

Abstract
Recent studies have shown that similar to cerebral gray matter (mainly composed of neuronal perikarya), white matter (composed of axons and glias) is vulnerable to ischemia. Edaravone, a free radical scavenger, has neuroprotective effects against focal cerebral ischemia even in humans. In this study, we investigated the time course and the severity of both gray and white matter damage following global cerebral ischemia by cardiac arrest, and examined whether edaravone protected the gray and the white matter. Male Sprague-Dawley rats were used. Global cerebral ischemia was induced by 5 minutes of cardiac arrest and resuscitation (CAR). Edaravone, 3 mg/kg, was administered intravenously either immediately or 60 minutes after CAR. The morphological damage was assessed by cresyl violet staining. The microtubule-associated protein 2 (a maker of neuronal perikarya and dendrites), the β amyloid precursor protein (the accumulation of which is a maker of axonal damage), and the ionized calcium binding adaptor molecule 1 (a marker of microglia) were stained for immunohistochemical analysis. Significant neuronal perikaryal damage and marked microglial activation were observed in the hippocampal CA1 region with little axonal damage one week after CAR. Two weeks after CAR, the perikaryal damage and microglial activation were unchanged, but obvious axonal damage occurred. Administration of edaravone 60 minutes after CAR significantly mitigated the perikaryal damage, the axonal damage, and the microglial activation. Our results show that axonal damage develops slower than perikaryal damage and that edaravone can protect both gray and white matter after CAR in rats.
Keywords: Cardiac arrest, Global Cerebral ischemia, Edaravone, Free radical scavenger, White matter damage
1. Introduction
Transient global cerebral ischemia, the most common clinical feature of which is circulatory arrest, leads to severe neuronal damage in selectively vulnerable brain areas, such as the hippocampal CA1 region, medium-sized neurons in the striatum, and the neocortical neurons in layers 3, 5 and 6, in both experimental animals and humans (Horn and Schlote, 1992; Kirino, 1982; Petito et al., 1987; Pulsinelli et al., 1982). A number of promising neuroprotective drugs for cerebral ischemia, especially for focal ischemia, have been proposed for preclinical testing, but almost all of these drugs have been found disappointing in clinical trials (Gladstone et al., 2002). One of the reasons why these neuroprotective drugs do not demonstrate obvious clinical benefits is the fact that preclinical studies have concentrated exclusively on protection of cerebral gray matter, i.e., neuronal perikarya. However, damage of white matter, which is composed of myelinated axons and glias, specifically oligodendrocytes, following acute cerebral ischemia and the effects of neuroprotective drugs on white matter have been largely neglected (Gladstone et al., 2002). Protecting only neuronal perikarya from ischemic insults is of little benefit to maintain neuronal function if their axons are not being protected, and thus Dewar et al. have emphasized that “total brain protection”, in which not only gray matter but also white matter should be protected, is important and necessary (Dewar et al., 1999).
Cerebral white matter is as vulnerable to ischemic insults as cerebral gray matter (Pantoni et al., 1996), although the pathophysiology and mechanisms of white matter damage differ in certain ways from those of gray matter damage (Dewar et al., 1999; Gladstone et al., 2002). For example, glutamatergic NMDA receptors are not expressed in axons and oligodendrocytes. As a consequence, blockade of Na+ channels and AMPA/kinate receptors is more important for white matter to maintain Ca2+ homeostasis during ischemia (Dewar et al., 1999). Actually, MK-801, which markedly reduces ischemic damage to neuronal perikarya, had no protective effect on axonal damage following middle cerebral artery occlusion (Yam et al., 2000).
There is increasing evidence that oxidative stress following ischemia-reperfusion contributes to neuronal damage (Liachenko et al., 2003). Edaravone (3-Methyl-1-phenyl-2-pyrazolin-5-one) is a free radical scavenger and clinically available in Japan (Yoneda et al., 2003). It has been demonstrated that edaravone has a neuroprotective effect on focal cerebral ischemia (Abe et al., 1988; Amemiya et al., 2005), while there is no consensus about edaravone’s neuroprotective effect on global cerebral ischemia (Jin et al., 2002; Otani et al., 2005). To our knowledge, there is no report on edaravone’s effect on cerebral white matter damage. We hypothesized that edaravone would be effective in protecting not only gray matter but also white matter against cerebral ischemia, because white matter is abundant in lipid which can be peroxidized following ischemia-reperfusion insults.
