

Graphical abstract
Highlights
► We review the case for redox involvement in chloroquine resistance in malaria. ► Whether chloroquine kills parasites by generating oxidative stress remains unclear. ► Glutathione levels modulate chloroquine response in a murine malaria species. ► A role for redox in human malaria chloroquine resistance is not firmly established.
Keywords: Plasmodium, Chloroquine resistance, Glutathione, Antioxidant defence
Abstract
Chloroquine (CQ) was once a very effective antimalarial drug that, at its peak, was consumed in the hundreds of millions of doses per year. The drug acts against the Plasmodium parasite during the asexual intra-erythrocytic phase of its lifecycle. Unfortunately, clinical resistance to this drug is now widespread. Questions remain about precisely how CQ kills malaria parasites, and by what means some CQ-resistant (CQR) parasites can withstand much higher concentrations of the drug than others that also fall in the CQR category. In this review we investigate the evidence for and against the proposal that CQ kills parasites by generating oxidative stress. Further, we examine a long-held idea that the glutathione system of malaria parasites plays a role in CQ resistance. We conclude that there is strong evidence that glutathione levels modulate CQ response in the rodent malaria species Plasmodium berghei, but that a role for redox in contributing to the degree of CQ resistance in species infectious to humans has not been firmly established.
1. Introduction
The deadliest malaria parasite of humans, Plasmodium falciparum, must acquire and digest haemoglobin (Hb) from the host erythrocyte in order to survive. Hb digestion takes place within the parasite’s internal digestive vacuole (DV), and results in the generation of large quantities of potentially toxic haem moieties. Chloroquine (CQ) is believed to inhibit the detoxification of haem in the DV.
P. falciparum parasites resistant to CQ emerged in the late 1950s, and throughout the remainder of the 20th century spread from a handful of founder locations to nearly every malaria-endemic region of the world (Wootton et al., 2002; Volkman et al., 2007). The primary determinant of CQ resistance in P. falciparum is mutations in the Chloroquine Resistance Transporter (PfCRT) (Fidock et al., 2000), a 49 kDa protein with 10 predicted transmembrane domains that resides in the membrane bounding the parasite’s DV (Fidock et al., 2000; Cooper et al., 2002). The weak-base nature of CQ results in the ‘trapping’ of the charged, protonated form of the drug in the acidic DV, and its accumulation therein to high concentrations. CQ-resistance-conferring mutations in PfCRT modify the substrate specificity of the protein such that it facilitates the escape of CQ, probably in its predominant diprotonated form, from the DV (Martin et al., 2009). This reduces the concentration of the drug at its site of action.
2. Chloroquine resistance – what determines the level?
Of the numerous mutations that occur in PfCRT, which occur as region-specific haplotypes, the K76T mutation is ubiquitous to CQR parasites. This mutation has been validated in multiple studies as a highly sensitive marker for CQ treatment failure (i.e. almost all patients who fail CQ treatment are infected with PfCRTK76T-bearing parasites) (Djimde et al., 2001; Picot et al., 2009). However, many patients infected with PfCRTK76T-expressing parasites respond adequately to CQ treatment (Wellems and Plowe, 2001). Pre-existing immunity and factors that influence the CQ concentration reached in the plasma most likely aid in the cure of some patients infected with PfCRTK76T-expressing parasites (Wellems and Plowe, 2001; Djimde et al., 2003). Parasite-associated factors that together determine the degree of mutant-PfCRT-mediated CQ resistance are also likely to play a role.
In field isolates, the K76T mutation is never found in isolation, and to date 16 variant residues have been identified in CQ-resistance-conferring forms of PfCRT (Ecker et al., in press). As a consequence, there are many different isoforms of the protein, and it is likely that these would be associated with different levels of susceptibility to CQ. To date, only two naturally occurring mutant forms of PfCRT (those expressed by the Southeast Asian Dd2 strain and the South American 7G8 strain) have been investigated in heterologous expression systems (Martin et al., 2009; Baro et al., 2011). Furthermore, the Dd2 and 7G8 alleles are, to date, the only natural variants for which data have been published on their effects on parasite drug response in the same genetic background (Sidhu et al., 2002). Recent studies on pfcrt variants from Southeast Asia and the Western Pacific confirm that distinct alleles can mediate substantially different degrees of reduced CQ susceptibility (Petersen and Fidock, in preparation).
There is ample evidence that the genetic background of a parasite also plays an important role in determining the level of CQ resistance imparted by mutant pfcrt. A study of the CQ susceptibility of those progeny of a genetic cross (between Dd2 and HB3) that had inherited the mutant Dd2 pfcrt allele revealed CQ IC90 values (i.e. the concentration of CQ required to inhibit parasite proliferation by 90%) ranging from 444 to 1141 nM (Ferdig et al., 2004). Mutations in another DV membrane-localised transporter, P-glycoprotein homologue 1 (Pgh1; encoded by pfmdr1), appear to modulate CQ response in some but not all genetic backgrounds (Reed et al., 2000; Sidhu et al., 2005; Sa et al., 2009; Patel et al., 2010). However, it is clear that parasites with identical pfcrt and pfmdr1 alleles can also vary considerably in their CQ responses (Chen et al., 2002; Ferdig et al., 2004; Sa et al., 2009). A recent transfection study in which wild-type pfcrt was replaced with the mutant 7G8 allele in three different CQ-sensitive (CQS) strains of P. falciparum provided direct evidence that the genetic background of a strain influences the extent to which mutant PfCRT affects CQ response (Valderramos et al., 2010).
The search for secondary determinants of CQ resistance has proven challenging. With the exception of pfmdr1, other genes potentially involved have thus far escaped detection (Mu et al., 2010), suggesting that the individual contributions of other CQ-response-modulating genes may be limited. A search for single nucleotide polymorphisms (SNPs) in predicted or known transporter proteins in 97 culture-adapted isolates (Mu et al., 2003) led to the identification of the P. falciparum Multidrug Resistance-Associated Protein homologue (PfMRP) as a potential secondary determinant of CQ resistance. PfMRP is, like Pgh1, a member of the ATP-binding Cassette (ABC) superfamily of transporter proteins. Two subsequent studies failed to confirm an association between mutations in PfMRP and CQ response (Anderson et al., 2005; Cojean et al., 2006). However, a study in which PfMRP was disrupted in the CQR W2 strain lent some support to the notion that PfMRP may play a role in CQ susceptibility, as the PfMRP knockout parasites accumulated more CQ than the parental strain and were rendered somewhat more susceptible to the drug (Raj et al., 2009).
A separate line of investigation involving a number of biochemical studies has implicated elements of the redox system in influencing parasite susceptibility to CQ. In the following sections we explore the hypothesis that differences in antioxidant power between different CQR strains of P. falciparum contribute to their varying degrees of CQ resistance. Variation in antioxidant power between parasite strains could arise from a number of distinct mechanisms, potentially complicating efforts to identify redox genes that could serve as secondary determinants of CQ resistance.
3. Haemoglobin digestion in the malaria parasite – an oxidative burden
In most eukaryotic cells, the mitochondrial electron transport chain is responsible in large part for the cell’s oxidative burden. Redox centres in this chain can ‘leak’ electrons to O2, resulting in the formation of reactive oxygen species (ROS) (Turrens, 2003). In the case of the malaria parasite, its intra-erythrocytic lifestyle has imparted an additional oxidative burden, arising from the digestion of the majority of the host cell’s ∼5 mM Hb (each molecule of which has four haem groups) as the parasite grows within the infected cell. Hb degradation occurs in large part, if not entirely, within the parasite’s DV (Klonis et al., 2007).
Hb digestion is essential for the parasite, providing it with amino acids for protein synthesis, creating space for its growth, and aiding the osmotic stability of the host cell (Lew et al., 2003; Goldberg, 2005). Hb digestion liberates toxic haem moieties, each of which contains an iron atom chelated in the centre of a porphyrin ring. In the acidic environment of the DV, the haem in oxyHb is oxidised from the Fe2+ to the Fe3+ state. This is accompanied by the production of , which in the acidic DV is thought to dismutate spontaneously to H2O2 and O2 (Becker et al., 2004). The parasite must destroy or neutralise the haem and H2O2, as both species give rise to a variety of toxic effects through their oxidation of biomolecules, and in the case of haem, through additional non-oxidative mechanisms (Fitch, 2004).
3.1. Oxidative stress and the parasite’s mechanisms for dealing with haem
The majority of the haem released in the parasite DV is thought to be sequestered into haemozoin (Hz) (Egan, 2008), an apparently inert substance that consists of chains of Fe3+-haem (“β-haematin”) dimers linked by hydrogen bonds (Pagola et al., 2000). However, one process may not be sufficient to neutralise completely the vast quantities of haem produced in the DV, and even if a small percentage of the haem were to escape incorporation into Hz this could give rise to a concentration that would be toxic to the parasite (Becker et al., 2004). The parasite may therefore have additional mechanisms of haem detoxification (Fig. 1). A number of parasite proteins have been reported to bind haem (Choi et al., 1999; Harwaldt et al., 2002; Campanale et al., 2003), but most of these proteins might also be inhibited by haem, making it difficult to discern whether this binding is a protective mechanism or an additional source of toxicity (Muller, 2004).
Fig. 1.
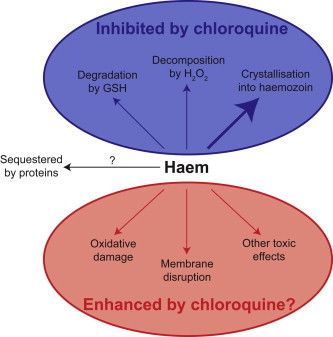
Possible mechanisms of haem toxicity and neutralisation in malaria parasites, and the proposed effect of chloroquine on these processes. GSH, reduced glutathione.
