

Abstract
Hematopoietic stem cells (HSCs) have the capacity to self-renew as well as to differentiate into all blood cell types, and they can reconstitute hematopoiesis in recipients with bone marrow ablation. In addition, transplantation therapy using HSCs is widely performed for the treatment of various incurable diseases such as hematopoietic malignancies and congenital immunodeficiency disorders. For the safe and successful transplantation of HSCs, their genetic and epigenetic integrities need to be maintained properly. Therefore, understanding the molecular mechanisms that respond to various cellular stresses in HSCs is important. The tumor suppressor protein, p53, has been shown to play critical roles in maintenance of “cell integrity” under stress conditions by controlling its target genes that regulate cell cycle arrest, apoptosis, senescence, DNA repair, or changes in metabolism. In this paper, we summarize recent reports that describe various biological functions of HSCs and discuss the roles of p53 associated with them.
1. Introduction
Adult stem cells have recently attracted significant public attention, mostly because they can be a source of donor cells for replacing cells in transplantation therapies to treat various incurable diseases. In fact, hematopoietic stem cell (HSC) transplantation is now routinely performed to treat patients with hematopoietic malignancies and other disorders of the blood and immune systems. Thus, understanding the regulatory molecular networks that regulate stem cells is very important to develop new strategies of treatment for intractable diseases.
Among adult stem cell types, HSCs are the most extensively studied because they are relatively easy to obtain from both healthy and diseased persons, compared with isolation of adult stem cells from other tissues. HSCs are considered a very important cell population capable of self-renewal and differentiating into and supplying all blood cell types for life, whereas other hematopoietic cells such as hematopoietic progenitor cells (HPCs) and more differentiated cells undergo transient proliferation and die within a limited time period. Moreover, HSCs are of interest because of their plasticity to become cell types of other tissues [1, 2].
Under steady-state conditions, most HSCs are in quiescence, a period in the G0 phase of the cell cycle, and proliferate very slowly [3]. Thus, elucidation of the regulatory molecular mechanisms that execute self-renewal as well as entry into and exit from the quiescence of HSCs is essential to understand the biology of HSCs.
Additionally, understanding the molecular pathways of HSCs under the stress conditions of DNA damage is also critical to better deal with suppression of the hematopoietic system by irradiation or the cytotoxic effects of anticancer drugs including arsenic trioxide, anthracycline, cisplatin, and bleomycin that are currently used for the treatment of cancer.
Recent reports have suggested that HSCs are controlled by various cell cycle regulators such as p53, p16Ink4a, and p19Arf under both steady and stress conditions [4, 5]. Among these regulators, p53 is extensively studied and well known as a major tumor suppressor involved in various critical cellular functions such as proliferation, cell cycle arrest, apoptosis, and DNA repair mechanisms [6, 7].
In this paper, we describe the molecular mechanisms for regulation of HSCs under both steady and stress conditions, and particularly the roles of p53 associated with HSC functions such as responses to cellular stresses, apoptosis, self-renewal, senescence, and plasticity in addition to leukemia stem cells (LSCs).
2. p53 as a DNA Damage Checkpoint Molecule
Somatic cells, including immature tissue stem cells, constantly receive intrinsic and extrinsic DNA damage caused by various stresses. To maintain the genomic integrity of stem cells as well as tissue homeostasis, checkpoint mechanisms that activate DNA damage repair are crucial [8]. Among the types of DNA damage, double strand breaks (DSBs), which can be caused by current therapeutic approaches such as ionizing radiation and chemotherapy, are the most cytotoxic type of DNA lesion [9].
To minimize adverse effects caused by DSBs, cells rapidly activate the DNA damage checkpoint pathway after exposure to such stresses. Upon DNA damage, the sensor protein ataxia-telangiectasia mutated (ATM) is activated, which phosphorylates various downstream target proteins and induces the cell cycle checkpoint response [10, 11]. After sensing DNA damage, activated ATM directly phosphorylates the tumor suppressor p53 at serine 15 within its amino-terminal transactivation domain. ATM also activates CHK2, a serine threonine kinase, which phosphorylates p53 at threonine 18 and serine 20. MDM2, an E3 ubiquitin ligase that targets p53, is also phosphorylated by ATM. These phosphorylations that modify p53 and MDM2 directly or indirectly by ATM lead to transcriptional activation and stabilization of p53 [11]. Accumulation of p53 following low or repairable levels of DNA damage leads to activation of p21Cip1 transcription, which inhibits cyclin-dependent kinases (CDKs) and induces a delay or arrest of the cell cycle [12, 13]. During this delay or arrest of cell cycle progression induced by the checkpoint mechanism, cells have an opportunity to repair DNA damage. When DNA damage is high or irreparable, p53 induces transcription of proapoptotic genes such as BAX, NOXA, and PUMA that eliminate damaged cells [14, 15].
Tumor suppressors p16Ink4a and p19Arf are also CDK inhibitors. p16Ink4a and p19Arf are distinct proteins translated from alternative reading frames of the same genomic locus called Cdkn2a. Whereas p16Ink4a binds to CDKs and prevents CDKs binding to cyclin D, which results in the inhibition of phosphorylation of the retinoblastoma (pRb) protein, p19Arf binds to Mdm2 and inhibits its ubiquitin ligase activity toward p53, resulting in promotion of p53 stabilization. These genes are negatively regulated by Bmi-1, which is strongly associated with HSC function as described below in the “p53 apoptosis pathway” section.
3. p53 Signaling against Reactive-Oxygen-Species- (ROS-) Induced Stresses
In living cells including HSCs, ROS such as superoxide anion (•O2−), singlet oxygen (1O2), hydrogen peroxide (H2O2), and hydroxyl radicals (•OH) are continuously generated owing to metabolic and other biochemical reactions as well as stresses caused by various extrinsic factors [16, 17]. While ROS play physiological roles as secondary messengers in intracellular signaling cascades, they also induce oxidative stress that can cause damage to cellular structures, including lipids, membranes, proteins, and DNA, which is thought to contribute toward cancer development [16–18].
Ito et al. reported that CD34− lineage− Sca1+ c-kit+ (CD34− LSK) HSCs can be separated into two fractions, cells with high and low ROS levels (ROShigh and ROSlow HSCs, resp.) and showed that ROSlow HSCs retain their long-term repopulating ability throughout serial transplantation assays, whereas this capacity decreases in serially transplanted ROShigh HSCs [19]. This decreased functional activity of ROShigh HSCs can be restored by treatment with the antioxidant N-acetyl-L-cysteine (NAC), which acts as an antioxidative agent by scavenging ROS, indicating that the ROS level affects HSC functions [19]. Indeed, it has been reported that ROS is an inducer of cell cycling by disrupting the maintenance of HSC quiescence [20].
