

Abstract
H4B is an essential catalytic cofactor of the mNOSs. It acts as an electron donor and activates the ferrous heme-oxygen complex intermediate during Arg oxidation (first step) and NOHA oxidation (second step) leading to nitric oxide and citrulline as final products. However, its role as a proton donor is still debated. Furthermore, its exact involvement has never been explored for other NOSs such as NOS-like proteins from bacteria. This article proposes a comparative study of the role of H4B between iNOS and bsNOS. In this work, we have used freeze-quench to stop the arginine and NOHA oxidation reactions and trap reaction intermediates. We have characterized these intermediates using multifrequency electron paramagnetic resonance. For the first time, to our knowledge, we report a radical formation for a nonmammalian NOS. The results indicate that bsNOS, like iNOS, has the capacity to generate a pterin radical during Arg oxidation. Our current electron paramagnetic resonance data suggest that this radical is protonated indicating that H4B may not transfer any proton. In the 2nd step, the radical trapped for iNOS is also suggested to be protonated as in the 1st step, whereas it was not possible to trap a radical for the bsNOS 2nd step. Our data highlight potential differences for the catalytic mechanism of NOHA oxidation between mammalian and bacterial NOSs.
Abbreviations used: Arg, L-arginine; EPR, electron paramagnetic resonance; FeIINO, ferrous heme-nitrosyl complex; FeIIO2, ferrous heme-oxygen complex; FeIIINO, ferric heme-nitrosyl complex; H4B, (6R)-5,6,7,8-tetrahydro-L-biopterin; HS-5c, high-spin hexacoordinated iron; NO, nitric oxide; NOHA, Nω-hydroxy-L-arginine; NOS, nitric oxide synthase; NOSoxy, oxygenase domain of NOS; bacNOS, bacterial NOS-like proteins; eNOS, endothelial nitric oxide synthase; iNOS, inducible nitric oxide synthase; mNOS, mammalian nitric oxide synthase; nNOS, neuronal nitric oxide synthase; bsNOS, NOS–like protein isolated from Bacillus subtilis; CPET, concerted proton electron transfer
Introduction
Nitric oxide is an essential molecule in various biological processes ranging from signaling to cytotoxic purposes (1). It is produced in mammals by NOSs. The mNOSs are homodimeric proteins (2). Each monomer consists of a reductase domain and an oxygenase domain. The reductase domain contains two flavins responsible for the electron transfer from NADPH to the oxygenase domain (3,4). The oxygenase domain bears the catalytic site that consists in a protoporphyrin IX with a cysteine thiolate as proximal ligand (5,6), the substrate binding site and the essential cofactor H4B (Scheme 1, (7)) binding site. H4B is crucial for the protein structure, especially for the inducible NOS (iNOS), as it induces structural reorganization, forces H2O out of the substrate cavity, and permits the ferric heme iron to become high-spin pentacoordinated (HS-5c FeIII) (8).
Scheme 1.
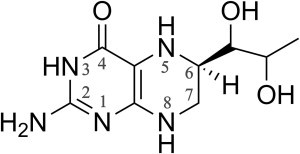
H4B chemical structure.
Like cytochrome P450 (CYP450), NOS are heme-thiolate proteins and are believed to exhibit a similar molecular mechanism (9–11). However H4B is not present for CYP450 and bears an essential redox role in NOS catalytic mechanism (Scheme 2). NOS produces NO through a two-step reaction. The Arg substrate is first oxidized into NOHA (12,13). The 2nd catalytic step, NOHA oxidation, leads to the release of NO and citrulline (14). The most accepted mechanism of Arg oxidation by mNOSs is represented on Scheme 2 A. FeIII is first reduced by an electron from the reductase domain. This event is followed by oxygen binding and the resulting complex is converted into a peroxo intermediate via an electron transfer from H4B. Two proton transfers are necessary for the heterolytic cleavage of the O–O bond. One of these protons could be given by the H4B considering that CPET is possible. The result is the formation of a Compound I species and water release. Compound I is responsible for Arg oxidation and NOHA formation (15,16). The biopterin radical is then reduced by an electron from the reductase domain (17). The 2nd catalytic step of NOS involves the same initial electron and proton transfer up to the formation of an (hydro)peroxo complex (Scheme 2 B). The subsequent course of events is yet unresolved. A nucleophilic attack of the peroxide intermediate on the NOHA guanidinium moiety is believed to lead to a tetrahedral complex (18), whose rearrangement results in the formation of a FeIIINO intermediate and then NO release (19).
Scheme 2.
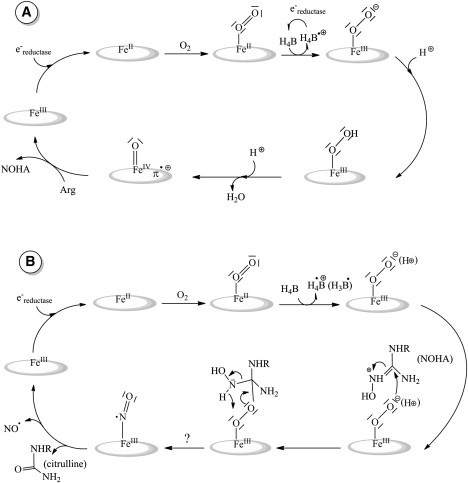
Proposed mechanisms of Arg (A) and NOHA (B) oxidations for mNOS. For both steps FeIII is first reduced by an e− from the reductase that is followed by oxygen binding. The oxo intermediate is converted into a (hydro)peroxo-complex via an electron transfer from H4B. In the 1st step (A) two proton transfers are necessary to achieve heterolytic cleavage of the O–O bond. This will result in the formation of a Compound I responsible for Arg oxidation and NOHA formation. For the 2nd step (B) NOHA oxidation is achieved after the nucleophilic attack of the peroxide intermediate on the NOHA guanidinium moiety leading to a tetrahedral complex. The course of events is unresolved yet but surely finishes with a FeIIINO intermediate responsible for NO release.
