and indicate the difference between the two conformations. The minimum of the monomer extracted from the antiparallel conformation is located at (Σα1 = 0.7 nm, Σα2 = 0.8 nm) in Fig. 8 A. Such low values reflect small helical hydrogen-bond distances, indicative of hydrogen bonds, and therefore, these regions have α-helical structure in this monomer.
Figure 8.
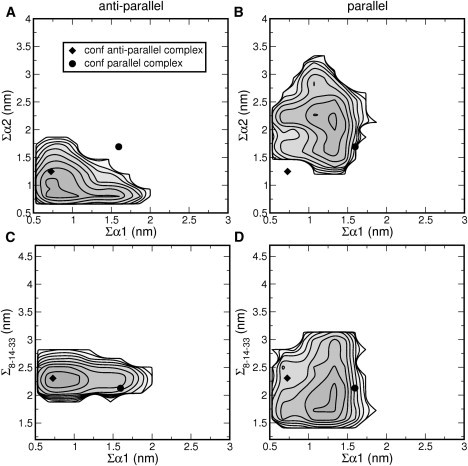
Log-probability plots of MD simulations of two different monomer conformations (A and C) extracted from the antiparallel complex and (B and D) extracted from the parallel complex. The profiles were calculated as a function of (A and B) the sum of helical hydrogen-bond distances in helix α1, Σα1, and helix α2, Σα2; and (C and D) Σα1 and the sum of distances between three hydrophobic groups: Leu8, Leu14, and Leu33, Σ8-14-33. (White areas) Not sampled. (Diamond) Starting conformation of the monomer originating from the antiparallel complex. (Circle) Parallel conformation of the monomer.
The minimum of the monomer obtained from the parallel conformation has higher values for the helical order parameters (Σα1 = 1.8 nm, Σα2 = 2.1 nm) (see Fig. 8 B), indicating that the helical structure in this orientation is less well retained. This monomer has a tighter hydrophobic core, as indicated by the minimum at (Σα1 = 1.8 nm, Σ8-14-33 = 1.7 nm) (see Fig. 8 D) in contrast to the minimum of the monomer originating from the antiparallel conformation at (Σα1 = 2.2 nm, Σ8-14-33 = 0.7 nm) (see Fig. 8 D). Interestingly, the log-probability plot of the monomer extracted from the parallel conformation also contains one additional minimum at (Σα1 = 2.5 nm, Σ8-14-33 = 0.7 nm), which is very similar to the minimum sampled in the simulations of the monomer obtained from the antiparallel complex. Clearly, these log-probability plots show that the sampling was not sufficient to achieve overlap in conformational space. Moreover, they show that the main difference between the monomer conformations in the antiparallel and the parallel form are located in helix α2. Finally, the profiles do show that the monomers are not confined to the conformations they adopted in the two different complexes and that, with longer sampling and/or more advanced simulation methods, convergence can be reached.
Discussion
Our results indicate that the antiparallel complex is the most stable conformation, as this complex stays intact regardless of ionic strength, sequence length, or mutation of Cys21 into serine. This is in agreement with the observations from small-angle x-ray scattering experiments (16). Despite the stability of the antiparallel conformations, introducing the C21S mutation may introduce a slight bias of H-NS toward adopting a parallel coiled-coil conformation. This effect is strongly dependent on the length of the sequence, as it is most pronounced for H-NS residues 2–58 and absent for H-NS residues 2-47. The Cys21-to-serine mutation is introduced to remove the possibility of disulfide bond formation between H-NS molecules, and thus prevent aggregation. With respect to DNA binding, melting, and characteristics in CD spectra, wild-type H-NS and the C21S mutant are similar (42).
Among H-NS homologs and paralogs such as StpA, cysteine is not a strictly conserved amino acid on position 21, and other likely residues for this position are leucine and phenylalanine (9). Nevertheless, this residue is one of the key characteristics to distinguish between H-NS and StpA (9). It is very interesting to note that StpA contains a phenylalanine at position 21 and is susceptible to proteolysis in vivo, whereas the StpA F21C mutant is not. This effect was attributed to the mutation changing the conformation to a more compact stable form (43). Based on that observation and the effects of the C21S mutation seen in our simulations, one could speculate that H-NS homologs and truncated variants, such as StpA or H-NSt not containing this cysteine residue, have a higher propensity to be in the parallel form.