DNA damage induced by ROS results in p53 upregulation, but p53 expression has not been detected in ROShigh HSCs and only slightly in ROSlow HSCs [21]. The reason for this low level of p53 in ROSlow HSCs is probably because a higher level of p53 decreases the intracellular ROS level to protect the genome from ROS-induced genomic damage by upregulating several antioxidant genes in ROSlow HSCs [22]. This role of p53 in downregulation of the ROS level in HSCs is enhanced by several molecules such as hypoxia inducible factor 1α (HIF-1α), an intrinsic transcription factor activated under a hypoxic condition [23]. HIF-1α stabilizes p53 in a hypoxic area of the bone marrow in vivo [24]. Indeed, HIF-1α deficiency causes an increase in ROS levels and a decrease in HSC numbers during various stresses including a serial transplantation assay, suggesting that HSCs maintain their normal functions via downregulation of ROS by the HIF-1α-p53 pathway [24].
Thus, p53 appears to play a central role in repressing ROS-induced stresses by upregulating antioxidant genes, which results in maintaining quiescence for the survival of HSCs.
4. p53 Apoptosis Pathway
As discussed in the above section, p53 functions in the survival and maintenance of HSCs, but it is also a critical regulator of apoptosis to eradicate HSCs in certain situations. It has been shown that the loss of Bmi-1 in HSCs results in impairment of HSC self-renewal owing to accumulation of p19Arf, which causes p53-dependent apoptosis [25].
Similarly, conditional deletion of Cbp/p300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain, 2 (Cited2) that controls proliferation of mouse embryonic fibroblasts (MEFs) and highly expressed in long-term (LT)-HSCs [26, 27], results in decreased HSC numbers [26]. This phenotype is rescued by additional deletion of p53, indicating that decreased HSC numbers in mice lacking Cited2 are caused by p53-induced cell death [26]. Moreover, inactivation of F-box and WD-40 domain protein 7 (Fbxw7), a subunit of the SKP1-CUL1-F-box protein (SCF)-type ubiquitin ligase complex, leads to impairment of the HSC repopulating capacity and a reduced HSC pool owing to active cell cycling and p53-dependent apoptosis [28, 29].
As discussed above, p53 has two opposing roles in regulating the fate of HSCs, which might depend on the level of damage to HSCs and the effect on p53 activity in each situation (Figure 1).
Figure 1.
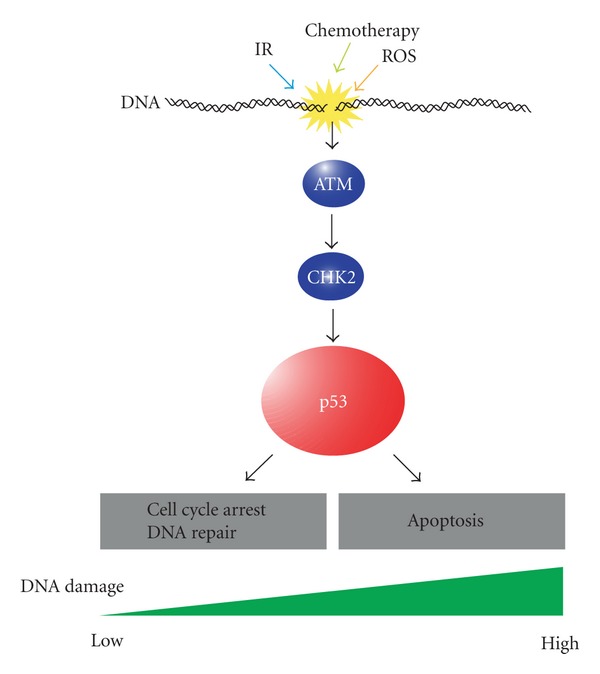
Functions of p53 in response to DNA damage. Genomic stresses caused by ionizing radiation (IR), chemotherapy, and reactive oxygen species (ROS) activate DNA damage check point pathway, which leads to the activation of ATM and CHK2 and subsequent stabilization of p53. Then, stabilized p53 induces cell cycle arrest and DNA repair when DNA damage is moderate, whereas it activates apoptosis pathway when DNA damage is extensive.
5. Roles of the Mdm2-p53 Pathway in HSCs
To examine the significance of the Mdm2-p53 interaction in more detail, p53 mutant protein (p53515C encoding p53R172P) that maintains the ability to induce senescence and cell cycle arrest, but not apoptosis, was analyzed in Mdm2−/−p53515C/515C mice [30]. Mdm2−/−p53515C/515C mice are born at normal Mendelian ratios but die by postnatal day 13 owing to hematopoietic failure [30]. Whereas the HSCs of Mdm2− /−p53515C/515C mice are normal in fetal livers at embryonic day (E)14.5, HSC numbers are significantly decreased in the bone marrow of E18.5 and postnatal mutant mice [30]. Moreover, ROS levels in fetal livers at E14.5 are low, but the bone marrow of E18.5 and postnatal Mdm2−/−p53515C/515C mice has higher ROS levels compared with that of Mdm2+/−p53515C/515C controls [30]. HSCs with a high ROS level in postnatal Mdm2−/−p53515C/515C mice show a reduced repopulating capacity, but this phenotype is rescued by NAC [30]. These results suggest that Mdm2 regulates appropriate hematopoiesis in postnatal mice by repressing ROS levels and regulating p53 activity.
6. Function of p53 in HSC Self-Renewal and Quiescence
It has been shown that hematopoiesis in p53−/− mice is almost normal [31]. However, p53 is preferentially expressed in HSC populations compared with that in various myeloid progenitor cells including common myeloid progenitors (CMPs), granulocyte/monocyte progenitors (GMPs), and megakaryocyte/erythroid progenitors (MEPs) [4]. Lin− Sca-1+ cells are thought to contain HSC population in mice, and cells in quiescence are detected as PyroninYlow cells [4]. Liu et al. have performed PyroninY staining of Lin− Sca-1+ cells from p53−/− mice and found a reduction of PyroninYlow cells in the population, indicating that loss of p53 leads to decreased HSCs in quiescence [4]. HSCs in quiescence are also identified as CD34− lineage− Sca1+ c-kit+ side population cells (CD34− LSK SP cells) based on their ability to efflux the fluorescent dye Hoechst 33342 [3, 32]. Liu et al. also found that CD34− LSK SP cells are decreased in adult bone marrow lacking p53, and proliferation of p53−/− CD34− LSK cells is increased significantly [4]. Furthermore, they identified growth factor independent-1 (Gfi1) and Necdin as p53 target genes that maintain HSC quiescence by comparing the expression profiles of wild-type and p53-null HSCs [4]. Therefore, p53 appears to play a role in promoting HSCs into quiescence, and HSCs tend to enter the cell cycle from quiescence in the absence of p53 (Figure 2). Consistent with this notion, it has been reported that loss of p53 function by the chemical p53 inhibitor, pifithrin β, promotes the proliferation of HSCs in vitro and in vivo [33].