Despite intensive investigations, this catalytic model remains mostly hypothetic (20). With regard to the 1st step (Arg hydroxylation), most of the reaction intermediates (such as Compound I) remain elusive. (Hydro)peroxo-intermediates have been trapped only in extreme conditions and in the absence of H4B (21,22). The H4B redox role has been demonstrated by radical trapping for the three mammalian NOS isoforms: iNOS, eNOS, and nNOS, respectively (23–25). Its formation is kinetically coupled to the FeIIO2 decay and to Arg hydroxylation (26). Although H4B is chemically able to realize a proton coupled electron transfer, this possibility has not been definitely settled. That is why the H4B protonation state is a key problem in the understanding of the NOSs catalytic cycle. The H4B radical that builds up during the 1st step of iNOSoxy has been studied in detail by EPR (27). Stoll et al. have demonstrated that the radical trapped is in the H4B⋅+ state during the 1st step, which suggests that the cofactor transfers only a single electron. During the 2nd step (NOHA oxidation), the H4B radical has also been observed (28) with kinetic correlation to the FeIIO2 decay. However, there was no spectroscopic characterization of the radical, its protonation state was not determined, and H4B remains a potential proton donor during the second catalytic step.
Some bacterial organisms exhibit NOS gene sequences in their genome. These bacNOS share the same oxygenase domain with mNOS (29). The three-dimensional structure of bacNOS is superimposable to the catalytic domain of mNOS, making it a good investigation model (30–32). However, these bacteria do not possess the H4B synthesis machinery (29) except maybe for Bacillus subtilis (33) but they are capable of producing H4B analogs, such as tetrahydrofolate, which also contain the pterin moiety. The pterin binding site in bacNOS is truncated and, unlike iNOS, no structural modifications are observed upon H4B binding. In addition, the redox role of H4B in bacNOS has never been proven. This questions the actual nature and role of biopterins in general in bacNOS catalysis. Because bacNOS are able to produce NO and this activity seems conditioned to the presence of H4B (34), H4B has been routinely used so far in the literature to study bacNOS proteins and compare them to mNOS. To address the redox role of H4B in bacNOS, our objective in this work was to comparatively explore the H4B radical state during catalysis for iNOSoxy and bsNOS. Radicals have been trapped by freeze-quench during FeIIO2 activation. Samples were studied with multifrequency EPR. The H4B radical formed during the iNOS 1st step has been compared to that formed during the 2nd step. In the same way the bsNOS capacity to form H4B radical has been investigated and compared to mNOSoxy.
Materials and Methods
Chemicals
All chemicals were purchased from Sigma or Aldrich (Sigma-Aldrich, St. Louis, MO). H4B and NOHA were purchased from Enzo Life Sciences (Enzo Life Sciences, Farmingdale, NY). NO was purchased from Messer (Messer France SA, Asnières, France).
Samples preparation
Wild-type bsNOS and iNOSoxy were overexpressed in Escherichia coli as described previously (34–36).
FeIII samples were prepared by conditioning the protein with Arg 5 mM or NOHA 5 mM and H4B 800 μM after two washing and concentrating cycles in 100 mM potassium phosphate buffer at pH 7.4. Samples were directly frozen into EPR tubes.
FeIINO samples were prepared from the ferric samples. Anaerobic FeIII NOS was obtained by 100 to 200 cycles of alternate vacuum and argon refilling, directly into an EPR tube. Ferric samples were reduced by an anaerobic dithionite solution (50 mM) added directly into the EPR tube using a gas-tight syringe (Hamilton, Reno, NV) in stoechiometric conditions. A small amount of NO saturated solution was added to form the FeIINO complex. The NO-saturated solutions were prepared by flushing NO gas through a previously degassed buffer.
For freeze-quench experiments, proteins were conditioned with substrate and cofactor after two washing and concentrating cycles. Anaerobic FeIII NOS 500 μM was obtained by 100 to 200 cycles of alternate vacuum and argon refilling, directly into a quartz cuvette. FeII samples were then obtained by reduction of FeIII NOS by the addition of a small volume of anaerobic dithionite solution (50 mM) directly into the cuvette by using a gas-tight syringe. Reduction was monitored by ultraviolet-visible absorption spectroscopy (Uvikon spectrometer from Serlabo Technologies, Entraigues-sur-la-Sorgue, France). To avoid unwanted reactions, the excess of dithionite was eliminated on PD 10 columns (Amersham Pharmacia Biotech, Uppsala, Sweden) or protein desalting spin columns (Thermo Scientific, Courtaboeuf, France) in a glove box. The ferrous protein sample was then quickly mixed at 10°C with an aerobic buffer by using a Freeze-Quench SFM-300 setup (BioLogic Science Instruments, Grenoble, France). The reaction was stopped at a given time by rapid freezing into an isopentane bath at 150 K. The sample was then collected into an EPR tube and frozen at 77 K. The final protein concentration was ∼200 μM. Time calibration on our freeze-quench setup was performed by using a known reaction between hemoglobin and azide (37).
For spectroscopic comparison, tetrahydrobiopterin in solution (CF3COOH:MeOH 6:1) was chemically oxidized by the addition of H2O2 (38). The mixture was immediately frozen in liquid nitrogen.
Some samples were annealed at temperatures above 77 K. This was achieved by transferring the sample to a methanol bath cooled with liquid nitrogen to a given temperature between 140 and 250 K for 1 min. The sample was then frozen back in liquid nitrogen.
EPR spectroscopy
9.4 GHz EPR (X-band) spectra were recorded on a Bruker ELEXSYS 500 spectrometer equipped with a standard TE cavity (Bruker, Wissenbourg, France) and an Oxford Instruments (Abingdon, UK) continuous flow liquid helium cryostat and a temperature control system. 285 GHz EPR spectra were recorded on a lab built transmission spectrometer, described previously (39). The absolute magnetic field was calibrated by using a manganese-doped magnesium oxide standard (40). The absolute error in g was 10−4. However, the relative accuracy was better than 2 × 10−5 in g. Sample temperature regulation was achieved by using a built-in helium flow cryostat. Simulations were performed by using the Easyspin software package (41) and routines written in the lab. The EPR parameters that were used for the simulations are reported in the corresponding tables.
Results
The NOS proteins were reduced and conditioned with saturating concentrations of substrate (Arg or NOHA) and cofactor under anaerobic atmosphere. The catalytic reaction was initiated by rapid mixing of the NOS samples to an air-saturated buffer and was stopped by freeze-quench (see experimental procedures). For iNOSoxy, the time used to stop the reaction was based on the maximum yield of radical reported in the literature: 120 ms for Arg oxidation (25) and 60 ms for NOHA oxidation (28). Additionally, we have used other freezing times to prepare iNOSoxy samples. In the case of bsNOS, various freezing times between 10 and up to 450 ms were tested. With Arg as substrate, we chose the freezing time that yielded the largest amount of H4B radical (35 ms in our case). With NOHA, the data shown here were obtained with a time of flight of 15 ms. Samples trapped by freeze-quench were analyzed by EPR.