In addition to Hz formation and haem sequestration by protein binding, two mechanisms by which haem might be degraded in the parasite have been described: glutathione-mediated degradation (Atamna and Ginsburg, 1995) and peroxidative decomposition (Loria et al., 1999). Both of these processes ultimately result in the release of iron from haem. Current experimental methods have precluded an unambiguous determination of the oxidation state of the released iron. However, irrespective of its oxidation state the liberated iron, in the presence of ROS, would almost certainly induce oxidative damage by participating in the Fenton reaction (Eq. (1)) or in the Haber–Weiss reaction (Eq. (2)), in which Fe3+ is reduced to Fe2+ so it can then participate in Fenton chemistry (Eq. (1)) (Liochev and Fridovich, 2002), resulting in the generation of hydroxyl radicals (•OH).
| (1) |
| (2) |
Hydroxyl radicals are highly reactive and in vivo often react close to their site of formation. Within the DV, the production of •OH would lead rapidly to the inactivation of proteins and to membrane damage by lipid peroxidation. Fenton chemistry is also especially damaging in vivo where iron levels are elevated, as is the case in the DV of the parasite. In environments such as this ongoing Fenton chemistry can be propagated by the release of additional iron from haem and its breakdown products. As a consequence, maintaining a favourable redox environment is likely to present a constant challenge for the parasite, even when not under chemotherapeutic attack.
4. Does chloroquine increase oxidative stress in the parasite?
CQ accumulates to high concentrations in the DV of the parasite through a combination of weak-base trapping and binding to haem, and the DV is thought to be its primary site of action (Fitch, 2004). The binding of CQ to haem has been reported to inhibit the incorporation of haem into Hz (Chou et al., 1980; Egan et al., 1994; Bray et al., 1998), the glutathione-mediated degradation of haem (Ginsburg et al., 1998), and the peroxidative decomposition of haem (Loria et al., 1999). It is believed that treatment of parasites with CQ results in a build-up of haem and CQ–haem complexes that are toxic to the parasite (Fitch, 2004). The precise molecular basis of CQ-induced toxicity remains to be elucidated.
It has been postulated that a CQ-mediated build-up of haem and CQ–haem complexes in the parasite may generate oxidative stress by enhancing the toxicity of the ROS generated in the DV during Hb degradation, and that this might cause or contribute to parasite death (de Almeida Ribeiro et al., 1997; Loria et al., 1999; Becker et al., 2004). However, it has also been suggested that inhibition of the (iron-releasing) process of glutathione-mediated haem degradation by CQ might lower oxidative stress in the parasite (Ginsburg et al., 1998).
Experimental evidence in favour of CQ giving rise to oxidative stress comes from Radfar et al. (2008), who showed that a 6 h treatment of erythrocytes infected with CQR Dd2 parasites with CQ at its 50% inhibitory concentration (IC50) led to an increase in the degree of oxidation of parasite proteins. The number of parasite proteins that were oxidised was also increased by CQ treatment (Radfar et al., 2008). Another study found that adding a sub-inhibitory concentration of CQ (that reduced parasite proliferation by ⩽10%) to parasites that were also exposed to sub-inhibitory concentrations of oxidative stress inducers (lactoperoxidase, glucose oxidase and H2O2), resulted in combinations that inhibited parasite growth by >25% (Malhotra et al., 1990). However, it was not clear whether such combinations were additive or synergistic, thus rendering the data difficult to interpret.
Evidence against the hypothesis that CQ causes oxidative stress comes from Atamna et al. (1994), who reported that the activity of the pentose phosphate pathway (which generates NADPH, the source of reducing equivalents for the glutathione- and thioredoxin (Trx)-based antioxidant defence systems) was decreased in infected erythrocytes and (to a lesser extent) isolated parasites after a 30 min incubation with CQ, albeit at a supra-pharmacological concentration (5 μM). In another study aimed at testing whether CQ acts through oxidative stress, parasite cultures were grown in an atmosphere comprised of 2% CO (which has a much higher affinity for Hb than O2), 5% CO2 and 93% N2; parasite susceptibility to CQ was then assessed (Monti et al., 2002). Under these conditions, carboxyHb rather than oxyHb would be digested in the parasite’s DV, and activated oxygen species should not form in the process. It was found that parasites propagated normally under these conditions and that their susceptibility to CQ was unchanged, suggesting that the production of ROS is not central to the mechanism by which CQ kills parasites (Monti et al., 2002).
Considering all the data, definitive evidence is still lacking on whether CQ increases the oxidative stress experienced by the parasite and, if so, on whether this contributes to CQ’s parasiticidal activity. The haem and CQ–haem complexes that accumulate upon treatment with CQ could conceivably kill parasites through oxidative stress and/or through non-oxidative means, for example by accumulating in and consequently disrupting membranes (Ginsburg et al., 1998; Loria et al., 1999) or through inhibitory interactions with proteins.
Even if CQ treatment does impose an additional oxidative burden, the parasite’s antioxidant defence mechanisms may be sufficient to negate it such that the parasite does not experience increased stress. Indeed, it has been reported that the expression of various parasite antioxidant genes can escalate rapidly in response to exogenously applied oxidative stress, and is increased in parasites grown in (antioxidant-defence-challenged) glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes (Akide-Ndunge et al., 2009). Studies on the effect of CQ treatment on the P. falciparum proteome (Prieto et al., 2008) and transcriptome (Gunasekera et al., 2003, 2007) did not uncover clear evidence for an up-regulation of antioxidant defence systems. However, a recent study focusing specifically on antioxidant defence genes reported an up-regulation of certain transcripts, including that for pfmrp, after CQ treatment (2 h at the IC50 concentration) in both CQS (3D7) and CQR (Dd2) parasites (Nogueira et al., 2010).
Recently Fu et al. (2010) described the application of a fluorescent ROS reporter [5-(and 6)-chloromethyl-2′,7′-dichlorodihydro-fluorescein diacetate acetyl ester] to analyse oxidative stress in P. falciparum-infected erythrocytes. It would be very informative in further studies to see whether CQ treatment causes a detectable increase in the fluorescence signal, indicating generation of ROS.
5. A link between the parasite’s glutathione system and chloroquine response?
Multiple lines of evidence have linked the antioxidant tripeptide glutathione to the parasite’s response to CQ. Many of the experiments involved were performed with the murine malaria parasite Plasmodium berghei, and it should be noted that current knowledge on mechanisms of CQ resistance reveal marked differences between murine parasite species and P. falciparum. The following sections summarise what is known about the glutathione system in malaria parasites (focusing on P. falciparum), and examine the literature concerning the link between glutathione levels and CQ response in both murine malaria parasites and P. falciparum. The focus of this review is the glutathione system of Plasmodium as a potential parasite factor that modulates CQ response; readers are referred to Becker et al. (2004) for a discussion of how the antioxidant status of the host affects clinical outcome in malaria.
5.1. The glutathione system of antioxidant defence
Glutathione (γ-l-glutamyl-l-cysteinyl-glycine; henceforth abbreviated as GSH when referring to the reduced form, GSSG when referring to oxidised glutathione disulphide, and not abbreviated when referring to both) plays a major role in cellular antioxidant defence. GSH functions as an electron donor and, through this mechanism, plays a role both in the maintenance of protein sulfhydryl moieties and in the reduction of hydrogen and lipid peroxides. GSH can also be conjugated to the electrophilic centres of hydrophobic drugs and toxic metabolites through the action of glutathione-S-transferase (GST) to form adducts that are then recognised for efflux by MRPs (Borst et al., 1999). Upon donating an electron, the resulting oxidised glutathione molecule is most likely to react with a second glutathione molecule (since glutathione is present at millimolar concentrations in the cell) to form GSSG, rather than reacting with another cellular component and causing oxidative damage. GSSG is converted back to GSH by glutathione reductase (GR) in a reaction that uses NADPH as the electron donor. The resulting NADP+ can be reduced back to NADPH by two enzymes in the pentose phosphate pathway: G6PD and 6-phosphogluconate dehydrogenase. Cells synthesise GSH in two ATP-dependent steps. γ-Glutamylcysteine synthetase (γ-GCS) catalyses the rate-limiting reaction between l-glutamate and l-cysteine to produce γ-l-glutamyl-l-cysteine. GSH synthetase (GS) then catalyses the addition of glycine to form GSH.
5.2. The Plasmodium falciparum glutathione system
Like most eukaryotic cells, malaria parasites have a functional glutathione system for their antioxidant defence (Fig. 2). Different cell types vary in their GSH:GSSG ratio but typically maintain it above 10:1 (Dalle-Done et al., 2009). Atamna and Ginsburg (1997) estimated a GSH:GSSG ratio of 285:1 for P. falciparum under normal culture conditions. Many components of the P. falciparum glutathione system have been identified and some have been studied extensively. The P. falciparum GSH biosynthesis enzymes, PfGCS and PfGS, have both been identified and their enzymatic activities characterised (Luersen et al., 1999, 2000; Meierjohann et al., 2002a,b). Neither enzyme has been localised to date. The absence of an apparent signal peptide in both enzymes is consistent with their being cytosolic, as is the case in mammalian cells (Dalle-Done et al., 2009). The biochemical and kinetic properties of P. falciparum GR have also been studied in depth, both with native PfGR isolated from parasites and with recombinant protein (reviewed in Becker et al., 2004). PfGR has been localised to both the cytosol and the apicoplast of the parasite (Kehr et al., 2010). The crystal structures of PfGR and PfGST have been solved (Fritz-Wolf et al., 2003; Sarma et al., 2003).
Fig. 2.
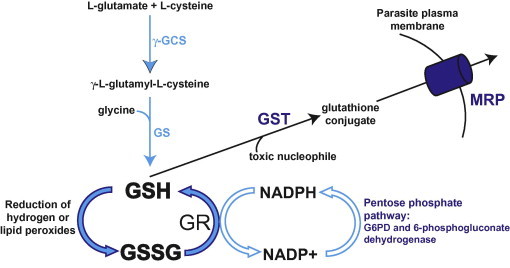
A schematic of the key components of the glutathione system in P. falciparum. G6PD, glucose-6-phosphate dehydrogenase; γ-GCS, γ-glutamylcysteine synthetase; GR, glutathione reductase; GS, glutathione synthetase; GSH, reduced glutathione; GSSG, oxidised glutathione disulphide; GST, glutathione-S-transferase; MRP, multidrug resistance-associated protein.