The aim of this study was to investigate firstly, the time course and the severity of both gray and white matter damage following global cerebral ischemia after cardiac arrest and secondly, the effect of edaravone on reducing gray and white matter damage.
2. Results
2. 1. Physiological Variables
Fig. 1 shows the schematic diagram depicting the experimental protocol. Fifty rats were randomly assigned to one of 5 groups (in each group, n = 10): Sham group, Isc1 group, Isc2 group, Edv0 group, and Edv60 group. Seven out of 50 rats (three rats in Isc1, two in Isc2, and two in Edv60 group were excluded from the study because of massive bleeding during operation.
Fig. 1. Experimental groups and protocol.

Fig. 1 shows the schematic diagram depicting the experimental protocol. In Sham group, rats were subjected to the same surgical procedure but did not undergo cardiac arrest and resuscitation (CAR). In Isc1 and Isc2 groups, rats were subjected to 5 minutes cardiac arrest and treated with saline immediately after CAR. Rats in Edv0 and Edv60 groups were subjected to 5 minutes cardiac arrest and given 3 mg/kg edaravone intravenously immediately after CAR or 60 minutes after CAR, respectively. The rats in the Sham and the Isc1 groups were killed one week after CAR. Rats in the Isc2, Edv0, and Edv60 groups were killed two weeks after CAR.
Table 1 summarizes the physiological variables before and after cardiac arrest and resuscitation (CAR). The pH, pO2, pCO2, and blood glucose were all within the normal physiological range before cardiac arrest and were not statistically different among the groups. Cardiac arrest time was not statistically different among the groups. After CAR, rats in the Isc2 group showed a significantly lower PaO2 than the Sham group.
Table 1.
Physiological variables
|
|
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| weight (g) | duration of cardiac arrest (minutes) | Before CAR
|
After CAR
|
||||||||
| pH | PaO2 (FiO2 0.6) (mmHg) | PaCO2 (mmHg) | Glucose (mg/dl) | pH | PaO2 (FiO2 0.6) (mmHg) | PaCO2 (mmHg) | Glucose (mg/dl) | ||||
|
|
|
|
|||||||||
| Sham | (n=10) | 248 ± 10 | - | 7.423 ± 0.01 | 226 ± 3 | 37 ± 2 | 133 ± 7 | 7.389 ± 0.01 | 208 ± 6 | 42 ± 2 | 126 ± 5 |
| Isc1 | (n=7) | 235 ± 6 | 5.26 ± 0.07 | 7.398 ± 0.02 | 217 ±17 | 44 ± 3 | 139 ± 14 | 7.324 ± 0.03 | 188 ± 13 | 47 ± 4 | 127 ± 6 |
| Isc2 | (n=8) | 265 ± 9 | 5.23 ± 0.06 | 7.422 ± 0.01 | 222 ± 10 | 40 ± 1 | 148 ± 10 | 7.274 ± 0.06 | 147 ± 18* | 55 ± 9 | 131 ± 9 |
| Edv0 | (n=10) | 245 ± 6 | 5.20 ± 0.04 | 7.420 ± 0.02 | 219 ± 21 | 40 ± 1 | 171 ± 11 | 7.335 ± 0.04 | 157 ± 20 | 49 ± 6 | 133 ± 7 |
| Edv60 | (n=8) | 248 ± 3 | 5.09 ± 0.07 | 7.452 ± 0.01 | 256 ± 15 | 37 ± 1 | 156 ± 11 | 7.359 ± 0.02 | 152 ± 12 | 43 ± 2 | 135 ± 11 |
|
|
|
|
|||||||||
P<0.05 compared with Sham group after CAR
CAR = cardiac arrest and resuscitation
2. 2. Neurologic Deficit Scores
Neurologic deficit scores (Jomura et al., 2007; Neumar et al., 1995) did not change after CAR in any group and showed almost full marks without paralysis (data is not shown).