The parallel conformation is also stabilized by the presence of ions as these shield the interactions between two highly charged regions at the C-terminal ends. This region of residues 48–58 mainly consist of alanine, glutamate, and positively charged amino acids (at neutral pH). This region seems to require only minor additional stabilization to remain intact, as the introduction of one additional hydrogen bond is enough to prevent dissociation of this region. The free energy barrier associated with the breaking of a hydrogen bond in a protein is ∼3 kBT (44). It is not uncommon for coiled-coil systems to have a different orientation as a result of slight changes in the sequence. Yadav et al. (45) showed that peptides derived from the GCN4 leucine zipper, that form a four-helical bundle, change from a parallel orientation to antiparallel upon mutation of only one glutamate residue into serine, per peptide. Moreover, SNARE complexes comprise helical bundles to which different proteins contribute occur in parallel and antiparallel forms, of which the parallel form is the most stable (46).
Relating the fact that the stability of the parallel complex strongly depends on the presence of NaCl to the function of H-NS as a salt-dependent regulator, allows for speculation about a possible mechanism for sensing differences in ionic strength by H-NS. At high ionic strength, the antiparallel and parallel conformations of the H-NS dimerization domains are in equilibrium. Upon a drop in ionic strength, this equilibrium is shifted toward the antiparallel conformation. This could be related to the recent observation that divalent ions can mediate a switch between two binding modes of H-NS in vitro (13). In vivo a conformational change and/or a switch between binding modes would likely affect the genome-wide binding of H-NS resulting in changes in global gene expression patterns. The salt-sensing mechanism that we propose assumes that the parallel and antiparallel conformation can be converted into one another passing through a monomeric intermediate. More advanced molecular simulation methods aimed at sampling rare events are required for testing this hypothesis.
Conclusion
We have shown that the antiparallel conformation of the H-NS dimerization domain remains intact under a wide variety of conditions. These conditions include truncation and extension of sequence, addition of NaCl, and the mutation of Cys21 to serine. Also, the coiled-coil core of the antiparallel conformation remains intact when simulated in isolation. In contrast, the parallel conformation is very sensitive to changes in sequence length, the C21S mutation, and the salt concentration. The C21S mutation stabilizes the parallel conformation for a specific sequence length containing residues 2–58, whereas this effect is lost upon truncation to 2–47. The coiled-coil core in parallel conformation unfolds and/or dissociates when simulated in isolation.
Addition of NaCl resulted in stabilization of the parallel conformation, which indicates that the parallel conformation could play a role in the salt-sensing mechanism of H-NS. Changes in salt could then alter the ratio between parallel and antiparallel conformations. Given the importance of the cysteine at position 21, the equilibrium in H-NS homologs or truncates lacking this residue might be shifted toward the parallel structure. Analogously, heterodimer formation with paralogs such as StpA or H-NSt might be a mechanism to shift the equilibrium to a different structure. Structures of the isolated dimerization domains provide important insight into the possible conformations of the full-length H-NS. It is, however, important to realize that the existing controversy around the orientations of the dimerization domain will be only fully resolved once studies addressing the structure of the full-length protein become available.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Dame R.T., Dorman C.J. Springer; Dordrecht, The Netherlands: 2010. Bacterial Chromatin. [Google Scholar]
- 2.Dame R.T., Kalmykowa O.J., Grainger D.C. Chromosomal macrodomains and associated proteins: implications for DNA organization and replication in Gram negative bacteria. PLoS Genet. 2011;7:e1002123. doi: 10.1371/journal.pgen.1002123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luijsterburg M.S., White M.F., Dame R.T. The major architects of chromatin: architectural proteins in bacteria, archaea and eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2008;43:393–418. doi: 10.1080/10409230802528488. [DOI] [PubMed] [Google Scholar]