PfMRP (discussed above as a potential modulator of CQ resistance) is present on the plasma membrane of the intra-erythrocytic P. falciparum parasite (Klokouzas et al., 2004; Raj et al., 2009). Erythrocytes infected with parasites in which the gene encoding PfMRP was disrupted were found to accumulate more radiolabelled GSH from the extracellular medium, consistent with an impairment of glutathione efflux from the parasite (Raj et al., 2009). Knockout parasites were also rendered more susceptible to multiple antimalarial drugs, including CQ. This led the authors to propose that PfMRP transports glutathione and various drugs out of the cell, possibly as glutathione-drug adducts. However, Atamna and Ginsburg (1997) had previously reported that there is no pathway in the parasite plasma membrane for the import of external GSH. Given the short time-scale of the GSH uptake experiments (with time points of <10 min showing a difference between knockout and wild-type parasites), the results of Raj et al. (2009) can probably not be explained by endocytic uptake of exogenous GSH. Perhaps the increased uptake of GSH seen by Raj et al. (2009) was into the host cell compartment and not the parasite. Further studies are required to resolve this issue.
No other parasite proteins have been implicated in glutathione transport to date and it is not known how the parasite’s glutathione is distributed among its different compartments. Presumably host cell glutathione is endocytosed by the parasite along with Hb; whether it would be susceptible to degradation by peptidases in the DV is not clear.
The parasite does not appear to possess a classical GSH peroxidase (an enzyme that catalyses the reduction of H2O2 and lipid peroxides by GSH). An enzyme that was originally thought to be a GSH peroxidase was subsequently shown to prefer the small redox-active protein Trx as the electron donor and has since been re-classified (Sztajer et al., 2001). However, it has been shown that PfGST displays a GSH-dependent peroxidase activity with H2O2 and certain hydroperoxides (Harwaldt et al., 2002). A classical glutaredoxin (PfGrx1) and a number of parasite glutaredoxin-like proteins have also been identified (reviewed in Rahlfs et al., 2003). These are small redox enzymes that use GSH as a cofactor to reduce various compounds and proteins. In a recent study, two of the glutaredoxin-like proteins were localised to the cytosol, while one was localised to the mitochondrion (Kehr et al., 2010).
5.3. Why would glutathione levels affect chloroquine response?
Two scenarios, which are not mutually exclusive, can be envisaged that might explain a link between parasite glutathione levels and response to CQ:
-
(1)
An increase in the glutathione concentration in the parasite might reduce the accumulation of CQ by: (a) the glutathione binding to haem (Atamna and Ginsburg, 1995) and thus possibly inhibiting the binding of CQ;(b) reducing the amount of free haem available for CQ binding via glutathione-mediated haem degradation (Ginsburg et al., 1998); and/or (c) increasing the formation of CQ–glutathione adducts that are transported out of the parasite, thus allowing the parasite to tolerate higher (external) CQ concentrations. It should be noted that an HPLC analysis of cells treated with radiolabelled CQ did not find any evidence for the formation of CQ adducts (Berger et al., 1995). However, the results of Raj et al. (2009) and (for P. berghei) Dubois et al. (1995) are consistent with the existence of CQ-adduct formation and efflux.
-
(2)
An increase in the glutathione concentration in the parasite might allow GSH to counter more of the oxidative damage potentially caused by the CQ-mediated build-up of haem and CQ–haem complexes, thus allowing the parasite to tolerate higher CQ concentrations.
5.4. Chloroquine and glutathione: studies in murine malaria parasites
In the first study of its kind, it was found that CQR P. berghei parasites selected for by CQ pressure in vivo had much higher GSH levels and greater GST activity than the CQS parasites from which they were derived (Dubois et al., 1995). GSH levels and GST activity were assessed in parasites that were ‘isolated’ from their host erythrocytes by ammonium chloride treatment. Furthermore, buthionine sulfoximine (BSO), an inhibitor of γ-GCS, decreased parasite GSH levels and rendered CQR parasites more sensitive to CQ in vivo (Dubois et al., 1995). A higher GST activity in CQR compared to CQS P. berghei (as well as Plasmodium yoelii) parasites was also reported in a subsequent study (Srivastava et al., 1999).
Following on from this, Platel et al. (1999) reported an inverse relationship between Hz content on one hand, and glutathione and GST levels (measured in isolated parasites) on the other, in P. berghei strains exhibiting varying levels of CQ resistance. The two most resistant lines provided an exception, as they differed markedly in their CQ IC50 values and Hz contents, yet displayed similar glutathione levels and GST activities. It should be noted that a reduction in Hz formation is a feature of CQ resistance in P. berghei but not in P. falciparum (Gligorijevic et al., 2006). The treatment of mice infected with CQR P. berghei with BSO, which was confirmed in this study to lower parasite glutathione levels, led to a marked increase in Hz levels. The authors proposed that GSH can detoxify haem in the DV in CQR P. berghei parasites, thereby preventing its incorporation into Hz and decreasing the potency of CQ (Platel et al., 1999).
Platel et al. (1999) also noted that CQ resistance in P. berghei was associated with an increased preference for reticulocytes, a finding not observed in P. falciparum. The authors examined reticulocyte glutathione levels and GST activity and found that both were much higher than in mature erythrocytes. Notably, CQS parasites that had invaded reticulocytes were found to have higher glutathione levels and GST activity (as assessed in parasites isolated from their host cells) as compared with CQS parasites that had invaded mature erythrocytes. This was accompanied by a reduction in both Hz formation and susceptibility to CQ. This is consistent with an earlier report of superior CQ potency against P. berghei parasites growing in mature erythrocytes compared to those growing in reticulocytes (Dei-Cas et al., 1984), and suggests that the uptake of glutathione from reticulocytes may make a substantial contribution to parasite glutathione levels and to the detoxification of haem in the DV. The shift in host cell preference is therefore likely to contribute to the observed correlation between CQ response and parasite glutathione levels in P. berghei.
A subsequent study found that mice infected with P. berghei or Plasmodium vinckei petteri were more responsive to CQ treatment when co-treated with drugs (acetaminophen and disulfiram) that have been shown to reduce glutathione levels or to decrease the GSH:GSSG ratio in tissues (Deharo et al., 2003). These parasites were somewhat less responsive to CQ when treated with N-acetylcysteine (NAC) (Deharo et al., 2003). NAC is converted by the cell to l-cysteine (the least available of the glutathione-component amino acids in at least some cell types), and might be expected to increase glutathione levels in Plasmodium. NAC alone appeared to exacerbate rodent malaria infections, thus complicating the interpretation of the CQ plus NAC data. Furthermore, the authors observed no major changes in glutathione levels in blood samples taken from mice treated with these putative GSH-depleting and GSH-elevating compounds (Deharo et al., 2003).
In another study with P. berghei, it was reported that a CQR line selected for by CQ pressure showed a 6-fold increase in the level of mRNA encoding γ-GCS compared to the CQS parental line (Perez-Rosado et al., 2002). No differences in pbγ-gcs sequence or copy number were observed. Glutathione levels were not measured but might be expected to increase upon overexpression of the rate-limiting enzyme in GSH synthesis, again speaking to a link between elevated glutathione levels and CQ resistance in P. berghei.
Safeukui et al. (2004) also reported that two CQR lines of P. berghei had higher GSH levels (as measured in saponin-isolated parasites) than a CQS line. However, the more resistant of the two CQR lines (which had been selected with a higher CQ concentration) did not have a higher GSH level than the other CQR line. In this study G6PD activity was found to be greater in the CQR parasites than in the CQS parasites. Dehydroepiandrosterone sulphate (DHEAS), an inhibitor of G6PD, was found to decrease G6PD activity and the GSH:GSSG ratio in CQR parasites, and rendered them more susceptible to CQ (Safeukui et al., 2004).
Taken together these studies provide strong evidence that glutathione levels modulate CQ response in P. berghei. In a surprising development, it was recently reported that the gene encoding γ-GCS (pbγ-gcs) can be disrupted in P. berghei parasites and is therefore not essential during the asexual blood stages (Vega-Rodriguez et al., 2009). However, pbγ-gcs was found to be essential for parasite development inside the mosquito. The disruption of pbγ-gcs led to a minor defect in the rate of proliferation of asexual blood stage parasites and a marked reduction, but not complete depletion, of parasite glutathione (Vega-Rodriguez et al., 2009). The authors suggested that the glutathione found in the knockout parasites was obtained from the host erythrocyte by endocytosis. The CQ susceptibility of the γ-GCS knockout parasites has not been reported.
GR can also be disrupted in P. berghei (Buchholz et al., 2010; Pastrana-Mena et al., 2010), demonstrating that this protein is also dispensable for asexual blood stage development. As was found for γ-GCS, GR was shown to be essential for oocyst development in the mosquito. Glutathione levels in the GR knockout parasites were not greatly reduced, and there was no effect of the gene disruption on the growth of asexual blood-stage parasites (Buchholz et al., 2010; Pastrana-Mena et al., 2010). CQ response was not tested. There is evidence that other parasite proteins can reduce GSSG (Kanzok et al., 2000; Becker et al., 2003), thereby potentially explaining the non-essential nature of GR. Attempts to generate parasites in which the genes encoding γ-GCS and GR were both disrupted were unsuccessful (Pastrana-Mena et al., 2010), suggesting that at least one of the two proteins must be present to protect the viability of asexual blood stage parasites.
Two studies have investigated whether the glutathione system is involved in the CQ response of another murine parasite, Plasmodium chabaudi. CQ resistance in this species has been linked to a mutation in a deubiquitinating enzyme (Hunt et al., 2007). Ferreira et al. (2004) reported that P. chabaudi strains that were CQS or had varying degrees of CQ resistance showed no difference in the sequence or mRNA expression level of five genes of the glutathione system. Furthermore, CQ treatment did not change the expression levels of these genes (Ferreira et al., 2004). However, He et al. (2009) reported that a CQR strain of P. chabaudi displayed higher glutathione levels, higher GST activity and lower GR activity (all assessed in isolated parasites) as compared to a CQS strain. Furthermore, three antiretroviral protease inhibitors that were shown to potentiate the activity of CQ in the CQR strain of P. chabaudi were found to decrease glutathione levels and GST activity (He et al., 2009). One of these compounds (ritonavir) also rendered the CQS strain more susceptible to CQ, and this compound (but not the others) caused a slight decrease in glutathione levels in this strain. Thus, the latter study indicates that glutathione may also be linked to CQ responses in P. chabaudi.