2. 3. Time Course and Severity of Neuronal Damage Induced by Global Cerebral Ischemia
Fig. 2 shows representative photomicrographs of the hippocampal CA1 region. In the Sham group, almost all neurons appeared healthy and showed round nuclei with dark cell bodies in cresyl violet staining (Fig. 2, A1). In contrast, there were numerous damaged neurons with pyknotic nuclei (the condensation of chromatin) and karyorhexis (the fragmentation of the nucleus) in the ischemic groups (Fig. 2, A2 and A3). The CAR caused a significant decrease in the number of surviving neurons in the hippocampal CA1 region (Fig. 3A). There was not significant difference between the number of surviving neurons in the Isc1 and Isc2 groups, which differed only in the recovery time, indicating that the neuronal perikaryal damage was fully established within a week after the CAR. Moreover, the ischemia caused an extensive decrease in microtubule-associated protein 2 (MAP2) expression in the hippocampal CA1 region (Fig. 2, B2 and B3). Fig. 3B shows the significant quantitative decrease in MAP2 expression in the CA1 region. The β amyloid precursor protein (βAPP) accumulation in the hippocampal CA1 region was quite low in the Sham and the Isc1 groups (Fig. 2, D1 and D2), but the global cerebral ischemia caused extensive βAPP accumulation 2 weeks after CAR (Fig. 2, D3 and Fig. 3C). Interestingly, βAPP accumulation following the global ischemia in axon-rich brain areas, such as the caudate putamen and the internal and external capsules, were sparse and some accumulation of βAPP was observed in the corpus callosum 2 weeks but not 1 week after CAR (Fig. 4, A2–3). Microglias were significantly activated at 1–2 weeks after CAR (Fig. 2, E1– 3).
Fig. 2. Histologic neuronal and axonal damages in the hippocampal CA1 region following global cerebral ischemia caused by cardiac arrest and resuscitation (CAR).

Representative sections of cresyl violet staining (A1–5) and immunostained sections of MAP2 (B1–5 and C1–5), βAPP (D1–5), and Iba-1 (E1–5) in the CA1 in the Sham, Isc1, Isc2, Edv0, and Edv60 groups are depicted. A1: Normal pyramidal neurons in the Sham group. A2 and A3: Typical appearance of neuronal damage 1 week (Isc1) and 2 weeks (Isc2) after CAR, respectively. A4 and A5: Reduction of neuronal damage by edaravone administered immediately (Edv0) and 60 min (Edv60) after CAR, respectively. B1: Normal MAP2 expression in the Sham group. A white square shows the region of predetermined area for the neuronal perikaryal and axonal damages evaluation. B2 and B3: Extensive decrease in MAP2 expression after CAR in the Isc1 and Isc2 groups, respectively. B4 and B5: Mitigation of decrease in MAP2 expression by edaravone in the Edv0 and Edv60 groups. C: MAP2 expression as in B at a higher magnification. D1 and D2: Normal detection level of the βAPP accumulation in the Sham and Isc1 groups, respectively, one week after CAR. D3: Extensive granular βAPP deposition 2 weeks after CAR in the Isc2 group. D4 and D5: Mitigation of βAPP accumulation in the edaravone-treated groups. E1: Scattered ramified microglias are found in the Sham group. E2 and E3: Dense accumulation of ameboid microglias 1 week and 2 weeks after CAR. E4: Microglial activation is slightly suppressed by edaravone administered immediately after CAR. E5: Microglial activation is markedly suppressed by edaravone administered 60 minutes after CAR. Scale Bar: 50 μm (A, C, D, and E); 1 mm (B).
Fig. 3. Quantified Analyses of neuronal and axonal damages in the hippocampal CA1 region following global cerebral ischemia caused by cardiac arrest and resuscitation.