- 4.Browning D.F., Grainger D.C., Busby S.J.W. Effects of nucleoid-associated proteins on bacterial chromosome structure and gene expression. Curr. Opin. Microbiol. 2010;13:773–780. doi: 10.1016/j.mib.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 5.Dillon S.C., Dorman C.J. Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 2010;8:185–195. doi: 10.1038/nrmicro2261. [DOI] [PubMed] [Google Scholar]
- 6.Dorman C.J. H-NS: a universal regulator for a dynamic genome. Nat. Rev. Microbiol. 2004;2:391–400. doi: 10.1038/nrmicro883. [DOI] [PubMed] [Google Scholar]
- 7.Hommais F., Krin E., Bertin P. Large-scale monitoring of pleiotropic regulation of gene expression by the prokaryotic nucleoid-associated protein, H-NS. Mol. Microbiol. 2001;40:20–36. doi: 10.1046/j.1365-2958.2001.02358.x. [DOI] [PubMed] [Google Scholar]
- 8.White-Ziegler C.A., Davis T.R. Genome-wide identification of H-NS-controlled, temperature-regulated genes in Escherichia coli K-12. J. Bacteriol. 2009;191:1106–1110. doi: 10.1128/JB.00599-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorman C.J., Hinton J.C.D., Free A. Domain organization and oligomerization among H-NS-like nucleoid-associated proteins in bacteria. Trends Microbiol. 1999;7:124–128. doi: 10.1016/s0966-842x(99)01455-9. [DOI] [PubMed] [Google Scholar]
- 10.Dame R.T., Wyman C., Goosen N. H-NS mediated compaction of DNA visualized by atomic force microscopy. Nucleic Acids Res. 2000;28:3504–3510. doi: 10.1093/nar/28.18.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dame R.T., Noom M.C., Wuite G.J.L. Bacterial chromatin organization by H-NS protein unraveled using dual DNA manipulation. Nature. 2006;444:387–390. doi: 10.1038/nature05283. [DOI] [PubMed] [Google Scholar]
- 12.Wiggins P.A., Dame R.T., Wuite G.J. Protein-mediated molecular bridging: a key mechanism in biopolymer organization. Biophys. J. 2009;97:1997–2003. doi: 10.1016/j.bpj.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y., Chen H., Yan J. A divalent switch drives H-NS/DNA-binding conformations between stiffening and bridging modes. Genes Dev. 2010;24:339–344. doi: 10.1101/gad.1883510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esposito D., Petrovic A., Ladbury J.E. H-NS oligomerization domain structure reveals the mechanism for high order self-association of the intact protein. J. Mol. Biol. 2002;324:841–850. doi: 10.1016/s0022-2836(02)01141-5. [DOI] [PubMed] [Google Scholar]
- 15.Bloch V., Yang Y., Kochoyan M. The H-NS dimerization domain defines a new fold contributing to DNA recognition. Nat. Struct. Biol. 2003;10:212–218. doi: 10.1038/nsb904. [DOI] [PubMed] [Google Scholar]
- 16.Arold S.T., Leonard P.G., Ladbury J.E. H-NS forms a superhelical protein scaffold for DNA condensation. Proc. Natl. Acad. Sci. USA. 2010;107:15728–15732. doi: 10.1073/pnas.1006966107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ueguchi C., Mizuno T. The Escherichia coli nucleoid protein H-NS functions directly as a transcriptional repressor. EMBO J. 1993;12:1039–1046. doi: 10.1002/j.1460-2075.1993.tb05745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amit R., Oppenheim A.B., Stavans J. Increased bending rigidity of single DNA molecules by H-NS, a temperature and osmolarity sensor. Biophys. J. 2003;84:2467–2473. doi: 10.1016/S0006-3495(03)75051-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ono S., Goldberg M.D., Ladbury J.E. H-NS is a part of a thermally controlled mechanism for bacterial gene regulation. Biochem. J. 2005;391:203–213. doi: 10.1042/BJ20050453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajkumari K., Kusano S., Gowrishankar J. Effects of H-NS and potassium glutamate on ΣS- and Σ70-directed transcription in vitro from osmotically regulated P1 and P2 promoters of proU in Escherichia coli. J. Bacteriol. 1996;178:4176–4181. doi: 10.1128/jb.178.14.4176-4181.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Porter M.E., Dorman C.J. A role for H-NS in the thermo-osmotic regulation of virulence gene expression in Shigella flexneri. J. Bacteriol. 1994;176:4187–4191. doi: 10.1128/jb.176.13.4187-4191.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frenkel D., Smit B. 2nd ed. Academic Press; San Diego, CA: 2002. Understanding Molecular Simulation. [Google Scholar]