Figure 2.
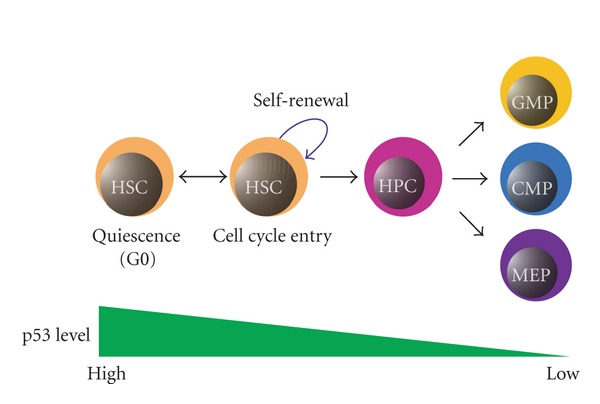
Regulation of quiescence and self-renewal in HSCs by p53. Maintenance of quiescence in HSCs needs proper expression of p53. Decrease in p53 expression promotes self-renewal and differentiation of HSCs.
HSCs lacking p53 are more successful at reconstituting bone marrow in a competitive repopulation assay, indicating that the repopulation capability of HSCs for the first transplantation of bone marrow is improved by inhibition of p53 [4, 34]. On the other hand, Chen et al. compared bone marrow reconstitution abilities between SLAM+ p53+/+ and SLAM+ p53−/− HSCs in a serial transplantation assay and found that lack of p53 reduces the repopulating ability of individual HSCs for serial transplantation, indicating that loss of p53 does not improve the long-term function of HSCs [35]. These results suggest that loss of p53 can improve self-renewal and bone marrow repopulating capabilities over a short term but cannot maintain these capabilities of HSCs over a long term, probably owing to the reduced population of quiescent HSCs that have a long-term repopulating ability.
HSC self-renewal is also affected by pathways activated by extracellular signaling molecules. Notch is a cell surface receptor that influences cell fate decisions such as cell differentiation, survival/apoptosis, and proliferation, which is activated by its ligand Jagged1 [36]. Activation of Notch leads to cleavage and release of the intracellular domain of Notch, which enters the nucleus and activates various transcription factors such as C promoter binding factor 1 (CBF1) [36]. Using transgenic Notch reporter mice, Duncan et al. reported that Notch pathway activation increases HSC self-renewal and decreases HSC differentiation in vivo [37]. However, Mancini et al. reported that HSC self-renewal does not require Jagged1-mediated Notch signaling, using conditional Jagged1−/− mice [38]. Thus, the requirement of Notch signaling for HSC self-renewal is still controversial [39, 40]. It should be noted that p53 is downregulated by Notch signaling [41], implying that an increase in HSC self-renewal by the activation of Notch signaling might be partly mediated by the suppression of p53 function.
HSC self-renewal is also regulated by Wnt signaling, which is activated by binding of the extracellular “Wnt” ligand to the cell surface receptors of the Frizzled family. When Wnt binds to a Frizzled family receptor, β-catenin is stabilized by release from its inhibitory complex consisting of axin, glycogen synthase kinase-3β (GSK-3β), and adenomatous polyposis coli (APC) (the APC/Axin/GSK-3β complex). The release of β-catenin from the APC/Axin/GSK-3β complex allows the translocation of β-catenin to the nucleus to interact and activate T cell factor and lymphoid enhancer factor (TCF/LEF) transcription factors [42, 43]. Reya et al. elucidated that β-catenin overexpression or treatment with soluble Wnt3a in culture promotes HSC self-renewal [44, 45]. Furthermore, activation of Wnt signaling in HSCs increases the expression of Notch1 and HoxB4, which strongly promotes HSC self-renewal [44, 45].
Additionally, microRNA-34 (miR-34) has been shown to bind to the untranslated region of β-catenin, leading to its downregulation [46]. Expression of miR-34 is induced by p53, and, therefore, p53 might downregulate Wnt signaling by miR-34-mediated inhibition of β-catenin, resulting in the suppression of self-renewal in HSCs.
Another essential signaling pathway involved in the regulation of stem cell self-renewal is Hedgehog signaling [47]. However, conditional knockout of Smoothened (Smo), an essential regulator of the Hedgehog pathway, does not show any dysfunction of HSCs, indicating that this pathway is dispensable for adult HSC self-renewal and other functions [48, 49].
The ability of HSCs to self-renew is also affected by the epigenetic status of chromatin structure regulated by components of the Polycomb complex, including Mel18, Rae28, and Bmi-1 [50]. Mice lacking Mel18 exhibit increased HoxB4 expression resulting in increased fetal HSC self-renewal [51]. In contrast, Rae28−/− mice exhibit decreased HSC self-renewal without affecting HoxB4 expression [25, 52]. Moreover, deficiency of Bmi-1 increases p16Ink4a and p19Arf levels, resulting in an increased p53 level, which leads to the suppression of HSC self-renewal [25]. Thus, Polycomb genes appear to regulate HSC self-renewal by various mechanisms.
In addition, another epigenetic regulator of chromatin, Mi-2β, a component of the chromatin remodeling nucleosome remodeling deacetylase (NuRD) complex, is involved in the regulation of HSC self-renewal [53]. Conditional inactivation of Mi-2β in bone marrow promotes HSC differentiation and inhibits HSC self-renewal [53].
The significance of the p53 pathway in regulation of HSC self-renewal by Polycomb or NuRD complexes is poorly understood, except for the molecular mechanism responsible for the inhibition of HSC self-renewal in Bmi-1−/− mice as described above.
CDK inhibitors involved in the G1 checkpoint of the cell cycle have also been shown to regulate HSC self-renewal [47]. Based on their sequence homology and specificity of action, CDK inhibitors are divided into two families: the Cip/Kip family including p21Cip1, p27Kip1, and p57Kip2, and the Ink4 family including p16Ink4a, p15Ink4b, p18Ink4c, and p19Ink4d [54].
Among the Cip/Kip family, p21Cip1 is upregulated by p53 in response to DNA damage, which induces cell cycle arrest by inhibiting CDKs as discussed above in Section 2 [12, 13, 55]. Cheng et al. showed that p21Cip1 plays an essential role in HSC quiescence and self-renewal by analyzing cells from B6/129 mixed background mice [56]. However, van Os et al. showed that HSC self-renewal is not impaired by analyzing pure B6 mice lacking p21Cip1 under normal conditions, whereas a deficiency of p21Cip1 decreases self-renewal in a competitive repopulation assay only when HSCs undergo irradiation stress [57]. The discrepancy regarding the importance of p21Cip1 for HSC self-renewal in a steady state between these two studies might be due to the difference in the genetic background of the mice used for analysis.
p57Kip2, another member of the Cip/Kip family, has been shown to be predominantly expressed in the LT-HSC population [58, 59]. Matsumoto et al. generated conditional p57Kip2-knockout mice and showed that p57Kip2 deficiency decreases HSC quiescence and self-renewal. In addition, loss of p57Kip2 results in upregulation of p53, leading to activation of the p53-dependent apoptosis pathway in HSCs, suggesting that p57Kip2 is required for the maintenance of both quiescence and self-renewal of the HSC pool in adult mice [59].