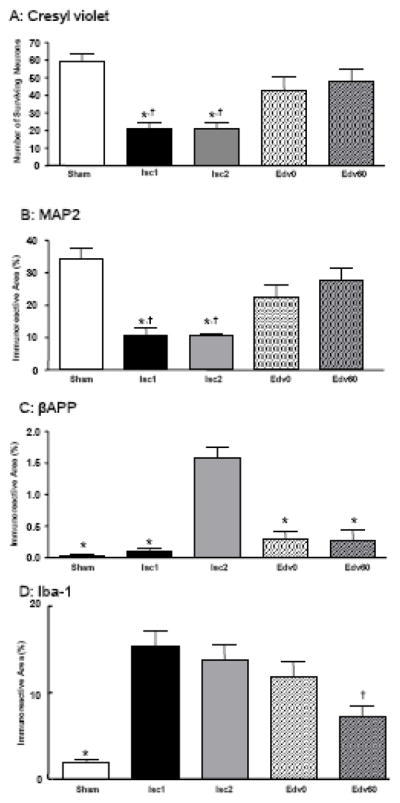
A: The extent of neuronal perikaryal damage is quantified by counting the number of surviving neurons in a predetermined hippocampal CA1 region. * P<0.05 compared with the Sham group; † P<0.05 compared with the Edv60 group. B: Neuronal perikaryal damage, quantified by the percentage of MAP2 immunoreactive areas in a predetermined hippocampal CA1 region. * P<0.05 compared with the Sham group; † P<0.05 compared with the Edv60 group. C: Axonal damage, quantified by the percentage of the βAPP immunoreactive areas in a predetermined CA1. * P<0.05 compared with the Isc2 group. D: Microglial activation, quantified by the percentage of the Iba-1 immunoreactive areas in a predetermined hippocampal CA1 region. * P<0.05 compared with the Isc1 and Isc2 groups; † P<0.05 compared with the Isc1 and Isc2 groups.
Fig. 4. Axonal damage in the corpus callosum following global cerebral ischemia caused by cardiac arrest and resuscitation (CAR).
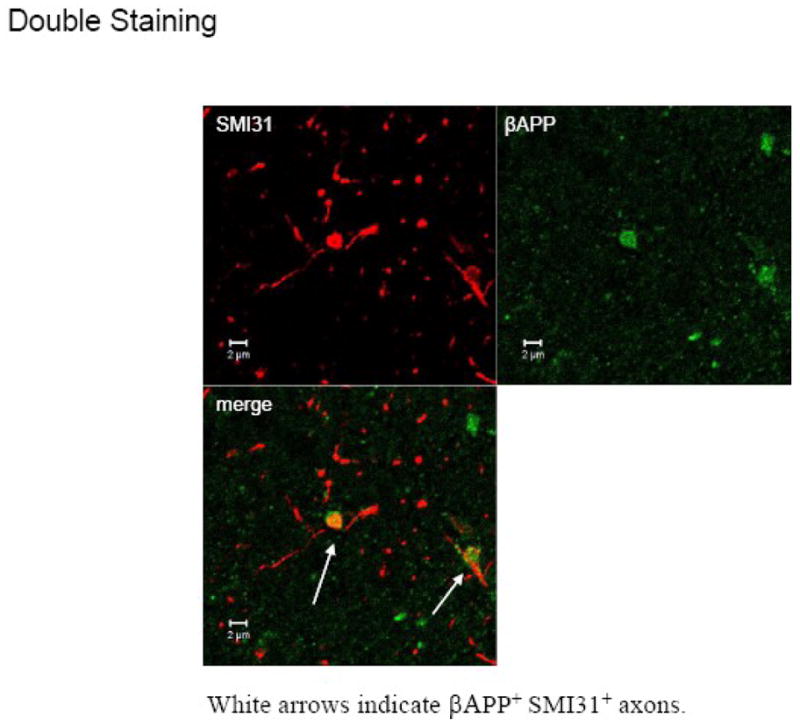
βAPP immunoreactivity in the corpus callosum. A1: Absence of βAPP accumulation in the Sham group. A2: Little βAPP accumulation 1 week after CAR. A3: Positive staining of βAPP is seen in axons 2 weeks after CAR (arrow). A4: βAPP accumulation is suppressed by edaravone administered 60 minutes after CAR. Scale Bar: 50 μm
2. 4. Effects of Edaravone on Neuronal Perikaryal Damage and Axonal Damage
Edaravone potently protected the brain against neuronal damage in the hippocampal CA1 region following global cerebral ischemia (Fig. 2, A4 and A5). The number of surviving neurons in the Edv60 group was significantly greater than in the Isc1 and Isc2 groups (Fig. 3, A). The number of surviving neurons in the Edv0 and Edv60 groups was not significantly different from that in the Sham group, indicating that edaravone restored the neuronal survivability to the control level (Fig. 3A). Furthermore, MAP2 staining confirmed that edaravone provided a significant neuroprotective effect against CAR (Fig. 2, B4 and B5). The bar graphs in Fig. 3B show that edaravone protected hippocampus against the MAP2 disruption nearly to the control level. In addition, edaravone significantly reduced the βAPP accumulation in the hippocampal CA1 region almost to the control (the Sham) level (Fig. 2, D1, D4, D5, and Fig. 3C). The βAPP accumulation in the corpus callosum was also attenuated by edaravone (Fig. 4, A3 and A4). The activation of microglias was significantly suppressed by edaravone (Fig. 2, E4 and E5 and Fig. 3D)
3. Discussion
The novel findings of this study are that axonal (white matter) damage develops slower than neuronal perikaryal (gray matter) damage, and that edaravone exerts significant protection against not only gray matter damage but also white matter damage induced by global cerebral ischemia.