5.5. Chloroquine and glutathione: studies in Plasmodium falciparum
CQ resistance in P. falciparum differs markedly from CQ resistance in P. berghei and P. chabaudi. In P. falciparum, CQ resistance has not been linked with a decrease in Hz formation (Gligorijevic et al., 2006) or a preferential invasion of reticulocytes. Rather, resistance is associated with a marked reduction in CQ accumulation (Fitch, 1970), which is thought to be mediated primarily by mutant-PfCRT-mediated CQ efflux from the DV (Martin et al., 2009). However, the degree of CQ resistance can differ among parasite lines that show no discernable difference in CQ accumulation (Lehane et al., 2011).
Nogueira et al. (2010) reported that the glutathione system can respond to CQ treatment and that this response can differ between strains, as evidenced by the up-regulation of certain transcripts (including pfmrp in the 3D7 and Dd2 strains and pfγ-gcs and pfg6pd in the 3D7 strain) after a 2 h exposure of the parasites to CQ, at a concentration equivalent to the IC50 of the strain in question. But does this response translate into an altered glutathione level, or modulate CQ susceptibility?
Ginsburg et al. (1998) investigated the effects of various agents that were reported to affect cellular glutathione levels on the in vitro CQ response of the CQR FCR3 strain of P. falciparum. Increasing glutathione levels in the infected erythrocyte by incubating cultures with either 15 mM l-cysteine (which yielded a ∼2.5-fold increase after a 2 h treatment) or 10 mM NAC (∼2-fold increase) was reported to increase resistance to CQ. The increases in the CQ IC50 values, as determined in 42 h assays with the same concentrations of l-cysteine and NAC, were ∼3-fold and ∼2.5-fold, respectively. Conversely, a 2 h treatment with either 50 μM 1-chloro-2,4-dinitrobenzene (CDNB, which conjugates GSH) or 6 mM BSO resulted in a decrease in the glutathione level to ∼70% of control levels. Drug assays with lower concentrations of these agents (4 μM CDNB and 3.5 mM BSO) resulted in a ∼30% reduction in the CQ IC50 value (Ginsburg et al., 1998).
Different results were obtained in our recent investigations into the effects of NAC and BSO (Lehane and Fidock, unpublished data). The addition of 10 mM NAC to the medium caused a marked acidification. When the pH was not adjusted back to its original value, there was indeed a 2.2–3.1-fold increase in the CQ IC50 value (tested in two CQR strains). However, when the pH was adjusted (with NaOH) back to the same value as that of medium lacking NAC, the increase in the CQ IC50 value in the presence of NAC was significantly less (1.3–1.5-fold). The pH of the medium is known to affect CQ accumulation and susceptibility in P. falciparum (Yayon et al., 1985). Furthermore, when tested at its approximate IC50 value (in our hands 30 μM, not 3.5 mM), BSO (present throughout 72 h drug assays) had no effect on parasite response to CQ (tested in two CQS and two CQR strains of P. falciparum).
In another study, Meierjohann et al. (2002b) investigated the sensitivity of a CQS P. falciparum strain, 3D7, and a CQR strain, Dd2, to BSO, methylene blue (proposed to inhibit parasite GR), and N,N1-bis(2-chloroethyl)-N-nitrosourea (BCNU; a compound that inhibits GR activity and also depletes GSH by reacting with it directly). 3D7 parasites were found to be more sensitive than Dd2 parasites to growth inhibition by BSO, methylene blue, and the oxidative stressor glucose oxidase, which catalyses the production of d-gluconate and H2O2 from d-glucose. However, the strains did not differ in their susceptibilities to BCNU (Meierjohann et al., 2002b). It should be noted that PfGR inhibition is probably not the mechanism by which methylene blue causes parasite death; GR knockout P. berghei parasites show the same sensitivity to this drug as the parental strain (Pastrana-Mena et al., 2010). Thus, the results for methylene blue are not necessarily related to differences in the glutathione systems of 3D7 and Dd2.
Similarly, it is possible that BSO kills P. falciparum parasites through a mechanism unrelated to its inhibition of the γ-GCS enzyme. In Trypanosoma brucei, it was shown that adding extra GSH to the medium did not prevent BSO-mediated parasite death, but did prevent the death of parasites in which γ-GCS expression levels were reduced by RNA interference (Huynh et al., 2003). This implies that BSO exerts its parasiticidal effect, at least against T. brucei, via a mechanism independent of its effect on glutathione concentration.
Luersen et al. (2000) investigated the activity of BSO against the FCBR strain of P. falciparum. The compound was reported to inhibit the proliferation of this strain with an IC50 value of 73 μM. At concentrations between 10 and 100 μM, BSO (after 24 h) was found to inhibit γ-GCS activity in both uninfected and infected erythrocytes. This translated into a marked reduction in the glutathione level in the infected erythrocytes but only a small reduction in the uninfected cells, consistent with there being a much higher turnover of glutathione in infected erythrocytes. The authors found that adding 1 mM GSH or GSH monoethyl ester to the medium decreased parasite susceptibility to BSO, but had no effect on GSH levels in the infected erythrocytes. However, higher concentrations of GSH and GSH monoethyl ester (15 mM) increased glutathione levels in BSO-treated infected cells. It is not clear how GSH and GSH monoethyl ester were able to reduce the antiplasmodial activity of BSO at concentrations below those needed to increase glutathione levels in the infected erythrocyte.
Meierjohann et al. (2002b) also reported that glutathione levels in erythrocytes infected with CQR Dd2 parasites were slightly higher than in those infected with CQS 3D7 parasites, although this was not statistically significant. However, Dd2 parasites isolated from their host cells by streptolysin O treatment were found to have significantly more glutathione (134 nmol per 1010 cells) than 3D7 parasites isolated in the same way (67 nmol per 1010 cells). A small difference such as this should be treated with caution, especially given that only one time point was investigated and the cells were only synchronised once per week (Meierjohann et al., 2002b). Of note, it has been shown that glutathione levels in erythrocytes infected with 3D7 parasites fluctuate somewhat during the course of the ∼48 h asexual cycle (Olszewski et al., 2009).
An investigation of individual components of the glutathione system in 3D7 parasites and Dd2 parasites also revealed some differences between the strains. GR activity was found to be significantly higher in 3D7 parasites than in Dd2 parasites, despite there being similar GR protein levels in the two strains (Meierjohann et al., 2002b). This suggests that the GR enzymes in the two strains may have different kinetic properties. Gilberger et al. (2000) had previously reported different kinetic properties for GRs from different strains, with the GR from K1 (a CQR strain) having a higher catalytic efficiency than that from the CQS 3D7 strain. The GRs from the two strains differed by three amino acids (Gilberger et al., 2000). More recently, a study in which GR activity was examined in eight different P. falciparum strains (four CQS and four CQR) revealed that while there were differences between strains, these were unrelated to the CQ susceptibility of the strain (Sarma et al., 2003).
Meierjohann et al. (2002b) also reported significant differences in γ-GCS activity between Dd2 and 3D7 parasites, this time with Dd2 parasites having the higher activity. The efflux of glutathione was found to be comparable for Dd2- and 3D7-infected erythrocytes. Based on these investigations, it was proposed that Dd2 parasites are more dependent on GSH synthesis to maintain their GSH levels than are 3D7 parasites, whereas in 3D7 parasites GSH levels are primarily determined by GR activity. Although these results are interesting, it must be remembered that 3D7 and Dd2 parasites have distinct genetic backgrounds, and thus any differences in their glutathione systems are not necessarily related to their different CQ susceptibilities.
Raj et al. (2009) also reported that manipulating GSH levels can influence CQ response. Addition of 2 mM GSH to the medium reduced CQ accumulation in the CQS 3D7 strain (by 48%), in the CQR W2 strain (by 11%) and in the W2 PfMRP knockout strain (by 30%). The addition of 2.5 mM GSH to the culture medium was also reported to cause a decrease in parasite susceptibility to CQ in all three strains (Raj et al., 2009). As mentioned above, these results appear to be at odds with the finding by Atamna and Ginsburg (1997) that GSH was not transported into the parasite across the parasite plasma membrane. Experiments in the Fidock laboratory, in which CQ drug assays were performed with four different strains of P. falciparum, revealed that the addition of 2 mM GSH acidified the medium, and that adjustment back to the original pH with NaOH nullified the small effect of GSH on CQ response (Lehane and Fidock, unpublished data). Taken together, this suggests that the pH, and consequently the protonation state of CQ, may have been responsible for the reduction in CQ accumulation and susceptibility observed by Raj et al. (2009) rather than a direct GSH-mediated effect.
In their study on the impact of antiretroviral inhibitors on CQ response, He et al. (2009) reported results for P. falciparum that were analogous to those observed for P. chabaudi (discussed above). The P. falciparum strains studied (3D7 and Dd2) were the same two that were investigated in Meierjohann et al. (2002b). Like Meierjohann et al., He et al. (2009) reported that glutathione levels were higher, and GR activity lower, in Dd2 parasites compared to 3D7 parasites. Three antiretroviral protease inhibitors that were reported to sensitise the Dd2 strain to CQ were found to decrease glutathione levels and GST activity in isolated Dd2 parasites. The most potent chemosensitiser, ritonavir, also decreased GR activity and glutathione peroxidase activity in Dd2 parasites. This agent also rendered 3D7 parasites somewhat more sensitive to CQ and caused a small decrease in glutathione levels and GR activity.
The putative association between GST activity and CQ response (as seen in murine parasites and reported for P. falciparum by He and colleagues) has been investigated in two other studies with P. falciparum. Rojpibulstit et al. (2004) reported that isolated CQR K1 parasites displayed higher GST activity than CQS T9/94 parasites. No kinetic differences were found, leading the authors to suggest that there might be a difference in GST expression levels between the strains (Rojpibulstit et al., 2004). However, a more comprehensive analysis involving four CQS and four CQR strains of P. falciparum found no association between GST activity and CQ resistance (Harwaldt et al., 2002).