On the other hand, Zou et al. isolated HSCs from p57Kip2−/− embryos, which are neonatal lethal [60, 61], and showed that loss of p57Kip2 results in a substantial reduction in the repopulating capacity of embryonic HSCs, but does not affect the number of HSCs in quiescence [58]. Moreover, deletion of p57Kip2 results in upregulation of p27Kip1 in embryonic HSCs, and loss of both p57Kip2 and p27Kip1 impairs maintenance of the quiescence and self-renewal of HSCs, which is more obvious compared with those of HSCs in mice lacking p57Kip2 alone, suggesting that p57Kip2 and p27Kip1 cooperate to maintain embryonic HSC quiescence and self-renewal, and p27Kip1 can partially compensate for the function of p57Kip2 [58]. The slight difference regarding the roles of p57Kip2 in the maintenance of quiescence in HSCs might be due to their origin.
Furthermore, loss of p27Kip1 alone in adult mice does not affect HSC self-renewal and quiescence, suggesting that p27Kip1 is not essential for HSC function under normal conditions [58, 62].
It has been shown that loss of other CDK inhibitors, such as p16Ink4a, p15Ink4b, or p18Ink4c in the Ink4 family, results in an increase in HSC self-renewal, although the function of p19Ink4d for HSC self-renewal remains unknown [5, 63–65], indicating that they are independent negative regulators of HSC self-renewal. Interestingly, mice with triple knockout of p16Ink4a, p19Arf, and p53 show a remarkable increase in HSC self-renewal. p19Arf binds to Mdm2 and inhibits the degradation of p53, thus p16Ink4a and the p19Arf-p53 pathway synergistically downregulate the self-renewal capacity of HSCs [5].
Interestingly, among CDK inhibitors, loss of Cip/Kip family members such as p21Cip1, p27Kip1, and p57Kip2 results in reduced HSC self-renewal, whereas loss of Ink4 family members such as p16Ink4a, p15Ink4b, and p18Ink4c leads to increased HSC self-renewal [5, 56–59, 62, 65, 66]. Although this difference regarding the roles of the two families in HSC self-renewal is interesting, its significance remains unknown.
7. Cellular Senescence, Organismal Aging, and p53 in HSCs
Although senescence and quiescence can be considered as analogous phenomena, they are different from each other. While senescence is programmed and essentially irreversible, quiescence is dependent upon environmental stimuli and is reversible [67]. Both are initiated by failure to progress through the G1 phase. Campisi have revealed that cellular senescence plays a critical role in tumor suppression in vivo [68].
Mutation and shortening of telomeres, a region of repeated nucleotide sequences at both ends of a chromosome, are important factors in cellular senescence [69, 70]. During DNA replication, DNA polymerase that synthesizes new DNA cannot completely replicate the telomere. Telomerase extends the telomere region to prevent telomere shortening. Telomerase consists of two essential components: a telomerase RNA component (Terc) and telomerase reverse transcriptase (Tert) [71]. Both components have been shown to be essential for telomerase activity. Terc−/− mice exhibit significantly shortened telomeres in HSCs, reduced regenerative capacity, and impaired hematopoiesis [71, 72]. These hematopoietic failures in Terc−/− mice are caused by activation of p53-dependent senescence in response to DNA damage caused by telomere shortening [71, 73, 74].
Mice expressing a truncated mutant of p53 lacking the first six exons (p53+/m mice) show hyperactive p53 activity, compared with that of wild-type mice, and exhibit an organismal aging phenotype such as reduced longevity, osteoporosis, generalized organ atrophy, and diminished stress tolerance [75]. Moreover, in older mice (18–20 months), the number of LT-HSCs in p53+/− mice increases compared with that in p53+/+ mice, and the number of LT-HSCs in p53+/m mice decreases compared with that in p53+/+ mice, suggesting that an increase or activation of p53 leads to cellular senescence and organismal aging in HSCs [76], although the contribution of cellular senescence to organismal aging is still controversial [77].
8. LSCs and p53
Cancer stem cells (CSCs) are defined as cells that can self-renew, produce various types of progeny cells with more differentiated characteristics, and have a strong ability to drive continued expansion of malignant cells [78–80]. These properties of CSCs have similarities with those that define normal tissue stem cells.
CSCs in leukemia are called LSCs [81], and LSCs in acute myeloid leukemia (AML) have been well characterized [82–84]. Bonnet and Dick have shown that a CD34+ CD38− rare subpopulation of leukemic cells is capable of initiating leukemia in nonobese-diabetic severe combined immunodeficient (NOD-SCID) mice, which is histologically very similar to the original AML [82]. Thus, LSCs share phenotypical similarities with normal HSCs, such as self-renewal and expression of the surface marker, CD34, although there are some differences such as the expression of the interleukin-3 receptor α (IL-3Rα) [85, 86].
The proliferation of LSCs and normal HSCs/HPCs has been shown to be regulated by the Polycomb group (PcG) gene Bmi-1 [87]. Lessard and Sauvageau have shown that Hoxa9 and Meis1a (AML-associated oncogenes) transduced fetal liver cells can form AML in sublethally irradiated syngeneic mice regardless of the presence of Bmi-1. However, Hoxa9-Meis1a transduced leukemic bone marrow cells lacking Bmi-1 cannot induce AML in secondary recipient mice, whereas control Hoxa9-Meis1a transduced leukemic bone marrow cells having Bmi-1 can induce AML, suggesting that Bmi-1 is important for LSCs to retain their capacity to initiate leukemias in vivo [87].
Additionally, in human acute promyelocytic leukemia (APL), expression of PML-RAR, a fusion type of oncogene, induces deacetylation and degradation of p53, leading to repressed p53 transcriptional activity and allowing APL cells to overcome p53-mediated stress responses that induce their eradication [88]. Interestingly, Viale et al. reported that PML-RAR expression in HSCs causes DNA damage and results in upregulation of p21 [89], leading to the cell cycle restriction and repair of damaged DNA [89]. The authors also suggested that the presence of moderate DNA damage, caused by oncogenes, and DNA repair activity enhanced by upregulated p21 increase the chance of mutagenesis in HSCs [89]. Thus, p21 and the associated DNA repair mechanisms appear to play critical roles in initiation and maintenance of LSCs in APL and may be appropriate targets for the treatment of this disease.