It has been demonstrated that damage to hippocampal pyramidal neurons is delayed and is generally not fully developed until 5–7 days after reperfusion in animal models of forebrain ischemia (Kirino, 1982; Pulsinelli et al., 1982). We confirmed that neuronal perikaryal damage in the hippocampal CA1 region was apparent and completed by 1 week after CAR, whereas axonal damage was absent one week after CAR and only becoming clearly evident 2 weeks after CAR. Based on these results, we recommend that, in in vivo studies, the observation period following cerebral ischemia should be extended to a minimum of 2 weeks. Otherwise it is difficult to assess the efficacy of a drug with a potential for so-called “total brain protection (Dewar et al., 1999)” against cerebral ischemia. In this study, we examined the neuronal perikaryal damage using both the conventional cresyl violet staining and the MAP2 immunohistochemistry. Neuronal perikarya and dendrites express MAP2, which binds to and stabilizes microtubules, and may help to regulate microtubule spacing (Matus, 1988). A loss of MAP2 expression can be used as a sensitive and reliable marker of neuronal damage (Arai et al., 1994; Kitagawa et al., 1989). To evaluate axonal damage, the accumulation of the βAPP was investigated. βAPP is usually produced within neurons at a concentration lying below the detection threshold (Beeson et al., 1994; Koo et al., 1990; Lin et al., 1999), and it accumulates in the axon, less often within the neuronal cell body, when the fast anterograde axonal transport is disrupted following brain damage, such as ischemia (Koo et al., 1990; Yam et al., 1998). It is generally accepted that βAPP accumulation is a good marker of axonal damage (Lin et al., 1999; van Groen et al., 2005).
White matter is traditionally considered less vulnerable than gray matter to ischemic insults (Marcoux et al., 1982). However, recent studies have shown that white matter is also vulnerable to ischemia (Dewar et al., 1999; Hirko et al., 2008; Pantoni et al., 1996; Pantoni and Garcia, 1997). Characteristic white matter lesions, sometimes in the absence of gray matter damage, are observed following expose of cyanide, or carbon monoxide (Pantoni et al., 1996). Such lesions also were observed after chronic cerebral hypoperfusion in a rat model (Miyamoto et al., 2001). In the present study, we cannot clarify whether the axonal damage in the CA1 is due to a direct effect of ischemia or a secondary damage of neuronal perikarya. However, we assume that the axonal damage is due to the direct effect of ischemia, because Wallerian degeneration in the mammalian central nervous system usually occurs very slowly, taking months to years to appear (Vargas and Barres, 2007). In addition, the evidence that there was a lot of βAPP accumulation in the CA1 stratum radiatum, which contains septal and commissural fibers from the contralateral hippocampus and Schaffer collateral fibers which are the projection forward from hippocampal CA3 region to the CA1 region, indicates that the axonal damage, at least partly, was induced directly by CAR but not secondarily after neuronal perikaryal damage because the CA3 neuronal perikarya were not damaged. Similar to βAPP accumulation in the CA1, βAPP accumulation in the corpus callosum, which is a representative white matter, was apparent at 2 weeks after the CAR and suppressed by edaravone.