- 23.Van Der Spoel D., Lindahl E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 24.Jorgensen W.L., Maxwell D.S., Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- 25.Kaminski G.A., Friesner R.A., Jorgensen W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations. J. Phys. Chem. B. 2001;105:6474–6487. [Google Scholar]
- 26.Fiser A., Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 27.Horn H.W., Swope W.C., Head-Gordon T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004;120:9665–9678. doi: 10.1063/1.1683075. [DOI] [PubMed] [Google Scholar]
- 28.Darden T., York D., Pedersen L. Particle mesh Ewald—an Nlog(N) method of Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 29.Essmann U., Perea L., Pedersen H.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 30.Hess B., Bekker B., Fraaije J. LINCS: a linear constraint solver. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 31.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 32.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- 33.Nose S., Klein M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983;50:1055–1076. [Google Scholar]
- 34.Stella S., Falconi M., Pon C.L. Environmental control of the in vivo oligomerization of nucleoid protein H-NS. J. Mol. Biol. 2006;355:169–174. doi: 10.1016/j.jmb.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 35.Trachman J.D., Maas W.K. Temperature regulation of heat-labile enterotoxin (LT) synthesis in Escherichia coli is mediated by an interaction of H-NS protein with the LT A-subunit DNA. J. Bacteriol. 1998;180:3715–3718. doi: 10.1128/jb.180.14.3715-3718.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donato G.M., Kawula T.H. Phenotypic analysis of random hns mutations differentiate DNA-binding activity from properties of fimA promoter inversion modulation and bacterial motility. J. Bacteriol. 1999;181:941–948. doi: 10.1128/jb.181.3.941-948.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cerdan R., Bloch V., Arold S.T. Crystal structure of the N-terminal dimerization domain of VicH, the H-NS-like protein of Vibrio cholerae. J. Mol. Biol. 2003;334:179–185. doi: 10.1016/j.jmb.2003.09.051. [DOI] [PubMed] [Google Scholar]
- 38.Leonard P.G., Parkinson G.N., Ladbury J.E. The absence of inorganic salt is required for the crystallization of the complete oligomerization domain of Salmonella typhimurium histone-like nucleoid-structuring protein. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010;66:421–425. doi: 10.1107/S1744309110004574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lupas A. Prediction and analysis of coiled-coil structures. Methods Enzymol. 1996;266:513–525. doi: 10.1016/s0076-6879(96)66032-7. [DOI] [PubMed] [Google Scholar]
- 40.Steinmetz M.O., Jelesarov I., Kammerer R.A. Molecular basis of coiled-coil formation. Proc. Natl. Acad. Sci. USA. 2007;104:7062–7067. doi: 10.1073/pnas.0700321104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frank S., Lustig A., Kammerer R.A. A distinct seven-residue trigger sequence is indispensable for proper coiled-coil formation of the human macrophage scavenger receptor oligomerization domain. J. Biol. Chem. 2000;275:11672–11677. doi: 10.1074/jbc.275.16.11672. [DOI] [PubMed] [Google Scholar]
- 42.Smyth C.P., Lundbäck T., Ladbury J.E. Oligomerization of the chromatin-structuring protein H-NS. Mol. Microbiol. 2000;36:962–972. doi: 10.1046/j.1365-2958.2000.01917.x. [DOI] [PubMed] [Google Scholar]
- 43.Johansson J., Uhlin B.E. Differential protease-mediated turnover of H-NS and StpA revealed by a mutation altering protein stability and stationary-phase survival of Escherichia coli. Proc. Natl. Acad. Sci. USA. 1999;96:10776–10781. doi: 10.1073/pnas.96.19.10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van der Spoel D., van Maaren P.J., Tîmneanu N. Thermodynamics of hydrogen bonding in hydrophilic and hydrophobic media. J. Phys. Chem. B. 2006;110:4393–4398. doi: 10.1021/jp0572535. [DOI] [PubMed] [Google Scholar]
- 45.Yadav M.K., Leman L.J., Ghadiri M.R. Coiled coils at the edge of configurational heterogeneity. Structural analyses of parallel and antiparallel homotetrameric coiled coils reveal configurational sensitivity to a single solvent-exposed amino acid substitution. Biochemistry. 2006;45:4463–4473. doi: 10.1021/bi060092q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weninger K., Bowen M.E., Brunger A.T. Single-molecule studies of SNARE complex assembly reveal parallel and antiparallel configurations. Proc. Natl. Acad. Sci. USA. 2003;100:14800–14805. doi: 10.1073/pnas.2036428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.