Interestingly, the behavior of LSCs is suggested to be associated with the drug resistance of certain types of leukemia [90]. For instance, CD34+ LSCs in chronic myelogenous leukemia (CML) in the G0 phase of the cell cycle (quiescence) are highly insensitive to Imatinib methylate (Gleevec or Glivec; previously known as STI-571 or CGP57148B) that targets the tyrosine kinase activity of BCR-ABL oncogene, whereas most dividing cells are eradicated by the drug [91–102]. Therefore, reduction of an LSC population in quiescence by the inhibition of senescence-inducing proteins such as p53 might be an effective strategy to negate the drug resistance of LSCs [103, 104].
9. Plasticity of HSCs
The notion that tissue-specific stem cells can only differentiate into cells of their tissue origin has been widely accepted, but several recent reports indicate that tissue-specific stem cells, including HSCs, can differentiate into cell types of various lineages [105, 106]. In 1998, Ferrari et al. described that unfractionated normal bone marrow cells transplanted into SCID mice with chemically induced muscle damage can contribute to muscle regeneration [107]. Similarly, Bittner et al. have performed transplantation of normal bone marrow cells into mice with experimental muscular dystrophy, a genetic disease with progressive weakness of skeletal muscles, and found that bone-marrow-derived cells are recruited to skeletal and cardiac muscles and differentiate into muscle cells, although the bone marrow subpopulation that engrafted in muscles was not clearly shown [108]. Orlic et al. reported that transplanted lineage-negative bone marrow cells expressing c-kit (Lin− c-kit+ cells) can contribute to myocardial regeneration in a mouse model of experimental myocardial infarction [109]. Other groups also indicate that bone marrow contains stem cells capable of differentiating into functional muscle cells [1, 2].
Bone marrow cells have also been suggested to contribute to the regeneration of liver cells. Lagasse et al. injected c-kithigh Thylow Lin− Sca-1+ (KTLS) HSCs intravenously into lethally irradiated mice with progressive liver failure and renal tubular damage owing to a lack of fumarylacetoacetate hydrolase (FAH) and found that KTLS HSCs can give rise to functional hepatocytes [110]. Such differentiation of a bone marrow population enriched with HSCs into mature hepatocytes in rodents has also been described by other studies [111, 112]. Moreover, the differentiation of bone-marrow-derived cells into mature hepatocytes has also been found in humans [113, 114].
Lin et al. showed that a small population of circulating endothelial progenitor cells (CEPs) derived from bone marrows exist in peripheral blood and contribute to postnatal neovascularization [115]. Asahara et al. performed transplantation of bone marrow mononuclear cells derived from transgenic mice expressing β-galactosidase (lacZ) driven by the endothelial cell-specific promoter (Flk1/LZ or Tie2/LZ) into lethally irradiated immunodeficient mice and examined neovascularization under various conditions by observing lacZ-positive cells. They concluded that bone-marrow-derived CEPs incorporate into and contribute to postnatal physiological and pathological neovascularization [116]. Krause et al. reported that adult bone marrow cells can differentiate into epithelial cells in the liver, lung, gastrointestinal tract, and skin [117].
As discussed above, although the plasticity of HSCs or cells in bone marrow is intriguing, the molecular mechanisms responsible for the transdifferentiation of HSCs or cells in bone marrow into cells of other tissue lineages remain unknown. One possibility is that the transdifferentiation of HSCs might occur by direct conversion of the epigenetic status of genes. Another possibility is that it might be induced by transient reprogramming of the genome (Figure 3). It has recently been shown that suppression of the p53-p21 pathway promotes the reprogramming efficiency of somatic cells by transfection of reprogramming factors such as Oct3/4 (also known as Pou5f1), Sox2, Klf4, and c-Myc in mice [118]. Thus, inhibition of p53 might facilitate the plasticity of HSCs or bone marrow cells.
Figure 3.
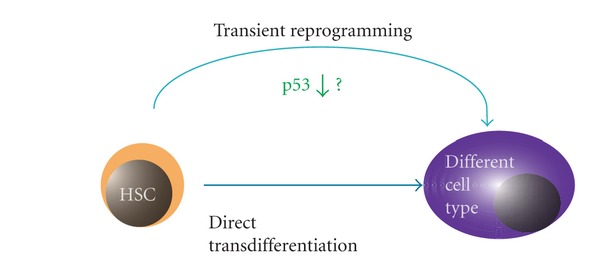
Possible mechanisms of HSC transdifferentiation. HSCs might directly transdifferentiate into another cell type (lower arrow) or through transient reprogramming in certain conditions. Loss of p53 might promote transient reprogramming for HSC transdifferentiation.
10. Conclusions
In this paper, we focused on recent advances in research regarding the roles of p53 associated with the regulation of HSCs and LSCs. It is surprising that one molecule plays roles in the various aspects of important normal cells as well as malignant cells (Figure 4). The importance of HSCs for various transplantation therapies of incurable diseases such as leukemias is obvious, and continuous efforts to elucidate the precise functions of p53 as a main regulator of HSCs will remain crucial and provide an insight into new strategies for treating various disorders.
Figure 4.
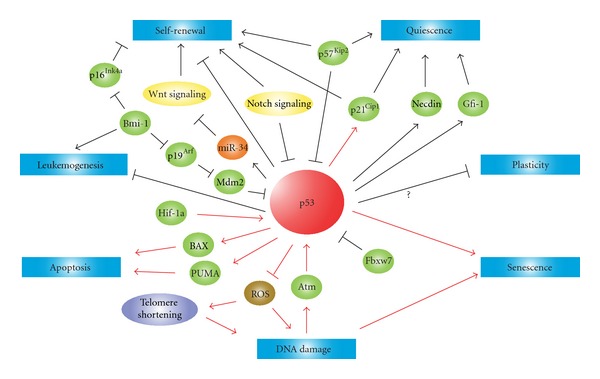
Schematic diagram of p53 roles in HSCs. p53 is involved in the control of response to DNA damage, self-renewal, quiescence, apoptosis, senescence, leukemogenesis, and plasticity in HSCs. Lines with an arrowhead indicate promotion and lines with a bar inhibition in steady state (black) or stress conditions (red).