5.6. Chloroquine and glutathione: summary
Several studies with P. berghei have correlated high glutathione levels with CQ resistance (Dubois et al., 1995; Platel et al., 1999; Safeukui et al., 2004). In all cases a substantial (>5-fold) difference was observed between the glutathione contents of isolated CQR and CQS parasites, and similar values for the glutathione concentrations of CQS and CQR strains were obtained in the different studies. As further evidence for the ability of glutathione to modulate CQ response in P. berghei, the γ-GCS inhibitor BSO was shown to lower glutathione levels and increase CQ susceptibility (Dubois et al., 1995), while growth in glutathione-rich reticulocytes was shown to elevate parasite glutathione levels and decrease CQ susceptibility (Platel et al., 1999).
In contrast, the case for the glutathione system’s involvement in P. falciparum CQ resistance is less robust. Two studies, both with the 3D7 (CQS) and Dd2 (CQR) strains of P. falciparum, reported a ∼2–2.5-fold higher glutathione level in the (isolated) CQR parasites compared to the CQS parasites (He et al., 2009; Meierjohann et al., 2002b). However, there was a >30-fold difference in the glutathione content estimates between the two studies; the reason for this is not clear. Ginsburg et al. (1998) reported that glutathione levels and the CQ IC50 value were lowered by ∼30% following 2 h exposures of parasites to millimolar concentrations of BSO (concentrations >45-fold higher than BSO IC50 values obtained by Luersen et al. (2000) and Lehane and Fidock (unpublished)). NAC (Ginsburg et al., 1998) and GSH (Raj et al., 2009) were reported to decrease parasite susceptibility to CQ but this may have resulted from the effects of these compounds on the pH of the medium (Lehane and Fidock, unpublished data). Despite the lack of definitive evidence to date, the idea that P. falciparum could bolster its glutathione levels to enhance its resistance to CQ is certainly plausible, especially in light of the findings for P. berghei.
6. Other antioxidant defence mechanisms and chloroquine response
In addition to the glutathione system, P. falciparum encodes the components of a Trx system. This system comprises Trx (a small protein that can provide reducing equivalents to peroxidases), Trx reductase (TrxR, which regenerates the reduced form of Trx from its oxidised form) and various Trx-dependent peroxidases (Tpx) (Muller, 2004). Several components of the P. falciparum Trx system were recently localised in blood-stage parasites (Kehr et al., 2010). TrxR, which localises to the cytosol and the mitochondrion (Kehr et al., 2010), is essential for erythrocytic-stage P. falciparum (Krnajski et al., 2002), but is dispensable for the blood stages of P. berghei (Buchholz et al., 2010). A recent study showed that P. falciparum imports a host Trx-dependent peroxiredoxin (hPrx-2) into its cytosol, and that this enzyme accounts for a significant fraction of Trx peroxidase activity in parasite extracts (Koncarevic et al., 2009).
P. falciparum also possesses two superoxide dismutases (SODs) (metalloproteins that catalyse the dismutation of into H2O2 and O2). One of these (SOD-1) localises to the cytosol and the other (SOD-2) localises to the mitochondrion of blood-stage parasites (Gratepanche et al., 2002; Sienkiewicz et al., 2004). Furthermore, the host cell SOD is taken up by the rodent parasite P. berghei with Hb (Fairfield et al., 1983). Whether SOD uptake from the host cell contributes to the dismutation of in the DV in P. falciparum is not clear (Muller, 2004).
To the best of our knowledge, only two studies have considered a possible link between any of these (non-glutathione-associated) antioxidant proteins and CQ response. An analysis of changes in protein abundance on treatment with CQ revealed that the amount of the human Trx-dependent peroxiredoxin imported by the parasite was increased in response to CQ pressure in both CQS (3D7 and HB3) and CQR (K1 and Dd2) strains, by a factor of 1.6–4.5 (Koncarevic et al., 2009). Nogueira et al. (2010) reported an increase in mRNA expression of pfFe-sod in 3D7 parasites, and of pftrxR and pftpx1 in Dd2 parasites, on exposure to IC50 levels of CQ for 2 h. Whether the changes in mRNA/protein abundance reported in the above studies translate into an increase in antioxidant power that enables a parasite to withstand higher concentrations of CQ remains to be seen.
7. Chloroquine action and resistance in Plasmodium vivax
Although widespread, Plasmodium vivax malaria, sometimes referred to as ‘benign’, has long been considered to be of secondary importance compared to P. falciparum malaria because it is less lethal. Nonetheless, there are an increasing number of reports of P. vivax malaria giving rise to severe symptoms (Baird, 2007; Tjitra et al., 2008) and its importance has clearly been underappreciated.
CQ survived as a first-line therapy for the treatment of P. vivax malaria well beyond its survival as an effective therapy for P. falciparum malaria, and it continues to be widely used. Nevertheless, following on from the first reports of CQR P. vivax in 1989 there are a growing number of reports of cases of CQR P. vivax malaria from South and Southeast Asia, as well as from South America (Suwanarusk et al., 2007).
The finding that in P. vivax, unlike in P. falciparum, the mature trophozoite-stage (Hb-digesting) form of the parasite is markedly less sensitive to CQ than younger, ring-stage parasites (Sharrock et al., 2008) raises the possibility that the mechanism by which CQ kills P. vivax might differ from that by which it kills P. falciparum. Similarly, the lack of a clear association between CQ resistance in P. vivax and mutations in the P. vivax orthologue of the CRT protein (Nomura et al., 2001; Suwanarusk et al., 2007; Barnadas et al., 2008; Orjuela-Sanchez et al., 2009) points to the mechanism of CQ resistance in P. vivax being different from that in P. falciparum.
The extent to which the redox system of the parasite might play a role in the mechanism of action of, or resistance to, CQ in P. vivax has not, to our knowledge, been investigated. Nor is it clear to what extent the intracellular parasite might draw on the redox machinery of its host cell. P. vivax, unlike P. falciparum, has a strong preference (if not an absolute requirement) for reticulocytes over the mature erythrocyte. Whether, as has been proposed for the reticulocyte-preferring murine parasite P. berghei (Platel et al., 1999), the intra-reticulocytic P. vivax parasite might draw on the redox system of its host cell to bolster its own oxidative defences, and whether this might play a role in CQ resistance in this parasite, would seem a worthwhile area for study.
8. Conclusion
Definitive studies are still required to test the hypothesis that oxidative stress is the lethal consequence of CQ’s binding to haem in the malaria parasite. Of note, a recent study with a fluorescent ROS reporter provided evidence that artemisinins generate oxidative stress within P. falciparum (Klonis et al., 2011). Similar experiments may well provide important insights into the mode of action of CQ. In P. berghei, it appears that CQ resistance is associated with higher intra-parasite glutathione levels, which serve to degrade CQ’s haem target. This association may be explained in part by the preferential invasion of glutathione-rich reticulocytes by CQR parasites. In P. falciparum, a link between glutathione levels and CQ response is not firmly established, and further work is required to determine whether the parasite’s redox system plays a role in determining the CQ resistance levels of PfCRTK76T-bearing parasites. One approach would be to analyse glutathione levels at different time points among a series of carefully synchronised strains with varying CQ susceptibilities. Furthermore, it would be informative to determine whether γ-GCS is dispensable for the P. falciparum blood stages. If blood-stage P. falciparum parasites in which the γ-gcs gene is disrupted prove to be viable and show decreased glutathione levels (as seen for P. berghei), then CQ drug assays on the knockout parasites would provide important insights into any role for glutathione in determining the CQ response of the human parasite. Whether the redox system of the parasite, or host cell, might play a role in the more recent phenomenon of CQ resistance in P. vivax remains an open question.
Acknowledgements
This work was supported by the Australian National Health and Medical Research Council (NHMRC) Overseas Biomedical Fellowship 585519 (to AML), the US National Institutes of Health Grant R01 A150234 (to DAF), an Investigator in Pathogenesis of Infectious Diseases Award from the Burroughs Wellcome Fund (to DAF) and NHMRC Grant 418055 (to KK). We thank Dr Richard Eastman for helpful suggestions.