Edaravone has been clinically used for the treatment of acute cerebral infarction in Japan since June 2001. Numerous animal studies have demonstrated the neuroprotective effects of edaravone against focal cerebral ischemia (Abe et al., 1988; Amemiya et al., 2005; Kawai et al., 1997), but there have been only a few reports which demonstrate that edaravone has neuroprotective effects against global (forebrain) cerebral ischemia (Otani et al., 2005; Watanabe et al., 1994; Yamamoto et al., 1997). We chose to use 3 mg/kg of edaravone, because this dose has been effective neuroprotection in rodent ischemic models (Abe et al., 1988; Amemiya et al., 2005; Watanabe et al., 1994). While the timing of edaravone administration was variable in previous studies (Abe et al., 1988; Amemiya et al., 2005; Otani et al., 2005; Watanabe et al., 1994), it was most often administered before or immediately after cerebral ischemia and reperfusion. In the present study, we demonstrated that edaravone, administered even 60 minutes after CAR, significantly protected not only gray matter but also white matter against ischemia.
Recent studies have shown that oxidative stress plays a pivotal role on brain damage following ischemia-reperfusion insult (Liachenko et al., 2003). Reactive oxygen species (ROS) induce lipid peroxidation, which can modify various cell membrane functions, such as neurotransmitter release and uptake, those of ion-channels, ion-motive ATPases, and GTP-binding proteins, leading to impaired mitochondrial function, resulting in apoptosis (Mattson, 1998). Our result showing greatly reduced axonal damage after edaravone treatment seems quite reasonable because white matter is abundant in lipid. The evidence that edaravone suppressed microglial activation suggests that edaravone exerted neuroprotection as a radical scavenger, because activated microglias produce ROS, which can cause neuronal damage and amplify the inflammatory response of microglias (Block et al., 2007). Although some authors have previously reported that free radical scavengers mitigate white matter damage in rodent brain ischemia models (Imai et al., 2001; Irving et al., 1997; Lin et al., 2006), they used radical scavengers other than edaravone and their cerebral ischemia models were different from ours. Moreover, as 60 minutes after CAR is a sufficiently long therapeutic time window in the clinical setting, edaravone seems to be a promising neuroprotective drug against global cerebral ischemia.
In conclusion, using a cardiac arrest model in rats, we demonstrated that white matter damage develops slower than gray matter damage in the hippocampal CA1 region, and edaravone, administered 60 minutes after CAR, significantly protected not only gray matter but also white matter. Our results indicate that the use of edaravone as a “total brain protection (Dewar et al., 1999)” drug against global cerebral ischemia in the clinical setting should be explored and evaluated.
4. Experimental Procedure
4.1. Animal Model
The study was approved by the Animal Research Committee at Kansai Medical University. Adult male Sprague-Dawley rats (Shimizu Laboratory Supplies Co., Ltd, Kyoto, Japan) weighing 210 to 290 g were used. Fifty rats were randomly assigned to one of five groups (Fig. 1). In the Sham group, rats were subjected to the same surgical procedure except for CAR. The Isc1 and Isc2 group rats were subjected to 5 minutes cardiac arrest and given physiologic saline intravenously immediately after CAR. The Edv0 and Edv60 group rats were subjected to 5 minutes cardiac arrest and treated with intravenous administration of 3 mg/kg edaravone immediately or 60 minutes after CAR, respectively.
Transient global ischemia was induced by the CAR technique as described previously (Jomura et al., 2007; Liachenko et al., 1998; Liachenko et al., 2001) with slight modifications. Under isoflurane anesthesia, rats were orotracheally intubated and mechanically ventilated. Rats were paralyzed by the intravenous administration of pancuronium bromide (1.5 mg/kg). Anesthesia was maintained with 1 to 2% isoflurane in 60% O2 unless otherwise stated. The temporalis muscle temperature was monitored and maintained at 36.5 ± 0.5 °C during the surgical procedure using a feedback controlled heating blanket. Before the induction of cardiac arrest, the arterial blood pH and gases were measured using an i-STAT portable clinical analyzer 300F (FUSO pharmaceutical industries, Ltd., Osaka, Japan). The arterial blood glucose was measured using Medisafe Mini MS-GK03V (Terumo Co., Tokyo, Japan).