Acknowledgments
The authors thank Michiko Ushijima for her administrative assistance and the members of Kenzaburo Tani's laboratory for providing constructive criticism and discussions. This work was supported by a grant from the Project for Realization of Regenerative Medicine (K. Tani, 08008010) and KAKENHI (T. Marumoto, 23590465) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
References
- 1.Gussoni E, Soneoka Y, Strickland CD, et al. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature. 1999;401(6751):390–394. doi: 10.1038/43919. [DOI] [PubMed] [Google Scholar]
- 2.Jackson KA, Majka SM, Wang H, et al. Regeneration of ischemic cardiac muscle and vascular endothelium by adult stem cells. Journal of Clinical Investigation. 2001;107(11):1395–1402. doi: 10.1172/JCI12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arai F, Hirao A, Ohmura M, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118(2):149–161. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Elf SE, Miyata Y, et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4(1):37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akala OO, Park IK, Qian D, Pihalja M, Becker MW, Clarke MF. Long-term haematopoietic reconstitution by Trp53−/−p16Ink4a-/-p19Arf-/- multipotent progenitors. Nature. 2008;453(7192):228–232. doi: 10.1038/nature06869. [DOI] [PubMed] [Google Scholar]
- 6.Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22(56):9030–9040. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 7.Marumoto T, Tashiro A, Friedmann-Morvinski D, et al. Development of a novel mouse glioma model using lentiviral vectors. Nature Medicine. 2009;15(1):110–116. doi: 10.1038/nm.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120(4):497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 9.Mohrin M, Bourke E, Alexander D, et al. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7(2):174–185. doi: 10.1016/j.stem.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poehlmann A, Roessner A. Importance of DNA damage checkpoints in the pathogenesis of human cancers. Pathology Research and Practice. 2010;206(9):591–601. doi: 10.1016/j.prp.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432(7015):316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 12.Wahl GM, Carr AM. The evolution of diverse biological responses to DNA damage: insights from yeast and p53. Nature Cell Biology. 2001;3(12):E277–E286. doi: 10.1038/ncb1201-e277. [DOI] [PubMed] [Google Scholar]
- 13.Kastan MB, Lim DS. The many substrates and functions of ATM. Nature Reviews Molecular Cell Biology. 2000;1(3):179–186. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 14.Tang D, Lotze MT, Kang R, Zeh HJ. Apoptosis promotes early tumorigenesis. Oncogene. 2011;30(16):1851–1854. doi: 10.1038/onc.2010.573. [DOI] [PubMed] [Google Scholar]
- 15.Ploner C, Kofler R, Villunger A. Noxa: at the tip of the balance between life and death. Oncogene. 2008;27(1):S84–S92. doi: 10.1038/onc.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loft S, Poulsen HE. Cancer risk and oxidative DNA damage in man. Journal of Molecular Medicine. 1996;74(6):297–312. doi: 10.1007/BF00207507. [DOI] [PubMed] [Google Scholar]
- 17.Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resistance Updates. 2004;7(2):97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-Biological Interactions. 2006;160(1):1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 19.Ito K, Hirao A, Arai F, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nature Medicine. 2006;12(4):446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- 20.Miyamoto K, Araki KY, Naka K, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1(1):101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 21.Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110(8):3056–3063. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nature Medicine. 2005;11(12):1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vukovic V, Haugland HK, Nicklee T, Morrison AJ, Hedley DW. Hypoxia-inducible factor-1α is an intrinsic marker for hypoxia in cervical cancer xenografts. Cancer Research. 2001;61(20):7394–7398. [PubMed] [Google Scholar]
- 24.Takubo K, Goda N, Yamada W, et al. Regulation of the HIF-1α level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7(3):391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 25.Park IK, Qian D, Kiel M, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423(6937):302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 26.Kranc KR, Schepers H, Rodrigues NP, et al. Cited2 is an essential regulator of adult hematopoietic stem cells. Cell Stem Cell. 2009;5(6):659–665. doi: 10.1016/j.stem.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kranc KR, Bamforth SD, Bragança J, Norbury C, Van Lohuizen M, Bhattacharya S. Transcriptional coactivator Cited2 induces Bmi1 and Mel18 and controls fibroblast proliferation via Ink4a/ARF. Molecular and Cellular Biology. 2003;23(21):7658–7666. doi: 10.1128/MCB.23.21.7658-7666.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakayama KI, Nakayama K. Regulation of the Cell Cycle by SCF-Type Ubiquitin Ligases. Elsevier; 2005. [DOI] [PubMed] [Google Scholar]
- 29.Matsuoka S, Oike Y, Onoyama I, et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes and Development. 2008;22(8):986–991. doi: 10.1101/gad.1621808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abbas HA, MacCio DR, Coskun S, et al. Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell. 2010;7(5):606–617. doi: 10.1016/j.stem.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lotem J, Sachs L. Hematopoietic cells from mice deficient in wild-type p53 are more resistant to induction of apoptosis by some agents. Blood. 1993;82(4):1092–1096. [PubMed] [Google Scholar]
- 32.Terunuma A, Jackson KL, Kapoor V, Telford WG, Vogel JC. Side population keratinocytes resembling bone marrow side population stem cells are distinct from label-retaining keratinocyte stem cells. Journal of Investigative Dermatology. 2003;121(5):1095–1103. doi: 10.1046/j.1523-1747.2003.12531.x. [DOI] [PubMed] [Google Scholar]
- 33.Leonova KI, Shneyder J, Antoch MP, et al. A small molecule inhibitor of p53 stimulates amplification of hematopoietic stem cells but does not promote tumor development in mice. Cell Cycle. 2010;9(7):1434–1443. doi: 10.4161/cc.9.7.11508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.TeKippe M, Harrison DE, Chen J. Expansion of hematopoietic stem cell phenotype and activity in Trp53-null mice. Experimental Hematology. 2003;31(6):521–527. doi: 10.1016/s0301-472x(03)00072-9. [DOI] [PubMed] [Google Scholar]
- 35.Chen J, Ellison FM, Keyvanfar K, et al. Enrichment of hematopoietic stem cells with SLAM and LSK markers for the detection of hematopoietic stem cell function in normal and Trp53 null mice. Experimental Hematology. 2008;36(10):1236–1243. doi: 10.1016/j.exphem.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Artavanis-Tsakonas S, Matsuno K, Fortini ME. Notch signaling. Science. 1995;268(5208):225–232. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- 37.Duncan AW, Rattis FM, DiMascio LN, et al. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nature Immunology. 2005;6(3):314–322. doi: 10.1038/ni1164. [DOI] [PubMed] [Google Scholar]
- 38.Mancini SJC, Mantei N, Dumortier A, Suter U, MacDonald HR, Radtke F. Jagged1-dependent Notch signaling is dispensable for hematopoietic stem cell self-renewal and differentiation. Blood. 2005;105(6):2340–2342. doi: 10.1182/blood-2004-08-3207. [DOI] [PubMed] [Google Scholar]