References
- Akide-Ndunge O.B., Tambini E., Giribaldi G., McMillan P.J., Muller S., Arese P., Turrini F. Co-ordinated stage-dependent enhancement of Plasmodium falciparum antioxidant enzymes and heat shock protein expression in parasites growing in oxidatively stressed or G6PD-deficient red blood cells. Malar. J. 2009;8:113. doi: 10.1186/1475-2875-8-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson T.J., Nair S., Qin H., Singlam S., Brockman A., Paiphun L., Nosten F. Are transporter genes other than the chloroquine resistance locus (pfcrt) and multidrug resistance gene (pfmdr) associated with antimalarial drug resistance? Antimicrob. Agents Chemother. 2005;49:2180–2188. doi: 10.1128/AAC.49.6.2180-2188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atamna H., Ginsburg H. Heme degradation in the presence of glutathione. A proposed mechanism to account for the high levels of non-heme iron found in the membranes of hemoglobinopathic red blood cells. J. Biol. Chem. 1995;270:24876–24883. doi: 10.1074/jbc.270.42.24876. [DOI] [PubMed] [Google Scholar]
- Atamna H., Ginsburg H. The malaria parasite supplies glutathione to its host cell - investigation of glutathione transport and metabolism in human erythrocytes infected with Plasmodium falciparum. Eur. J. Biochem. 1997;250:670–679. doi: 10.1111/j.1432-1033.1997.00670.x. [DOI] [PubMed] [Google Scholar]
- Atamna H., Pascarmona G., Ginsburg H. Hexose-monophosphate shunt activity in intact Plasmodium falciparum-infected erythrocytes and in free parasites. Mol. Biochem. Parasitol. 1994;67:79–89. doi: 10.1016/0166-6851(94)90098-1. [DOI] [PubMed] [Google Scholar]
- Baird J.K. Neglect of Plasmodium vivax malaria. Trends Parasitol. 2007;23:533–539. doi: 10.1016/j.pt.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Barnadas C., Ratsimbasoa A., Tichit M., Bouchier C., Jahevitra M., Picot S., Menard D. Plasmodium vivax resistance to chloroquine in Madagascar: clinical efficacy and polymorphisms in pvmdr1 and pvcrt-o genes. Antimicrob. Agents Chemother. 2008;52:4233–4240. doi: 10.1128/AAC.00578-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baro N.K., Pooput C., Roepe P.D. Analysis of chloroquine resistance transporter (CRT) isoforms and orthologues in S. cerevisiae yeast. Biochemistry. 2011;50:6701–6710. doi: 10.1021/bi200922g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker K., Kanzok S.M., Iozef R., Fischer M., Schirmer R.H., Rahlfs S. Plasmoredoxin, a novel redox-active protein unique for malarial parasites. Eur. J. Biochem. 2003;270:1057–1064. doi: 10.1046/j.1432-1033.2003.03495.x. [DOI] [PubMed] [Google Scholar]
- Becker K., Tilley L., Vennerstrom J.L., Roberts D., Rogerson S., Ginsburg H. Oxidative stress in malaria parasite-infected erythrocytes: host-parasite interactions. Int. J. Parasitol. 2004;34:163–189. doi: 10.1016/j.ijpara.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Berger B.J., Martiney J., Slater A.F., Fairlamb A.H., Cerami A. Chloroquine resistance is not associated with drug metabolism in Plasmodium falciparum. J. Parasitol. 1995;81:1004–1008. [PubMed] [Google Scholar]
- Borst P., Evers R., Kool M., Wijnholds J. The multidrug resistance protein family. Biochim. Biophys. Acta. 1999;1461:347–357. doi: 10.1016/s0005-2736(99)00167-4. [DOI] [PubMed] [Google Scholar]
- Bray P.G., Mungthin M., Ridley R.G., Ward S.A. Access to hematin: the basis of chloroquine resistance. Mol. Pharmacol. 1998;54:170–179. doi: 10.1124/mol.54.1.170. [DOI] [PubMed] [Google Scholar]
- Buchholz K., Putrianti E.D., Rahlfs S., Schirmer R.H., Becker K., Matuschewski K. Molecular genetics evidence for the in vivo roles of the two major NADPH-dependent disulfide reductases in the malaria parasite. J. Biol. Chem. 2010;285:37388–37395. doi: 10.1074/jbc.M110.123323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanale N., Nickel C., Daubenberger C.A., Wehlan D.A., Gorman J.J., Klonis N., Becker K., Tilley L. Identification and characterization of heme-interacting proteins in the malaria parasite, Plasmodium falciparum. J. Biol. Chem. 2003;278:27354–27361. doi: 10.1074/jbc.M303634200. [DOI] [PubMed] [Google Scholar]
- Chen N., Russell B., Fowler E., Peters J., Cheng Q. Levels of chloroquine resistance in Plasmodium falciparum are determined by loci other than pfcrt and pfmdr1. J. Infect. Dis. 2002;185:405–407. doi: 10.1086/338470. [DOI] [PubMed] [Google Scholar]
- Choi C.Y., Cerda J.F., Chu H.A., Babcock G.T., Marletta M.A. Spectroscopic characterization of the heme-binding sites in Plasmodium falciparum histidine-rich protein 2. Biochemistry. 1999;38:16916–16924. doi: 10.1021/bi991665k. [DOI] [PubMed] [Google Scholar]
- Chou A.C., Chevli R., Fitch C.D. Ferriprotoporphyrin IX fulfills the criteria for identification as the chloroquine receptor of malaria parasites. Biochemistry. 1980;19:1543–1549. doi: 10.1021/bi00549a600. [DOI] [PubMed] [Google Scholar]
- Cojean S., Noel A., Garnier D., Hubert V., Le Bras J., Durand R. Lack of association between putative transporter gene polymorphisms in Plasmodium falciparum and chloroquine resistance in imported malaria isolates from Africa. Malar. J. 2006;5:24. doi: 10.1186/1475-2875-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper R.A., Ferdig M.T., Su X.Z., Ursos L.M., Mu J., Nomura T., Fujioka H., Fidock D.A., Roepe P.D., Wellems T.E. Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol. Pharmacol. 2002;61:35–42. doi: 10.1124/mol.61.1.35. [DOI] [PubMed] [Google Scholar]
- Dalle-Done I., Rossi R., Colombo G., Giustarini D., Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem. Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- de Almeida Ribeiro M.C., Augusto O., da Costa Ferreira A.M. Influence of quinoline-containing antimalarials in the catalase activity of ferriprotoporphyrin IX. J. Inorg. Biochem. 1997;65:15–23. [Google Scholar]
- Deharo E., Barkan D., Krugliak M., Golenser J., Ginsburg H. Potentiation of the antimalarial action of chloroquine in rodent malaria by drugs known to reduce cellular glutathione levels. Biochem. Pharmacol. 2003;66:809–817. doi: 10.1016/s0006-2952(03)00396-4. [DOI] [PubMed] [Google Scholar]
- Dei-Cas E., Slomianny C., Prensier G., Vernes A., Colin J.J., Verhaeghe A., Savage A., Charet P. Preferential action of chloroquine on Plasmodium within mature erythrocytes. Pathol. Biol. 1984;32:1019–1023. [PubMed] [Google Scholar]
- Djimde A., Doumbo O.K., Cortese J.F., Kayentao K., Doumbo S., Diourte Y., Dicko A., Su X.Z., Nomura T., Fidock D.A., Wellems T.E., Plowe C.V. A molecular marker for chloroquine-resistant falciparum malaria. N. Engl. J. Med. 2001;344:257–263. doi: 10.1056/NEJM200101253440403. [DOI] [PubMed] [Google Scholar]
- Djimde A.A., Doumbo O.K., Traore O., Guindo A.B., Kayentao K., Diourte Y., Niare-Doumbo S., Coulibaly D., Kone A.K., Cissoko Y., Tekete M., Fofana B., Dicko A., Diallo D.A., Wellems T.E., Kwiatkowski D., Plowe C.V. Clearance of drug-resistant parasites as a model for protective immunity in Plasmodium falciparum malaria. Am. J. Trop. Med. Hyg. 2003;69:558–563. [PubMed] [Google Scholar]
- Dubois V.L., Platel D.F., Pauly G., Tribouley-Duret J. Plasmodium berghei: implication of intracellular glutathione and its related enzyme in chloroquine resistance in vivo. Exp. Parasitol. 1995;81:117–124. doi: 10.1006/expr.1995.1099. [DOI] [PubMed] [Google Scholar]
- Ecker, A., Lehane, A.M., Fidock, D.A., in press. Molecular markers of Plasmodium resistance to antimalarials. In: Staines, H.M., Krishna, S. (Eds), Treatment and prevention of malaria: antimalarial drug chemistry, action and use. Birkhauser Verlag, Basel.
- Egan T.J. Haemozoin formation. Mol. Biochem. Parasitol. 2008;157:127–136. doi: 10.1016/j.molbiopara.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Egan T.J., Ross D.C., Adams P.A. Quinoline anti-malarial drugs inhibit spontaneous formation of beta-haematin (malaria pigment) FEBS Lett. 1994;352:54–57. doi: 10.1016/0014-5793(94)00921-x. [DOI] [PubMed] [Google Scholar]
- Fairfield A.S., Meshnick S.R., Eaton J.W. Malaria parasites adopt host cell superoxide dismutase. Science. 1983;221:764–766. doi: 10.1126/science.6348944. [DOI] [PubMed] [Google Scholar]
- Ferdig M.T., Cooper R.A., Mu J., Deng B., Joy D.A., Su X.Z., Wellems T.E. Dissecting the loci of low-level quinine resistance in malaria parasites. Mol. Microbiol. 2004;52:985–997. doi: 10.1111/j.1365-2958.2004.04035.x. [DOI] [PubMed] [Google Scholar]
- Ferreira I.D., Nogueira F., Borges S.T., do Rosario V.E., Cravo P. Is the expression of genes encoding enzymes of glutathione (GSH) metabolism involved in chloroquine resistance in Plasmodium chabaudi parasites? Mol. Biochem. Parasitol. 2004;136:43–50. doi: 10.1016/j.molbiopara.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Fidock D.A., Nomura T., Talley A.K., Cooper R.A., Dzekunov S.M., Ferdig M.T., Ursos L.M., Sidhu A.B., Naude B., Deitsch K.W., Su X.Z., Wootton J.C., Roepe P.D., Wellems T.E. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell. 2000;6:861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch C.D. Plasmodium falciparum in owl monkeys: drug resistance and chloroquine binding capacity. Science. 1970;169:289–290. doi: 10.1126/science.169.3942.289. [DOI] [PubMed] [Google Scholar]
- Fitch C.D. Ferriprotoporphyrin IX, phospholipids, and the antimalarial actions of quinoline drugs. Life Sci. 2004;74:1957–1972. doi: 10.1016/j.lfs.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Fritz-Wolf K., Becker A., Rahlfs S., Harwaldt P., Schirmer R.H., Kabsch W., Becker K. X-ray structure of glutathione S-transferase from the malarial parasite Plasmodium falciparum. Proc. Natl. Acad. Sci. USA. 2003;100:13821–13826. doi: 10.1073/pnas.2333763100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Tilley L., Kenny S., Klonis N. Dual labeling with a far red probe permits analysis of growth and oxidative stress in P. falciparum-infected erythrocytes. Cytometry A. 2010;77:253–263. doi: 10.1002/cyto.a.20856. [DOI] [PubMed] [Google Scholar]