X-band EPR spectra obtained on iNOSoxy and bsNOS samples for steps 1 and 2 are shown in Fig. 1. In all cases, the samples exhibit a rhombic EPR signal characteristic of a HS-5c FeIII species showing that the active site has been at least partially oxidized. In the case of the iNOSoxy 1st step (Arg as initial substrate, Fig. 1 a), the effective g-values of the freeze-quench sample agree with a ferric heme with NOHA occupying the substrate cavity (see Table 1). This supports the fact that Arg has been oxidized in the portion of the proteins that display a ferric heme. Similarly, the bsNOS sample initially loaded with Arg also displays a ferric heme EPR signal in agreement with the presence of NOHA in the substrate cavity (Fig. 1 c, Table 1). In the case of the 2nd catalytic step (NOHA as initial substrate), a similar high-spin ferric heme signal is also observed for both iNOSoxy and bsNOS. However, in this case, the effective g-values (Table 1) do not allow distinguishing conclusively between the presence and the absence of NOHA in the substrate binding pocket. It should be noted that the samples obtained from freeze-quench correspond to a trapped state that might have slightly different effective g-values from a prepared native relaxed state due to geometry constraints.
Figure 1.

9 GHz EPR spectra of iNOSoxy and bsNOS freeze-quench samples. iNOSoxy/Arg stopped at 120 ms (a), iNOSoxy/NOHA stopped at 60 ms (b), bsNOS/Arg stopped at 35 ms (c), and bsNOS/NOHA stopped at 15 ms (d). Measurement parameters: microwave frequency 9.496 GHz, microwave power 4 mW, field modulation amplitude 1 mT, temperature 10 K.
Table 1.
Effective g-values of the HS FeIII heme for iNOSoxy and bsNOS
| Samples | g1eff | g2eff | g3eff | E/D |
|---|---|---|---|---|
| iNOSoxy H4B + Arg at 120 ms | 7.63 | 4.05 | 1.80 | 0.076 |
| iNOSoxy H4B + NOHA at 60 ms | 7.66 | 4.05 | 1.80 | 0.078 |
| bsNOS H4B + Arg at 35 ms | 7.71 | 3.97 | 1.79 | 0.081 |
| bsNOS H4B + NOHA at 15 ms | 7.72 | 3.97 | 1.79 | 0.081 |
| iNOSoxy H4B/Arg | 7.57 | 4.13 | 1.81 | 0.073 |
| iNOSoxy H4B/NOHA | 7.66 | 4.04 | 1.80 | 0.078 |
| iNOSoxy H4B/no substrate | 7.68 | 3.98 | 1.79 | 0.079 |
| bsNOS H4B/Arg | 7.64 | 4.07 | 1.80 | 0.077 |
| bsNOS H4B/NOHA | 7.73 | 3.97 | 1.78 | 0.082 |
| bsNOSoxy H4B/no substrate | 7.69 | 3.99 | 1.78 | 0.080 |
Freeze-quench samples (top part) are compared to prepared ferric samples (bottom part) with and without substrates. g-values are given ± 0.05.
In addition to the rhombic HS FeIII signal, the X-band EPR spectra exhibits an additional signal in the g = 2 region (around 340 mT), which is expanded in Fig. 2. The iNOSoxy 1st step sample (Fig. 2 a) exhibits a radical signal attributed to the biopterin cofactor that has been recently well characterized (27). We recorded the EPR spectra at 70 K or above to alleviate the effect of the dipolar coupling to the nearby high-spin heme species and to better resolve the hyperfine pattern of the radical EPR signal (27). In the case of bsNOS, a radical signal is also observed during the 1st step of catalysis (Fig. 2 c). The spectrum exhibits a similar linewidth and hyperfine coupling pattern compared to that of the iNOSoxy 1st step thus supporting its attribution to a biopterin radical. Further evidence for the attribution of the signal to a biopterin radical in this bacNOS has been obtained by high-field EPR (see below). For the first time, to our knowledge, such a radical is trapped and directly observed for a bacterial NOS-like protein.
Figure 2.

g = 2 region of the 9 GHz EPR spectra of iNOSoxy and bsNOS freeze-quench samples. iNOSoxy/Arg stopped at 120 ms (a); iNOSoxy/NOHA stopped at 60 ms (b); bsNOS/Arg stopped at 35 ms (c); bsNOS/NOHA stopped at 15 ms (black line), simulation (gray line) (d). Measurement parameters: microwave frequency 9.496 GHz, microwave power 1 mW (a–c) and 0.06 mW (d), field modulation amplitude 0.4 mT (a–c) and 1 mT (d), temperature 70 K (a–c), and 10 K (d), accumulation 20 scans (a–c) and 5 scans (d).
The EPR signature of the freeze-quenched sample for the second catalytic step of iNOSoxy (Fig. 2 b) is different than that of the first step. Indeed, the g = 2 region of the EPR spectrum shows a mixture of two signals: a radical similar to that observed in the 1st step and an additional signal visible on the low field side. The position of this signal is similar to what is observed for FeIINO. We have therefore recorded EPR spectra for various FeIINO NOS samples (see Fig. S2 in the Supporting Material and Table 2). The additional signal observed for the freeze-quench 2nd step iNOS sample (Fig. 2 b and Fig. 3 a, black line), corresponds well with the signal of an iNOS FeIINO sample with NOHA in the substrate cavity (Fig. 3 a, gray line). The difference EPR spectrum between these two samples yields a radical signal (Fig. 3 b) that is superimposable to that observed for the 1st step. In addition, the spectrum obtained on a sample frozen at an earlier time (15 ms, see Fig. 4 b) clearly show different relative amounts of radical and FeIINO species compared to those of the 60 ms sample (Fig. 4 a). The annealing of the 15 ms sample at different temperatures shows an increase of the radical signal before it starts to decrease (Fig. 4 c). The 240 K annealing point essentially shows a nitrosyl species signal left. These results indicate that the yields of FeIINO and radical species observed experimentally are not correlated and that the signals of these two species do not decrease concomitantly. Therefore, the nitrosyl heme complex observed here cannot be responsible for the reduction of the biopterin radical back into H4B.
Table 2.