- 39.Pajcini KV, Speck NA, Pear WS. Notch signaling in mammalian hematopoietic stem cells. Leukemia. 2011;25:1525–1532. doi: 10.1038/leu.2011.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzuki T, Chiba S. Notch signaling in hematopoietic stem cells. International Journal of Hematology. 2005;82(4):285–294. doi: 10.1532/IJH97.05115. [DOI] [PubMed] [Google Scholar]
- 41.Beverly LJ, Felsher DW, Capobianco AJ. Suppression of p53 by Notch in lymphomagenesis: implications for initiation and regression. Cancer Research. 2005;65(16):7159–7168. doi: 10.1158/0008-5472.CAN-05-1664. [DOI] [PubMed] [Google Scholar]
- 42.Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes and Development. 1997;11(24):3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- 43.Willert K, Nusse R. β-catenin: a key mediator of Wnt signaling. Current Opinion in Genetics and Development. 1998;8(1):95–102. doi: 10.1016/s0959-437x(98)80068-3. [DOI] [PubMed] [Google Scholar]
- 44.Reya T, Duncan AW, Ailles L, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423(6938):409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 45.Antonchuk J, Sauvageau G, Humphries RK. HOXB4-induced expansion of adult hematopoietic stem cells ex vivo. Cell. 2002;109(1):39–45. doi: 10.1016/s0092-8674(02)00697-9. [DOI] [PubMed] [Google Scholar]
- 46.Kim NH, Kim HS, Kim N-G, et al. p53 and MicroRNA-34 are suppressors of canonical Wnt signaling. Science Signaling. 2011;4(197):p. ra71. doi: 10.1126/scisignal.2001744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Molofsky AV, Pardal R, Morrison SJ. Diverse mechanisms regulate stem cell self-renewal. Current Opinion in Cell Biology. 2004;16(6):700–707. doi: 10.1016/j.ceb.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 48.Hofmann I, Stover EH, Cullen DE, et al. Hedgehog signaling is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem Cell. 2009;4(6):559–567. doi: 10.1016/j.stem.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao J, Graves S, Koch U, et al. Hedgehog signaling is dispensable for adult hematopoietic stem cell function. Cell Stem Cell. 2009;4(6):548–558. doi: 10.1016/j.stem.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jacobs JJL, van Lohuizen M. Polycomb repression: from cellular memory to cellular proliferation and cancer. Biochimica et Biophysica Acta. 2002;1602(2):151–161. doi: 10.1016/s0304-419x(02)00052-5. [DOI] [PubMed] [Google Scholar]
- 51.Kajiume T, Ninomiya Y, Ishihara H, Kanno R, Kanno M. Polycomb group gene mel-18 modulates the self-renewal activity and cell cycle status of hematopoietic stem cells. Experimental Hematology. 2004;32(6):571–578. doi: 10.1016/j.exphem.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 52.Ohta H, Sawada A, Kim JY, et al. Polycomb group gene rae28 is required for sustaining activity of hematopoietic stem cells. Journal of Experimental Medicine. 2002;195(6):759–770. doi: 10.1084/jem.20011911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoshida T, Hazan I, Zhang J, et al. The role of the chromatin remodeler Mi-2β in hematopoietic stem cell self-renewal and multilineage differentiation. Genes and Development. 2008;22(9):1174–1189. doi: 10.1101/gad.1642808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes and Development. 1999;13(12):1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 55.Bunz F, Dutriaux A, Lengauer C, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282(5393):1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 56.Cheng T, Rodrigues N, Shen H, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287(5459):1804–1809. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 57.van Os R, Kamminga LM, Ausema A, et al. A limited role for p21cip1/Waf1 in maintaining normal hematopoietic stem cell functioning. Stem Cells. 2007;25(4):836–843. doi: 10.1634/stemcells.2006-0631. [DOI] [PubMed] [Google Scholar]
- 58.Zou P, Yoshihara H, Hosokawa K, et al. p57Kip2 and p27Kip1 cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell. 2011;9(3):247–261. doi: 10.1016/j.stem.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 59.Matsumoto A, Takeishi S, Kanie T, et al. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2011;9(3):262–271. doi: 10.1016/j.stem.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 60.Yan Y, Frisén J, Lee MH, Massagué J, Barbacid M. Ablation of the CDK inhibitor p57KIP2 results in increased apoptosis and delayed differentiation during mouse development. Genes and Development. 1997;11(8):973–983. doi: 10.1101/gad.11.8.973. [DOI] [PubMed] [Google Scholar]
- 61.Zhang P, Liégeois NJ, Wong C, et al. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature. 1997;387(6629):151–158. doi: 10.1038/387151a0. [DOI] [PubMed] [Google Scholar]
- 62.Cheng T, Rodrigues N, Dombkowski D, Stier S, Scadden DT. Stem cell repopulation efficiency but not pool size is governed by p27kip1. Nature Medicine. 2000;6(11):1235–1240. doi: 10.1038/81335. [DOI] [PubMed] [Google Scholar]
- 63.Ko RM, Kim HG, Wolff L, Klug CA. Roles of p15Ink4b and p16Ink4a in myeloid differentiation and RUNX1-ETO-associated acute myeloid leukemia. Leukemia Research. 2008;32(7):1101–1111. doi: 10.1016/j.leukres.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park BJ, Kang JW, Lee SW, et al. The haploinsufficient tumor suppressor p18 upregulates p53 via interactions with ATM/ATR. Cell. 2005;120(2):209–221. doi: 10.1016/j.cell.2004.11.054. [DOI] [PubMed] [Google Scholar]
- 65.Stepanova L, Sorrentino BP. A limited role for p16Ink4a and p19Arf in the loss of hematopoietic stem cells during proliferative stress. Blood. 2005;106(3):827–832. doi: 10.1182/blood-2004-06-2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yuan Y, Shen H, Franklin DS, Scadden DT, Cheng T. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nature Cell Biology. 2004;6(5):436–442. doi: 10.1038/ncb1126. [DOI] [PubMed] [Google Scholar]
- 67.Rinesh K. Principles of Animal Cell Culture: Student Compendium. International Book Distributing; 2008. [Google Scholar]
- 68.Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends in Cell Biology. 2001;11(11):S27–S31. doi: 10.1016/s0962-8924(01)02151-1. [DOI] [PubMed] [Google Scholar]
- 69.Bodnar AG, Ouellette M, Frolkis M, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279(5349):349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 70.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345(6274):458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 71.Choudhury AR, Ju Z, Djojosubroto MW, et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nature Genetics. 2007;39(1):99–105. doi: 10.1038/ng1937. [DOI] [PubMed] [Google Scholar]