- Gilberger T.W., Schirmer R.H., Walter R.D., Muller S. Deletion of the parasite-specific insertions and mutation of the catalytic triad in glutathione reductase from chloroquine-sensitive Plasmodium falciparum 3D7. Mol. Biochem. Parasitol. 2000;107:169–179. doi: 10.1016/s0166-6851(00)00188-2. [DOI] [PubMed] [Google Scholar]
- Ginsburg H., Famin O., Zhang J., Krugliak M. Inhibition of glutathione-dependent degradation of heme by chloroquine and amodiaquine as a possible basis for their antimalarial mode of action. Biochem. Pharmacol. 1998;56:1305–1313. doi: 10.1016/s0006-2952(98)00184-1. [DOI] [PubMed] [Google Scholar]
- Gligorijevic B., McAllister R., Urbach J.S., Roepe P.D. Spinning disk confocal microscopy of live, intraerythrocytic malarial parasites. 1 Quantification of hemozoin development for drug sensitive versus resistant malaria. Biochemistry. 2006;45:12400–12410. doi: 10.1021/bi061033f. [DOI] [PubMed] [Google Scholar]
- Goldberg D.E. Hemoglobin Degradation. In: Sullivan D.J., Krishna S., editors. Malaria: drugs, Disease and Post-genomic Biology. Springer; Berlin: 2005. pp. 275–291. [Google Scholar]
- Gratepanche S., Menage S., Touati D., Wintjens R., Delplace P., Fontecave M., Masset A., Camus D., Dive D. Biochemical and electron paramagnetic resonance study of the iron superoxide dismutase from Plasmodium falciparum. Mol. Biochem. Parasitol. 2002;120:237–246. doi: 10.1016/s0166-6851(02)00004-x. [DOI] [PubMed] [Google Scholar]
- Gunasekera A.M., Myrick A., Le Roch K., Winzeler E., Wirth D.F. Plasmodium falciparum: genome wide perturbations in transcript profiles among mixed stage cultures after chloroquine treatment. Exp. Parasitol. 2007;117:87–92. doi: 10.1016/j.exppara.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Gunasekera A.M., Patankar S., Schug J., Eisen G., Wirth D.F. Drug-induced alterations in gene expression of the asexual blood forms of Plasmodium falciparum. Mol. Microbiol. 2003;50:1229–1239. doi: 10.1046/j.1365-2958.2003.03787.x. [DOI] [PubMed] [Google Scholar]
- Harwaldt P., Rahlfs S., Becker K. Glutathione S-transferase of the malarial parasite Plasmodium falciparum: characterization of a potential drug target. Biol. Chem. 2002;383:821–830. doi: 10.1515/BC.2002.086. [DOI] [PubMed] [Google Scholar]
- He Z., Chen L., You J., Qin L., Chen X. Antiretroviral protease inhibitors potentiate chloroquine antimalarial activity in malaria parasites by regulating intracellular glutathione metabolism. Exp. Parasitol. 2009;123:122–127. doi: 10.1016/j.exppara.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Hunt P., Afonso A., Creasey A., Culleton R., Sidhu A.B., Logan J., Valderramos S.G., McNae I., Cheesman S., do Rosario V., Carter R., Fidock D.A., Cravo P. Gene encoding a deubiquitinating enzyme is mutated in artesunate- and chloroquine-resistant rodent malaria parasites. Mol. Microbiol. 2007;65:27–40. doi: 10.1111/j.1365-2958.2007.05753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh T.T., Huynh V.T., Harmon M.A., Phillips M.A. Gene knockdown of gamma-glutamylcysteine synthetase by RNAi in the parasitic protozoa Trypanosoma brucei demonstrates that it is an essential enzyme. J. Biol. Chem. 2003;278:39794–39800. doi: 10.1074/jbc.M306306200. [DOI] [PubMed] [Google Scholar]
- Kanzok S.M., Schirmer R.H., Turbachova I., Iozef R., Becker K. The thioredoxin system of the malaria parasite Plasmodium falciparum. Glutathione reduction revisited. J. Biol. Chem. 2000;275:40180–40186. doi: 10.1074/jbc.M007633200. [DOI] [PubMed] [Google Scholar]
- Kehr S., Sturm N., Rahlfs S., Przyborski J.M., Becker K. Compartmentation of redox metabolism in malaria parasites. PLoS Pathog. 2010;6:e1001242. doi: 10.1371/journal.ppat.1001242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klokouzas A., Tiffert T., van Schalkwyk D., Wu C.P., van Veen H.W., Barrand M.A., Hladky S.B. Plasmodium falciparum expresses a multidrug resistance-associated protein. Biochem. Biophys. Res. Commun. 2004;321:197–201. doi: 10.1016/j.bbrc.2004.06.135. [DOI] [PubMed] [Google Scholar]
- Klonis N., Crespo-Ortiz M.P., Bottova I., Abu-Bakar N., Kenny S., Rosenthal P.J., Tilley L. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc. Natl. Acad. Sci. USA. 2011;108:11405–11410. doi: 10.1073/pnas.1104063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klonis N., Tan O., Jackson K., Goldberg D., Klemba M., Tilley L. Evaluation of pH during cytostomal endocytosis and vacuolar catabolism of haemoglobin in Plasmodium falciparum. Biochem. J. 2007;407:343–354. doi: 10.1042/BJ20070934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koncarevic S., Rohrbach P., Deponte M., Krohne G., Prieto J.H., Yates J., Rahlfs S., Becker K. The malarial parasite Plasmodium falciparum imports the human protein peroxiredoxin 2 for peroxide detoxification. Proc. Natl. Acad. Sci. USA. 2009;106:13323–13328. doi: 10.1073/pnas.0905387106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krnajski Z., Gilberger T.W., Walter R.D., Cowman A.F., Muller S. Thioredoxin reductase is essential for the survival of Plasmodium falciparum erythrocytic stages. J. Biol. Chem. 2002;277:25970–25975. doi: 10.1074/jbc.M203539200. [DOI] [PubMed] [Google Scholar]
- Lehane A.M., van Schalkwyk D.A., Valderramos S.G., Fidock D.A., Kirk K. Differential drug efflux or accumulation does not explain variation in the chloroquine response of Plasmodium falciparum strains expressing the same isoform of mutant PfCRT. Antimicrob. Agents Chemother. 2011;55:2310–2318. doi: 10.1128/AAC.01167-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew V.L., Tiffert T., Ginsburg H. Excess hemoglobin digestion and the osmotic stability of Plasmodium falciparum-infected red blood cells. Blood. 2003;101:4189–4194. doi: 10.1182/blood-2002-08-2654. [DOI] [PubMed] [Google Scholar]
- Liochev S.I., Fridovich I. The Haber-Weiss cycle – 70 years later: an alternative view. Redox Rep. 2002;7:55–57. doi: 10.1179/135100002125000190. [DOI] [PubMed] [Google Scholar]
- Loria P., Miller S., Foley M., Tilley L. Inhibition of the peroxidative degradation of haem as the basis of action of chloroquine and other quinoline antimalarials. Biochem. J. 1999;339:363–370. [PMC free article] [PubMed] [Google Scholar]
- Luersen K., Walter R.D., Muller S. The putative gamma-glutamylcysteine synthetase from Plasmodium falciparum contains large insertions and a variable tandem repeat. Mol. Biochem. Parasitol. 1999;98:131–142. doi: 10.1016/s0166-6851(98)00161-3. [DOI] [PubMed] [Google Scholar]
- Luersen K., Walter R.D., Muller S. Plasmodium falciparum-infected red blood cells depend on a functional glutathione de novo synthesis attributable to an enhanced loss of glutathione. Biochem. J. 2000;346:545–552. [PMC free article] [PubMed] [Google Scholar]
- Malhotra K., Salmon D., Le Bras J., Vilde J.L. Potentiation of chloroquine activity against Plasmodium falciparum by the peroxidase–hydrogen peroxide system. Antimicrob. Agents Chemother. 1990;34:1981–1985. doi: 10.1128/aac.34.10.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R.E., Marchetti R.V., Cowan A.I., Howitt S.M., Broer S., Kirk K. Chloroquine transport via the malaria parasite’s chloroquine resistance transporter. Science. 2009;325:1680–1682. doi: 10.1126/science.1175667. [DOI] [PubMed] [Google Scholar]
- Meierjohann S., Walter R.D., Muller S. Glutathione synthetase from Plasmodium falciparum. Biochem. J. 2002;363:833–838. doi: 10.1042/0264-6021:3630833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meierjohann S., Walter R.D., Muller S. Regulation of intracellular glutathione levels in erythrocytes infected with chloroquine-sensitive and chloroquine-resistant Plasmodium falciparum. Biochem. J. 2002;368:761–768. doi: 10.1042/BJ20020962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti D., Basilico N., Parapini S., Pasini E., Olliaro P., Taramelli D. Does chloroquine really act through oxidative stress? FEBS Lett. 2002;522:3–5. doi: 10.1016/s0014-5793(02)02881-8. [DOI] [PubMed] [Google Scholar]
- Mu J., Ferdig M.T., Feng X., Joy D.A., Duan J., Furuya T., Subramanian G., Aravind L., Cooper R.A., Wootton J.C., Xiong M., Su X.Z. Multiple transporters associated with malaria parasite responses to chloroquine and quinine. Mol. Microbiol. 2003;49:977–989. doi: 10.1046/j.1365-2958.2003.03627.x. [DOI] [PubMed] [Google Scholar]
- Mu J., Myers R.A., Jiang H., Liu S., Ricklefs S., Waisberg M., Chotivanich K., Wilairatana P., Krudsood S., White N.J., Udomsangpetch R., Cui L., Ho M., Ou F., Li H., Song J., Li G., Wang X., Seila S., Sokunthea S., Socheat D., Sturdevant D.E., Porcella S.F., Fairhurst R.M., Wellems T.E., Awadalla P., Su X.Z. Plasmodium falciparum genome-wide scans for positive selection, recombination hot spots and resistance to antimalarial drugs. Nat. Genet. 2010;42:268–271. doi: 10.1038/ng.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S. Redox and antioxidant systems of the malaria parasite Plasmodium falciparum. Mol. Microbiol. 2004;53:1291–1305. doi: 10.1111/j.1365-2958.2004.04257.x. [DOI] [PubMed] [Google Scholar]