Simulation parameters for the 10 K EPR spectra of bsNOS and iNOS FeIINO samples
| g1 (g1 strain) | g2 (g2 strain) | g3 (g3 strain) | A1 (MHz) | A2 (MHz) | A3 (MHz) | |
|---|---|---|---|---|---|---|
| bsNOS H4B/NOHA at 120 ms | 2.073 (0.0180) | 2.006 (0.0000) | 1.974 (0.0117) | 22 | 53 | 25 |
| bsNOS H4B/NOHA | 2.073 (0.0180) | 2.007 (0.0000) | 1.974 (0.0117) | 22 | 58 | 20 |
| bsNOS H4B/Arg (54) | 2.082 (0.0075) | 2.005 (0.0010) | 1.967 (0.0054) | 29 | 59 | 33 |
| bsNOS H4B/citrulline | 2.083 (0.0080) | 2.005 (0.0000) | 1.971 (0.0100) | 30 | 58 | 35 |
| bsNOS H4B/- | 2.082 (0.0050) | 2.004 (0.0000) | 1.972 (0.0025) | 28 | 55 | 29 |
| iNOS H4B/NOHA at 60 ms | 2.070 (0.0160) | 2.009 (0.0032) | 1.984 (0.0140) | 40 | 52 | 14 |
| iNOS H4B/NOHA | 2.073 (0.0160) | 2.009 (0.0032) | 1.984 (0.0140) | 40 | 52 | 14 |
| iNOS H4B/Arg | 2.084 (0.0180) | 2.005 (0.0000) | 1.972 (0.0108) | 30 | 56 | 34 |
| iNOS H4B/Citrulline | 2.084 (0.0080) | 2.005 (0.0000) | 1.974 (0.0122) | 30 | 57 | 37 |
| iNOS H4B/- | 2.084 (0.0080) | 2.005 (0.0000) | 1.974 (0.0117) | 30 | 58 | 36 |
Freeze-quench samples (120 and 60 ms samples) are compared to samples prepared with different substrates. The hyperfine couplings (Ai) correspond to the coupling with the nitrogen nucleus of the NO ligand.
Figure 3.

g = 2 region of the 9 GHz EPR spectra of the 60 ms iNOSoxy + NOHA freeze-quench sample. iNOSoxy/NOHA stopped at 60 ms (black line), scaled spectrum of a prepared iNOSoxy FeIINO sample with NOHA as substrate (gray line) (a); difference spectrum between the last two (b). Measurement parameters: microwave frequency 9.496 GHz, microwave power 1 mW, field modulation amplitude 0.4 mT, temperature 70 K, accumulation 20 scans.
Figure 4.
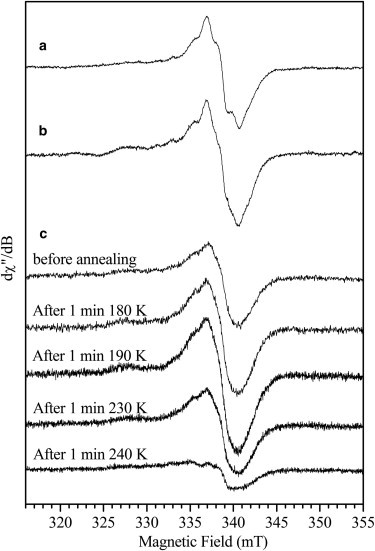
g = 2 region of the 9 GHz EPR spectra of iNOSoxy + NOHA freeze-quench samples. iNOSoxy/NOHA stopped at 60 ms (a); iNOSoxy/NOHA stopped at 15 ms (b). iNOSoxy/NOHA stopped at 15 ms and spectra after annealing (see experimental procedures) (c). Measurement parameters: microwave frequency 9.496 GHz, microwave power 1 mW (a and b) and 0.25 mW (c), field modulation amplitude 0.4 mT (a and b) and 1 mT (c), temperature 70 K (a and b) and 10 K (c), accumulation 20 scans (a and b) and 2 scans (c).
By contrast to iNOSoxy, no radical was observed by freeze-quench experiments for the 2nd step in the case of bsNOS (Figs. 1 d and 2 d), whatever the trapping time used between 10 ms up to 450 ms (see Fig. S1). The spectrum recorded in the g = 2 region exhibits instead a signal characteristic of a FeIINO species in very low quantity. The g-values obtained by simulations (see Fig. S3 and Table 2) are close to those normally obtained for the nitrosyl species when NOHA is present in the distal pocket, as was the case for the FeIINO species observed for the 2nd step of iNOSoxy.
The g-anisotropy of organic radicals is very small but can be resolved by using high-field EPR. The 285 GHz EPR spectra recorded on the same freeze-quench samples analyzed with 9 GHz EPR are shown in Fig. 5 (black lines) along with their simulations (gray lines). Due to a significant decrease of intensity with temperature at this frequency, it was not possible to record the high-field spectra at higher temperatures in our samples. The parameters for the simulations are reported in Table 3. For comparison, we obtained a spectrum in organic solvent for an isolated tetrahydrobiopterin radical oxidized chemically. Noteworthy, the three turning points of the corresponding high-field spectrum (Fig. 5 d) are well resolved and the linewidth narrower compared to what is observed on the protein samples. This is due to the absence of magnetic interaction between the radical and the heme in addition to good glass properties of the organic solvent. The similarities in g-values between the isolated biopterin radical and those observed in the NOS protein samples allows to attribute experimentally the protein radicals to the cofactor (Table 3). The radical obtained with iNOSoxy during Arg oxidation exhibits g-values (gx = 2.0043, gy = 2.0038, and gz = 2.0021) in good agreement with those reported recently in the literature (27). The 285 GHz high-field spectrum for the radical generated in the bsNOS 1st step (Fig. 5 c) is virtually identical to that of the iNOSoxy 1st step. Similarly, the high-field EPR spectrum recorded on the freeze-quenched sample for the 2nd step of iNOSoxy (Fig. 5 b) shows a radical signal identical to that observed for the 1st step.
Figure 5.

285 GHz EPR spectra of iNOSoxy and bsNOS freeze-quench samples. Black lines: experimental spectra for iNOSoxy/Arg stopped at 120 ms (a), iNOSoxy/NOHA stopped at 60 ms (b), bsNOS/Arg stopped at 35 ms (c), and H4B chemically oxidized (d). Gray lines: simulated spectra. Measurement parameters: spectrometer frequency 285 GHz, microwave power 0.25 μW, field modulation amplitude 2 mT, temperature 4 K. Simulation parameters are given in Table 2.