- 72.Rudolph KL, Chang S, Lee HW, et al. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96(5):701–712. doi: 10.1016/s0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- 73.D’Adda Di Fagagna F, Reaper PM, Clay-Farrace L, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426(6963):194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 74.Karlseder J, Broccoli D, Yumin D, Hardy S, De Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283(5406):1321–1325. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- 75.Tyner SD, Venkatachalam S, Choi J, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415(6867):45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 76.Dumble M, Moore L, Chambers SM, et al. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood. 2007;109(4):1736–1742. doi: 10.1182/blood-2006-03-010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311(5765):p. 1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- 78.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 79.Jordan CT, Guzman ML, Noble M. Cancer stem cells. The New England Journal of Medicine. 2006;355(12):1253–1261. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- 80.Jordan CT. Cancer stem cell biology: from leukemia to solid tumors. Current Opinion in Cell Biology. 2004;16(6):708–712. doi: 10.1016/j.ceb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 81.Buss EC, Ho AD. Leukemia stem cells. International Journal of Cancer. 2011;129(10):2328–2336. doi: 10.1002/ijc.26318. [DOI] [PubMed] [Google Scholar]
- 82.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature Medicine. 1997;3(7):730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 83.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 84.Appelbaum FR, Rowe JM, Radich J, Dick JE. Acute myeloid leukemia. Hematology. 2001:62–86. doi: 10.1182/asheducation-2001.1.62. [DOI] [PubMed] [Google Scholar]
- 85.Jordan CT, Upchurch D, Szilvassy SJ, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stems cells. Leukemia. 2000;14(10):1777–1784. doi: 10.1038/sj.leu.2401903. [DOI] [PubMed] [Google Scholar]
- 86.Testa U, Riccioni R, Militi S, et al. Elevated expression of IL-3Rα in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood. 2002;100(8):2980–2988. doi: 10.1182/blood-2002-03-0852. [DOI] [PubMed] [Google Scholar]
- 87.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423(6937):255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 88.Insinga A, Monestiroli S, Ronzoni S, et al. Impairment of p53 acetylation, stability and function by an oncogenic transcription factor. The EMBO Journal. 2004;23(5):1144–1154. doi: 10.1038/sj.emboj.7600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Viale A, De Franco F, Orleth A, et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature. 2009;457(7225):51–56. doi: 10.1038/nature07618. [DOI] [PubMed] [Google Scholar]
- 90.Krause DS, Van Etten RA. Right on target: eradicating leukemic stem cells. Trends in Molecular Medicine. 2007;13(11):470–481. doi: 10.1016/j.molmed.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ben-Neriah Y, Daley GQ, Mes-Masson AM. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 1986;233(4760):212–214. doi: 10.1126/science.3460176. [DOI] [PubMed] [Google Scholar]
- 92.Buchdunger E, Zimmermann J, Mett H, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Research. 1996;56(1):100–104. [PubMed] [Google Scholar]
- 93.Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210(bcr/abl) gene of the Philadelphia chromosome. Science. 1990;247(4944):824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 94.Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. The New England Journal of Medicine. 2001;344(14):1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 95.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293(5531):876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 96.Graham SM, Jørgensen HG, Allan E, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- 97.Heisterkamp N, Stam K, Groffen J. Structural organization of the bcr gene and its role in the Ph’ translocation. Nature. 1985;315(6022):758–761. doi: 10.1038/315758a0. [DOI] [PubMed] [Google Scholar]
- 98.le Coutre P, Tassi E, Varella-Garcia M, et al. Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood. 2000;95(5):1758–1766. [PubMed] [Google Scholar]
- 99.Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247(4946):1079–1082. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- 100.Mahon FX, Deininger MWN, Schultheis B, et al. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood. 2000;96(3):1070–1079. [PubMed] [Google Scholar]
- 101.Shtivelman E, Lifshitz B, Gale RP, Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature. 1985;315(6020):550–554. doi: 10.1038/315550a0. [DOI] [PubMed] [Google Scholar]
- 102.Weisberg E, Griffin JD. Mechanism of resistance to the ABL tyrosine kinase inhibitor STI571 in BCR/ABL-transformed hematopoietic cell lines. Blood. 2000;95(11):3498–3505. [PubMed] [Google Scholar]
- 103.Komarova NL, Wodarz D. Effect of cellular quiescence on the success of targeted CML therapy. PLoS ONE. 2007;2(10, article e990) doi: 10.1371/journal.pone.0000990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rice KN, Jamieson CHM. Molecular pathways to CML stem cells. International Journal of Hematology. 2010;91(5):748–752. doi: 10.1007/s12185-010-0615-8. [DOI] [PubMed] [Google Scholar]
- 105.Jiang Y, Jahagirdar BN, Reinhardt RL, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418(6893):41–49. doi: 10.1038/nature00870. [DOI] [PubMed] [Google Scholar]
- 106.Nakajima H. Role of transcription factors in differentiation and reprogramming of hematopoietic cells. The Keio Journal of Medicine. 2011;60(2):47–55. doi: 10.2302/kjm.60.47. [DOI] [PubMed] [Google Scholar]
- 107.Ferrari G, Cusella-De Angelis G, Coletta M, et al. Muscle regeneration by bone marrow-derived myogenic progenitors. Science. 1998;279(5356):1528–1530. doi: 10.1126/science.279.5356.1528. [DOI] [PubMed] [Google Scholar]
- 108.Bittner RE, Schöfer C, Weipoltshammer K, et al. Recruitment of bone-marrow-derived cells by skeletal and cardiac muscle in adult dystrophic mdx mice. Anatomy and Embryology. 1999;199(5):391–396. doi: 10.1007/s004290050237. [DOI] [PubMed] [Google Scholar]
- 109.Orlic D, Kajstura J, Chimenti S, et al. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410(6829):701–705. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 110.Lagasse E, Connors H, Al-Dhalimy M, et al. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nature Medicine. 2000;6(11):1229–1234. doi: 10.1038/81326. [DOI] [PubMed] [Google Scholar]
- 111.Petersen BE, Bowen WC, Patrene KD, et al. Bone marrow as a potential source of hepatic oval cells. Science. 1999;284(5417):1168–1170. doi: 10.1126/science.284.5417.1168. [DOI] [PubMed] [Google Scholar]
- 112.Theise ND, Badve S, Saxena R, et al. Derivation of hepatocytes from bone marrow cells in mice after radiation-induced myeloablation. Hepatology. 2000;31(1):235–240. doi: 10.1002/hep.510310135. [DOI] [PubMed] [Google Scholar]
- 113.Theise ND, Nimmakayalu M, Gardner R, et al. Liver from bone marrow in humans. Hepatology. 2000;32(1):11–16. doi: 10.1053/jhep.2000.9124. [DOI] [PubMed] [Google Scholar]
- 114.Alison MR, Poulsom R, Jeffery R, et al. Hepatocytes from non-hepatic adult stem cells. Nature. 2000;406(6793):p. 257. doi: 10.1038/35018642. [DOI] [PubMed] [Google Scholar]
- 115.Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. Journal of Clinical Investigation. 2000;105(1):71–77. doi: 10.1172/JCI8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Asahara T, Masuda H, Takahashi T, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circulation Research. 1999;85(3):221–228. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 117.Krause DS, Theise ND, Collector MI, et al. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105(3):369–377. doi: 10.1016/s0092-8674(01)00328-2. [DOI] [PubMed] [Google Scholar]
- 118.Hong H, Takahashi K, Ichisaka T, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460(7259):1132–1135. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]