- Nogueira F., Diez A., Radfar A., Perez-Benavente S., do Rosario V.E., Puyet A., Bautista J.M. Early transcriptional response to chloroquine of the Plasmodium falciparum antioxidant defence in sensitive and resistant clones. Acta Trop. 2010;114:109–115. doi: 10.1016/j.actatropica.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Nomura T., Carlton J.M., Baird J.K., del Portillo H.A., Fryauff D.J., Rathore D., Fidock D.A., Su X., Collins W.E., McCutchan T.F., Wootton J.C., Wellems T.E. Evidence for different mechanisms of chloroquine resistance in 2 Plasmodium species that cause human malaria. J. Infect. Dis. 2001;183:1653–1661. doi: 10.1086/320707. [DOI] [PubMed] [Google Scholar]
- Olszewski K.L., Morrisey J.M., Wilinski D., Burns J.M., Vaidya A.B., Rabinowitz J.D., Llinas M. Host-parasite interactions revealed by Plasmodium falciparum metabolomics. Cell Host Microbe. 2009;19:191–199. doi: 10.1016/j.chom.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orjuela-Sanchez P., de Santana Filho F.S., Machado-Lima A., Chehuan Y.F., Costa M.R., Alecrim M.G., del Portillo H.A. Analysis of single-nucleotide polymorphisms in the crt-o and mdr1 genes of Plasmodium vivax among chloroquine-resistant isolates from the Brazilian Amazon region. Antimicrob. Agents Chemother. 2009;53:3561–3564. doi: 10.1128/AAC.00004-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagola S., Stephens P.W., Bohle D.S., Kosar A.D., Madsen S.K. The structure of malaria pigment beta-haematin. Nature. 2000;404:307–310. doi: 10.1038/35005132. [DOI] [PubMed] [Google Scholar]
- Pastrana-Mena R., Dinglasan R.R., Franke-Fayard B., Vega-Rodriguez J., Fuentes-Caraballo M., Baerga-Ortiz A., Coppens I., Jacobs-Lorena M., Janse C.J., Serrano A.E. Glutathione reductase-null malaria parasites have normal blood stage growth but arrest during development in the mosquito. J. Biol. Chem. 2010;285:27045–27056. doi: 10.1074/jbc.M110.122275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J.J., Thacker D., Tan J.C., Pleeter P., Checkley L., Gonzales J.M., Deng B., Roepe P.D., Cooper R.A., Ferdig M.T. Chloroquine susceptibility and reversibility in a Plasmodium falciparum genetic cross. Mol. Microbiol. 2010;78:770–787. doi: 10.1111/j.1365-2958.2010.07366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Rosado J., Gervais G.W., Ferrer-Rodriguez I., Peters W., Serrano A.E. Plasmodium berghei: analysis of the gamma-glutamylcysteine synthetase gene in drug-resistant lines. Exp. Parasitol. 2002;101:175–182. doi: 10.1016/s0014-4894(02)00138-8. [DOI] [PubMed] [Google Scholar]
- Picot S., Olliaro P., de Monbrison F., Bienvenu A.L., Price R.N., Ringwald P. A systematic review and meta-analysis of evidence for correlation between molecular markers of parasite resistance and treatment outcome in falciparum malaria. Malar. J. 2009;8:89. doi: 10.1186/1475-2875-8-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platel D.F., Mangou F., Tribouley-Duret J. Role of glutathione in the detoxification of ferriprotoporphyrin IX in chloroquine resistant Plasmodium berghei. Mol. Biochem. Parasitol. 1999;98:215–223. doi: 10.1016/s0166-6851(98)00170-4. [DOI] [PubMed] [Google Scholar]
- Prieto J.H., Koncarevic S., Park S.K., Yates J., Becker K. Large-scale differential proteome analysis in Plasmodium falciparum under drug treatment. PLoS One. 2008;3:e4098. doi: 10.1371/journal.pone.0004098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radfar A., Diez A., Bautista J.M. Chloroquine mediates specific proteome oxidative damage across the erythrocytic cycle of resistant Plasmodium falciparum. Free Radic. Biol. Med. 2008;44:2034–2042. doi: 10.1016/j.freeradbiomed.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Rahlfs S., Nickel C., Deponte M., Schirmer R.H., Becker K. Plasmodium falciparum thioredoxins and glutaredoxins as central players in redox metabolism. Redox Rep. 2003;8:246–250. doi: 10.1179/135100003225002844. [DOI] [PubMed] [Google Scholar]
- Raj D.K., Mu J., Jiang H., Kabat J., Singh S., Sullivan M., Fay M.P., McCutchan T.F., Su X.Z. Disruption of a Plasmodium falciparum multidrug resistance-associated protein (PfMRP) alters its fitness and transport of antimalarial drugs and glutathione. J. Biol. Chem. 2009;284:7687–7696. doi: 10.1074/jbc.M806944200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed M.B., Saliba K.J., Caruana S.R., Kirk K., Cowman A.F. Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature. 2000;403:906–909. doi: 10.1038/35002615. [DOI] [PubMed] [Google Scholar]
- Rojpibulstit P., Kangsadalampai S., Ratanavalachai T., Denduangboripant J., Chavalitshewinkoon-Petmitr P. Glutathione-S-transferases from chloroquine-resistant and -sensitive strains of Plasmodium falciparum: what are their differences? Southeast Asian J. Trop. Med. Public Health. 2004;35:292–299. [PubMed] [Google Scholar]
- Sa J.M., Twu O., Hayton K., Reyes S., Fay M.P., Ringwald P., Wellems T.E. Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proc. Natl. Acad. Sci. USA. 2009;106:18883–18889. doi: 10.1073/pnas.0911317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safeukui I., Mangou F., Malvy D., Vincendeau P., Mossalayi D., Haumont G., Vatan R., Olliaro P., Millet P. Plasmodium berghei: dehydroepiandrosterone sulfate reverses chloroquino-resistance in experimental malaria infection; correlation with glucose 6-phosphate dehydrogenase and glutathione synthesis pathway. Biochem. Pharmacol. 2004;68:1903–1910. doi: 10.1016/j.bcp.2004.05.049. [DOI] [PubMed] [Google Scholar]
- Sarma G.N., Savvides S.N., Becker K., Schirmer M., Schirmer R.H., Karplus P.A. Glutathione reductase of the malarial parasite Plasmodium falciparum: crystal structure and inhibitor development. J. Mol. Biol. 2003;328:893–907. doi: 10.1016/s0022-2836(03)00347-4. [DOI] [PubMed] [Google Scholar]
- Sharrock W.W., Suwanarusk R., Lek-Uthai U., Edstein M.D., Kosaisavee V., Travers T., Jaidee A., Sriprawat K., Price R.N., Nosten F., Russell B. Plasmodium vivax trophozoites insensitive to chloroquine. Malar. J. 2008;7:94. doi: 10.1186/1475-2875-7-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu A.B., Valderramos S.G., Fidock D.A. Pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol. Microbiol. 2005;57:913–926. doi: 10.1111/j.1365-2958.2005.04729.x. [DOI] [PubMed] [Google Scholar]
- Sidhu A.B., Verdier-Pinard D., Fidock D.A. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002;298:210–213. doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sienkiewicz N., Daher W., Dive D., Wrenger C., Viscogliosi E., Wintjens R., Jouin H., Capron M., Muller S., Khalife J. Identification of a mitochondrial superoxide dismutase with an unusual targeting sequence in Plasmodium falciparum. Mol. Biochem. Parasitol. 2004;137:121–132. doi: 10.1016/j.molbiopara.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Srivastava P., Puri S.K., Kamboj K.K., Pandey V.C. Glutathione-S-transferase activity in malarial parasites. Trop. Med. Int. Health. 1999;4:251–254. doi: 10.1046/j.1365-3156.1999.00387.x. [DOI] [PubMed] [Google Scholar]
- Suwanarusk R., Russell B., Chavchich M., Chalfein F., Kenangalem E., Kosaisavee V., Prasetyorini B., Piera K.A., Barends M., Brockman A., Lek-Uthai U., Anstey N.M., Tjitra E., Nosten F., Cheng Q., Price R.N. Chloroquine resistant Plasmodium vivax: in vitro characterisation and association with molecular polymorphisms. PLoS One. 2007;2:e1089. doi: 10.1371/journal.pone.0001089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztajer H., Gamain B., Aumann K.D., Slomianny C., Becker K., Brigelius-Flohe R., Flohe L. The putative glutathione peroxidase gene of Plasmodium falciparum codes for a thioredoxin peroxidase. J. Biol. Chem. 2001;276:7397–7403. doi: 10.1074/jbc.M008631200. [DOI] [PubMed] [Google Scholar]
- Tjitra E., Anstey N.M., Sugiarto P., Warikar N., Kenangalem E., Karyana M., Lampah D.A., Price R.N. Multidrug-resistant Plasmodium vivax associated with severe and fatal malaria: a prospective study in Papua, Indonesia. PLoS Med. 2008;5:e128. doi: 10.1371/journal.pmed.0050128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valderramos S.G., Valderramos J.C., Musset L., Purcell L.A., Mercereau-Puijalon O., Legrand E., Fidock D.A. Identification of a mutant PfCRT-mediated chloroquine tolerance phenotype in Plasmodium falciparum. PLoS Pathog. 2010;6:e1000887. doi: 10.1371/journal.ppat.1000887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Rodriguez J., Franke-Fayard B., Dinglasan R.R., Janse C.J., Pastrana-Mena R., Waters A.P., Coppens I., Rodriguez-Orengo J.F., Srinivasan P., Jacobs-Lorena M., Serrano A.E. The glutathione biosynthetic pathway of Plasmodium is essential for mosquito transmission. PLoS Pathog. 2009;5:e1000302. doi: 10.1371/journal.ppat.1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman S.K., Sabeti P.C., DeCaprio D., Neafsey D.E., Schaffner S.F., Milner D.A., Daily J.P., Sarr O., Ndiaye D., Ndir O., Mboup S., Duraisingh M.T., Lukens A., Derr A., Stange-Thomann N., Waggoner S., Onofrio R., Ziaugra L., Mauceli E., Gnerre S., Jaffe D.B., Zainoun J., Wiegand R.C., Birren B.W., Hartl D.L., Galagan J.E., Lander E.S., Wirth D.F. A genome-wide map of diversity in Plasmodium falciparum. Nat. Genet. 2007;39:113–119. doi: 10.1038/ng1930. [DOI] [PubMed] [Google Scholar]
- Wellems T.E., Plowe C.V. Chloroquine-resistant malaria. J. Infect. Dis. 2001;184:770–776. doi: 10.1086/322858. [DOI] [PubMed] [Google Scholar]
- Wootton J.C., Feng X., Ferdig M.T., Cooper R.A., Mu J., Baruch D.I., Magill A.J., Su X.Z. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature. 2002;418:320–323. doi: 10.1038/nature00813. [DOI] [PubMed] [Google Scholar]
- Yayon A., Cabantchik Z.I., Ginsburg H. Susceptibility of human malaria parasites to chloroquine is pH dependent. Proc. Natl. Acad. Sci. USA. 1985;82:2784–2788. doi: 10.1073/pnas.82.9.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]