Table 3.
Simulation parameters for the high-field EPR spectra of the H4B radicals
| g1 (g-strain) | g2 (g-strain) | g3 (g-strain) | Azz (MHz) | |
|---|---|---|---|---|
| iNOSoxy H4B/Arg at 120 ms | 2.0043 (0.00003) | 2.0038 (0.00018) | 2.0021 (0.0012) | 63 |
| iNOSoxy H4B/NOHA at 60 ms | 2.0042 (0.00003) | 2.0036 (0.00018) | 2.0021 (0.0012) | 63 |
| bsNOS H4B/Arg at 35 ms | 2.0043 (0.00003) | 2.0036 (0.00018) | 2.0021 (0.0012) | 66 |
| H4B chemical oxidation | 2.0045 | 2.0036 | 2.0022 | 61 |
| iNOSoxy H4B/Arg (27) | 2.0043 | 2.0035 | 2.0021 | 63 |
Hyperfine coupling (Azz) corresponds to the coupling of the N5 nitrogen nucleus of H4B.
For these three radicals, the similarity in g-values (measured from the 285 GHz spectra) and above all hyperfine coupling patterns (comparison of the 9 GHz spectra) strongly support the same protonation state. More precisely, the hyperfine coupling of the N5 nucleus is significantly affected for the neutral radical compared to the protonated radical (27); therefore, a change of protonation state would affect the hyperfine splitting dominating the 9 GHz spectra. This is not observed experimentally (see Fig. 2 and Fig. S4 for samples in D2O), which indicates that all three observed radicals are in the H4B⋅+ state.
Discussion
H4B is believed to play a crucial role in the NOSs molecular mechanism on one hand for its implication in the electron transfer processes required for oxygen activation (23,42) and FeIINO oxidation (28), and on the other hand as a potential proton donor. The pterin radical has been well characterized for the 1st step of iNOSoxy by EPR combined to the density functional theory calculations and demonstrated to be in its protonated form H4B⋅+ (27). During NOS catalysis it has been proposed that the biopterin cofactor acts not only as an electron donor but also as a proton donor in a CPET (21,43,44). If the debate remains open due to the unknown initial protonation state of the pterin and the possibility of ultrafast initial CPET as discussed by Stoll (27), the fact that the trapped radical is H4B⋅+ tend to support a model in which the pterin acts only as an electron donor. In earlier EPR work, the radical trapped for the 1st catalytic step of nNOS and eNOS was already suggested to be protonated (26,45). By analyzing the EPR signals observed both for the heme and the radical, we show that the buildup of the H4B⋅+ radical is correlated to the formation of NOHA-bound ferric species, which indicates that H4B electron transfer is associated to Arg hydroxylation not only for iNOSoxy but also for bsNOS in the 1st step of catalysis.
Investigations on the role of H4B for the iNOSoxy 2nd step are less extended. The trapping of H4B radical during NOHA oxidation has been reported only once (28), but its protonation state has neither been determined nor compared to the 1st step. We completed the characterization of this radical using the same methodological approach for the 2nd catalytic step of iNOSoxy. The comparison of the EPR spectra obtained for this radical relative to that of the 1st step indicates that its protonation state is identical in both steps. Therefore, the biopterin seems to transfer only one electron, and no proton, in the course of NOHA oxidation. The absence of proton transfer from the pterin cofactor, combined with the modification of the water-guanidinium-FeIIO2 H-bond network (46–48), drastically restrains the number of possible proton donors. This result is an additional support for a mechanism that does not involve the heterolytic cleavage of NOS ferric heme-peroxide intermediate.
Compared to the 1st step, the reduction of the H4B⋅+ radical in iNOSoxy is greatly accelerated for the 2nd step (0.71 s−1 vs. 8.3 s−1 (26,28)), which implies that it may exert an oxidative role at the end of the catalytic cycle. Various regeneration pathways for the pterin radical have been proposed, including reduction by the FeIINO intermediate (Scheme 3). The same FeIINO complex, obtained with Angeli's salt, was shown to speed up the H4B⋅+ reduction, supporting an electron transfer from the nitrosyl complex to the pterin radical (49). Thus, the FeIINO oxidation pathway is believed to account for the decay of the pterin radical. However, this should have allowed monitoring the related FeIINO→FeIIINO oxidation reaction, whereas no FeIINO buildup has ever been reported for iNOSs in stopped-flow or freeze-quench experiments. In addition, the reported rates of FeIIINO appearance (36.7 s−1) and pterin radical decay (8.3 s−1) do not match (28), which suggests additional reaction intermediates. Noteworthy, we observed the presence of a FeIINO intermediate along with the H4B⋅+ radical. This is somewhat similar to what has been observed in eNOS loaded with 4-amino-H4B and NOHA by Sørlie and co-workers (44). FeIINO was observed for different freezing times (15 ms and 60 ms) but in different quantities and without any correlation with the pterin radical buildup. Considering the extent of FeIINO buildup and the presence of NOHA in the substrate cavity of the nitrosyl complex, this could arise from some minor side reactions between released NO and residual ferrous heme. In any case the results observed here regarding uncorrelated amounts of radical and nitrosyl species do not support a model for the 2nd step in which the reduction of the radical would be provided by a ferrous heme-nitrosyl intermediate to form FeIIINO that then releases NO.
Scheme 3.
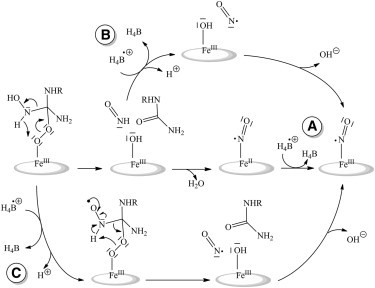
Proposed mechanisms for H4B⋅+ regeneration during NOHA oxidation. (A) After the recombination of NO− with NOS ferric heme, the H4B radical reoxidizes FeIINO into FeIIINO. (B) Direct reaction of the H4B radical with the nitroxyl product. (C) The H4B radical may be able to participate to the tetrahedral complex rearrangement directly leading to NO⋅.
Another aspect of our work deals with the actual role of H4B in bacNOS mechanism. Although bacNOS are commonly used as a study model for mNOSs, they have not been shown to share the same catalytic mechanism. Although H4B (but also tetrahydrofolate) increases the rate of bacNOS FeIIO2 decay and enhances their nitrite production (34), this effect is not necessarily related to a redox role. Furthermore, the effect of the presence of H4B on the FeIIO2 decay is much greater for iNOSoxy than for bsNOS: 120 times faster with H4B for iNOSoxy (28) versus 13 times for bsNOS (34). Radical trapping by freeze-quench allowed us to investigate the formation of a pterin radical during bsNOS catalysis and to compare its properties with those of the radical trapped for iNOSoxy. The radical we trapped for bsNOS during Arg oxidation suggests the capacity of the pterin to transfer one electron to the FeIIO2 species. The protonation state of the radical is the same for both the bsNOS and iNOSoxy 1st steps. These results strongly suggest that bacNOS and mNOS share a common electron transfer process when Arg is the substrate. They support the conservation of the 1st Arg-hydroxylation step and validate the use of bacNOS as a study model for more complex mNOSs.
By contrast, results obtained for the 2nd catalytic step revealed strong differences between iNOSoxy and bsNOS. Indeed, we did not observe any radical build-up for bsNOS 2nd step for a time ranging from 15 to 450 ms. There are a few possibilities for the absence of radical formation in this case: i), First it could arise from extremely fast H4B⋅+ reduction kinetics. The decay of the FeIIO2 species has been reported to be ∼8 s−1 for the 2nd step of bsNOS (34,50) and should correspond to the formation rate of the biopterin radical. Assuming an identical radical decay rate for bsNOS and iNOSoxy (8.3 s−1 (28)), the calculated maximum yield of the radical should be 36% for a reaction time of 120 ms, which should be detected by using our protocol. Therefore, the lack of radical signal would mean a much faster reduction rate (estimated to at least 8 to 10 times that of iNOSoxy 2nd step) so that the maximum radical yield is low enough not to be detected. Although this seems unlikely, it could derive from an increased solvent accessibility to the pterin pocket in the case of bsNOS. However, in that case it should also affect the reduction kinetics for the 1st step and impede the buildup of the radical, which is not the case. ii), We could also envision the absence of oxidative turnover. Indeed, the g-values of ferric bsNOS at 15 ms do not allow excluding the presence of NOHA still in the substrate cavity. Our protocol (initial excess of NOHA and desalting on column) cannot preclude the rebinding of some NOHA as substrate after reaction. This is supported by the fact that the g-values for the FeIINO species observed are in agreement with the presence of NOHA. At the same time, the presence of some FeIINO species argues for the occurrence of some NOHA oxidation. In addition, the measurement of nitrite and nitrate concentrations with the Griess reagent for the 2nd step freeze-quench samples also supports turnover of NOS. iii), The absence of radical trapping for bsNOS 2nd step might as well simply indicate the absence of electron transfer from H4B. This raises the question of the redox role of H4B in the 2nd catalytic step of bacNOS. Indeed, H4B is not formally required to produce NO (2,51). Some chemical considerations and density functional theory calculations plead for an electron or H⋅ transfer directly from the NOHA (52,53). These argumentations could be pertinent for NOS of bacteria for which, noticeably enough, H4B is intrinsically absent. The effect of H4B on bacNOS activity should be reassessed when one considers that this activity (NO or nitrite production) is negligible compared to iNOS production (only 8%). In any case, our results stress the strong discrepancy between iNOSoxy and bsNOS in the role of H4B in the 2nd catalytic step. Further work ongoing in our laboratory aims at elucidating the origin of these differences that could shed some light on the catalytic mechanism and give insights into the role of the bsNOS protein.
Conclusion
Our report furthers the essential investigation of the redox role of H4B in the catalytic activity of various NOS enzymes. We showed that the nature of the pterin radical is similar for both iNOSoxy catalytic steps. Our results are in agreement with an absence of the direct role of H4B in the proton transfer sequences. For the first time, to our knowledge, we showed that bsNOS is able to generate a similar pterin radical as for iNOSoxy. Our results suggest that bacterial and mammalian NOSs share the same mechanism for the 1st catalytic step (Arg hydroxylation). On the opposite, we failed to trap a radical in bsNOS 2nd catalytic step. This supports the hypothesis that the 2nd catalytic step of bsNOS and iNOSoxy probably differs. The possible specificity of bsNOS molecular mechanism and catalytic activity raises again the question of the actual biological functioning and role of bacterial NOS-like proteins.
Acknowledgments
We thank Sun Un for the use of the High-Field EPR spectrometer and useful discussions and Dennis Stuehr for discussions.
Supporting Material
References
- 1.Ignarro L.J. Nitric oxide as a unique signaling molecule in the vascular system: a historical overview. J. Physiol. Pharmacol. 2002;53:503–514. [PubMed] [Google Scholar]
- 2.Alderton W.K., Cooper C.E., Knowles R.G. Nitric oxide synthases: structure, function and inhibition. Biochem. J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stuehr D.J., Tejero J., Haque M.M. Structural and mechanistic aspects of flavoproteins: electron transfer through the nitric oxide synthase flavoprotein domain. FEBS J. 2009;276:3959–3974. doi: 10.1111/j.1742-4658.2009.07120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roman L.J., Martásek P., Masters B.S. Intrinsic and extrinsic modulation of nitric oxide synthase activity. Chem. Rev. 2002;102:1179–1190. doi: 10.1021/cr000661e. [DOI] [PubMed] [Google Scholar]
- 5.White K.A., Marletta M.A. Nitric oxide synthase is a cytochrome P-450 type hemoprotein. Biochemistry. 1992;31:6627–6631. doi: 10.1021/bi00144a001. [DOI] [PubMed] [Google Scholar]
- 6.Crane B.R., Arvai A.S., Tainer J.A. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 7.Wei C.C., Crane B.R., Stuehr D.J. Tetrahydrobiopterin radical enzymology. Chem. Rev. 2003;103:2365–2383. doi: 10.1021/cr0204350. [DOI] [PubMed] [Google Scholar]
- 8.Presta A., Siddhanta U., Stuehr D.J. Comparative functioning of dihydro- and tetrahydropterins in supporting electron transfer, catalysis, and subunit dimerization in inducible nitric oxide synthase. Biochemistry. 1998;37:298–310. doi: 10.1021/bi971944c. [DOI] [PubMed] [Google Scholar]
- 9.Sono M., Stuehr D.J., Dawson J.H. Identification of nitric oxide synthase as a thiolate-ligated heme protein using magnetic circular dichroism spectroscopy. Comparison with cytochrome P-450-CAM and chloroperoxidase. J. Biol. Chem. 1995;270:19943–19948. doi: 10.1074/jbc.270.34.19943. [DOI] [PubMed] [Google Scholar]
- 10.Sono M., Roach M.P., Dawson J.H. Heme-containing oxygenases. Chem. Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 11.Meunier B., de Visser S.P., Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes. Chem. Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 12.Marletta M.A., Hurshman A.R., Rusche K.M. Catalysis by nitric oxide synthase. Curr. Opin. Chem. Biol. 1998;2:656–663. doi: 10.1016/s1367-5931(98)80098-7. [DOI] [PubMed] [Google Scholar]
- 13.Stuehr D.J., Kwon N.S., Wiseman J. N omega-hydroxy-L-arginine is an intermediate in the biosynthesis of nitric oxide from L-arginine. J. Biol. Chem. 1991;266:6259–6263. [PubMed] [Google Scholar]
- 14.Kwon N.S., Nathan C.F., Stuehr D.J. L-citrulline production from L-arginine by macrophage nitric oxide synthase. The ureido oxygen derives from dioxygen. J. Biol. Chem. 1990;265:13442–13445. [PubMed] [Google Scholar]
- 15.Poulos T.L. Structural biology of heme monooxygenases. Biochem. Biophys. Res. Commun. 2005;338:337–345. doi: 10.1016/j.bbrc.2005.07.204. [DOI] [PubMed] [Google Scholar]
- 16.Li H., Poulos T.L. Structure-function studies on nitric oxide synthases. J. Inorg. Biochem. 2005;99:293–305. doi: 10.1016/j.jinorgbio.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 17.Wei C.C., Wang Z.Q., Stuehr D.J. Catalytic reduction of a tetrahydrobiopterin radical within nitric-oxide synthase. J. Biol. Chem. 2008;283:11734–11742. doi: 10.1074/jbc.M709250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woodward J.J., Chang M.M., Marletta M.A. The second step of the nitric oxide synthase reaction: evidence for ferric-peroxo as the active oxidant. J. Am. Chem. Soc. 2009;131:297–305. doi: 10.1021/ja807299t. [DOI] [PubMed] [Google Scholar]
- 19.Boggs S., Huang L., Stuehr D.J. Formation and reactions of the heme-dioxygen intermediate in the first and second steps of nitric oxide synthesis as studied by stopped-flow spectroscopy under single-turnover conditions. Biochemistry. 2000;39:2332–2339. doi: 10.1021/bi9920228. [DOI] [PubMed] [Google Scholar]
- 20.Santolini J. The molecular mechanism of mammalian NO-synthases: a story of electrons and protons. J. Inorg. Biochem. 2011;105:127–141. doi: 10.1016/j.jinorgbio.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 21.Davydov R., Ledbetter-Rogers A., Hoffman B.M. EPR and ENDOR characterization of intermediates in the cryoreduced oxy-nitric oxide synthase heme domain with bound L-arginine or N(G)-hydroxyarginine. Biochemistry. 2002;41:10375–10381. doi: 10.1021/bi0260637. [DOI] [PubMed] [Google Scholar]
- 22.Davydov R., Sudhamsu J., Hoffman B.M. EPR and ENDOR characterization of the reactive intermediates in the generation of NO by cryoreduced oxy-nitric oxide synthase from Geobacillus stearothermophilus. J. Am. Chem. Soc. 2009;131:14493–14507. doi: 10.1021/ja906133h. [DOI] [PubMed] [Google Scholar]
- 23.Bec N., Gorren A.C., Lange R. Reaction of neuronal nitric-oxide synthase with oxygen at low temperature. Evidence for reductive activation of the oxy-ferrous complex by tetrahydrobiopterin. J. Biol. Chem. 1998;273:13502–13508. doi: 10.1074/jbc.273.22.13502. [DOI] [PubMed] [Google Scholar]
- 24.Hurshman A.R., Krebs C., Marletta M.A. Formation of a pterin radical in the reaction of the heme domain of inducible nitric oxide synthase with oxygen. Biochemistry. 1999;38:15689–15696. doi: 10.1021/bi992026c. [DOI] [PubMed] [Google Scholar]
- 25.Wei C.C., Wang Z.Q., Stuehr D.J. Rapid kinetic studies link tetrahydrobiopterin radical formation to heme-dioxy reduction and arginine hydroxylation in inducible nitric-oxide synthase. J. Biol. Chem. 2001;276:315–319. doi: 10.1074/jbc.M008441200. [DOI] [PubMed] [Google Scholar]
- 26.Wei C.C., Wang Z.Q., Stuehr D.J. The three nitric-oxide synthases differ in their kinetics of tetrahydrobiopterin radical formation, heme-dioxy reduction, and arginine hydroxylation. J. Biol. Chem. 2005;280:8929–8935. doi: 10.1074/jbc.M409737200. [DOI] [PubMed] [Google Scholar]
- 27.Stoll S., NejatyJahromy Y., Britt R.D. Nitric oxide synthase stabilizes the tetrahydrobiopterin cofactor radical by controlling its protonation state. J. Am. Chem. Soc. 2010;132:11812–11823. doi: 10.1021/ja105372s. [DOI] [PubMed] [Google Scholar]
- 28.Wei C.C., Wang Z.Q., Stuehr D.J. A tetrahydrobiopterin radical forms and then becomes reduced during Nomega-hydroxyarginine oxidation by nitric-oxide synthase. J. Biol. Chem. 2003;278:46668–46673. doi: 10.1074/jbc.M307682200. [DOI] [PubMed] [Google Scholar]
- 29.Sudhamsu J., Crane B.R. Bacterial nitric oxide synthases: what are they good for? Trends Microbiol. 2009;17:212–218. doi: 10.1016/j.tim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Adak S., Bilwes A.M., Stuehr D.J. Cloning, expression, and characterization of a nitric oxide synthase protein from Deinococcus radiodurans. Proc. Natl. Acad. Sci. USA. 2002;99:107–112. doi: 10.1073/pnas.012470099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pant K., Bilwes A.M., Crane B.R. Structure of a nitric oxide synthase heme protein from Bacillus subtilis. Biochemistry. 2002;41:11071–11079. doi: 10.1021/bi0263715. [DOI] [PubMed] [Google Scholar]
- 32.Bird L.E., Ren J., Stammers D.K. Crystal structure of SANOS, a bacterial nitric oxide synthase oxygenase protein from Staphylococcus aureus. Structure. 2002;10:1687–1696. doi: 10.1016/s0969-2126(02)00911-5. [DOI] [PubMed] [Google Scholar]
- 33.Crane B.R., Sudhamsu J., Patel B.A. Bacterial nitric oxide synthases. Annu. Rev. Biochem. 2010;79:445–470. doi: 10.1146/annurev-biochem-062608-103436. [DOI] [PubMed] [Google Scholar]
- 34.Adak S., Aulak K.S., Stuehr D.J. Direct evidence for nitric oxide production by a nitric-oxide synthase-like protein from Bacillus subtilis. J. Biol. Chem. 2002;277:16167–16171. doi: 10.1074/jbc.M201136200. [DOI] [PubMed] [Google Scholar]
- 35.Ghosh D.K., Wu C., Stuehr D.J. Characterization of the inducible nitric oxide synthase oxygenase domain identifies a 49 amino acid segment required for subunit dimerization and tetrahydrobiopterin interaction. Biochemistry. 1997;36:10609–10619. doi: 10.1021/bi9702290. [DOI] [PubMed] [Google Scholar]
- 36.Gachhui R., Ghosh D.K., Stuehr D.J. Mutagenesis of acidic residues in the oxygenase domain of inducible nitric-oxide synthase identifies a glutamate involved in arginine binding. Biochemistry. 1997;36:5097–5103. doi: 10.1021/bi970331x. [DOI] [PubMed] [Google Scholar]
- 37.Brittain T. The ferric form of the three human embryonic hemoglobins and their reactions with azide ions. J. Inorg. Biochem. 2000;81:99–103. doi: 10.1016/s0162-0134(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 38.Bobst A. Trapping of radicals during the oxidation of tetrahydrofolic acid and tetrahydropterine. Helv. Chim. Acta. 1967;50:2222–2225. [Google Scholar]
- 39.Dorlet P., Rutherford A.W., Un S. Orientation of the tyrosyl D, pheophytin anion, and semiquinone QA⋅- radicals in photosystem II determined by high-field electron paramagnetic resonance. Biochemistry. 2000;39:7826–7834. doi: 10.1021/bi000175l. [DOI] [PubMed] [Google Scholar]
- 40.Burghaus O., Plato M., Lubitz W. 3-mm High-field EPR on semiquinone radical anions Q⋅- related to photosynthesis and on the primary donor P⋅+ and acceptor QA⋅- in reaction centers of Rhodobacter sphaeroides R-26. J. Phys. Chem. 1993;97:7639–7647. [Google Scholar]
- 41.Stoll S., Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 42.Gorren A.C., Bec N., Mayer B. Low-temperature optical absorption spectra suggest a redox role for tetrahydrobiopterin in both steps of nitric oxide synthase catalysis. Biochemistry. 2000;39:11763–11770. doi: 10.1021/bi0007775. [DOI] [PubMed] [Google Scholar]
- 43.Gorren A.C., Mayer B. Nitric-oxide synthase: a cytochrome P450 family foster child. Biochim. Biophys. Acta. 2007;1770:432–445. doi: 10.1016/j.bbagen.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 44.Sorlie M., Gorren A.C., Mayer B. Single-turnover of nitric-oxide synthase in the presence of 4-amino-tetrahydrobiopterin: proposed role for tetrahydrobiopterin as a proton donor. J. Biol. Chem. 2003;278:48602–48610. doi: 10.1074/jbc.M305682200. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt P.P., Lange R., Andersson K.K. Formation of a protonated trihydrobiopterin radical cation in the first reaction cycle of neuronal and endothelial nitric oxide synthase detected by electron paramagnetic resonance spectroscopy. J. Biol. Inorg. Chem. 2001;6:151–158. doi: 10.1007/s007750000185. [DOI] [PubMed] [Google Scholar]
- 46.Chartier F.J., Couture M. Substrate-specific interactions with the heme-bound oxygen molecule of nitric-oxide synthase. J. Biol. Chem. 2007;282:20877–20886. doi: 10.1074/jbc.M701800200. [DOI] [PubMed] [Google Scholar]
- 47.Li D., Kabir M., Yeh S.R. Substrate- and isoform-specific dioxygen complexes of nitric oxide synthase. J. Am. Chem. Soc. 2007;129:6943–6951. doi: 10.1021/ja070683j. [DOI] [PubMed] [Google Scholar]
- 48.Pant K., Crane B.R. Nitrosyl-heme structures of Bacillus subtilis nitric oxide synthase have implications for understanding substrate oxidation. Biochemistry. 2006;45:2537–2544. doi: 10.1021/bi0518848. [DOI] [PubMed] [Google Scholar]
- 49.Woodward J.J., Nejatyjahromy Y., Marletta M.A. Pterin-centered radical as a mechanistic probe of the second step of nitric oxide synthase. J. Am. Chem. Soc. 2010;132:5105–5113. doi: 10.1021/ja909378n. [DOI] [PubMed] [Google Scholar]
- 50.Wang Z.Q., Wei C.C., Stuehr D.J. A conserved Val to Ile switch near the heme pocket of animal and bacterial nitric-oxide synthases helps determine their distinct catalytic profiles. J. Biol. Chem. 2004;279:19018–19025. doi: 10.1074/jbc.M311663200. [DOI] [PubMed] [Google Scholar]
- 51.Zhu Y., Silverman R.B. Revisiting heme mechanisms. A perspective on the mechanisms of nitric oxide synthase (NOS), Heme oxygenase (HO), and cytochrome P450s (CYP450s) Biochemistry. 2008;47:2231–2243. doi: 10.1021/bi7023817. [DOI] [PubMed] [Google Scholar]
- 52.Cho K.B., Gauld J.W. Second half-reaction of nitric oxide synthase: computational insights into the initial step and key proposed intermediate. J. Phys. Chem. B. 2005;109:23706–23714. doi: 10.1021/jp054864o. [DOI] [PubMed] [Google Scholar]
- 53.Robinet J.J., Cho K.B., Gauld J.W. A density functional theory investigation on the mechanism of the second half-reaction of nitric oxide synthase. J. Am. Chem. Soc. 2008;130:3328–3334. doi: 10.1021/ja072650+. [DOI] [PubMed] [Google Scholar]
- 54.Brunel A., Wilson A., Santolini J. The proximal hydrogen bond network modulates Bacillus subtilis nitric-oxide synthase electronic and structural properties. J. Biol. Chem. 2011;286:11997–12005. doi: 10.1074/jbc.M110.195446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.