Cardiac arrest was induced by an ultra-short-acting β1 -blocker, esmolol (7 mg), followed by the stoppage of mechanical ventilation. At this point, a sharp decrease in the mean arterial pressure to less than 10 mmHg was considered as the induction of cardiac arrest. Strictly 5 minutes after the induction of cardiac arrest, resuscitation was performed by retrograde infusion of oxygenated blood mixed with a resuscitation mixture containing heparin (8 U/ml), sodium bicarbonate (0.05 mEq/ml), and epinephrine (8.5 μg/ml), at the same time ventilation was started with 100% O2 for 10 minutes. The rats were ventilated for at least 60 minutes and were extubated when sustained spontaneous breathing was observed. Rats were returned to the cages and followed up for one or two weeks.
4. 2. Neurologic Deficit Scores
Functional damage and recovery were evaluated using neurologic deficit scores (NDS), which ranges from a score of 500 indicating a neurologically normal rat, to 0 for brain death (Jomura et al., 2007; Neumar et al., 1995).
4. 3. Histology Analysis
Rats were observed for either 1 week (Sham and Isc1 groups) or 2 weeks (Isc2, Edv0, and Edv60 groups) after the surgery. Under deep isoflurane anesthesia, rats were perfused with buffered 10% formalin phosphate and the brains were extracted. The brain sections containing the dorsal hippocampal region were embedded in paraffin and sliced into 5-μm-thick coronal sections. The sections were stained with cresyl violet and the hippocampal CA1 regions were photographed. For immunohistochemical staining, brain sections were deparaffinized and incubated with mouse monoclonal antibodies against MAP2 (Sigma-Aldrich, St. Louis, MO) or βAPP (Zymed, South San Francisco, CA), or with a rabbit polyclonal antibody against Iba-1 (Wako Pure Chemical, Osaka, Japan). The sections were subsequently incubated with biotinylated anti-mouse IgG for monoclonal antibodies or biotinylated anti-rabbit IgG for Iba-1 (Vector Laboratories, Burlingame, CA), and then incubated with an avidin-biotin peroxidase complex solution (Vector Laboratories). The immunoreactive products were visualized with a solution of 0.02% 3, 3′-diaminobenzidine tetrahydrochloride containing 0.005% H2O2 in 0.05 M Tris buffer (pH 7.6).
Morphometry and Statistical Analysis
The neuronal and axonal damages were determined in a predetermined area of 0.0374 mm2 in the hippocampal CA1 region in each hemisphere (square in Fig. 2, B1). To quantify the histological damage, morphologically normal neurons stained with cresyl violet were counted by two observers blind to the treatment and showed as the number of surviving neurons. The βAPP, MAP2, and Iba-1 expression positive areas were calculated with the use of a computer-assisted image analysis system (NIH Image J) attached to a light microscope and a high-resolution color video camera (Miyamoto et al., 2001).
Statistical comparison of physiological variables, histological data, NDS, and body weight on the same day among groups was made by one-way analysis of variance (ANOVA) followed by the Bonferroni post hoc test using Graph Pad Prizm (GraphPad Software, Inc., San Diego, CA). All data were expressed as mean ± SEM. A P value < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research B-19791092 and C-19591825 from the Japan Society for the Promotion of Science. Yan Xu acknowledges the support from the National Institutes of Health of the United States (Grant # R01NS036124). Edaravone was kindly donated by Mitsubishi Tanabe Pharma Corporation (Osaka, Japan).
Abbreviations
- CAR
cardiac arrest and resuscitation
- MAP2
microtubule-associated protein 2
- βAPP
beta amyloid precursor protein
- Iba-1
ionized calcium binding adaptor molecule 1
- NDS
neurologic deficit score
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could a3ect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe K, Yuki S, Kogure K. Strong attenuation of ischemic and postischemic brain edema in rats by a novel free radical scavenger. Stroke. 1988;19:480–5. doi: 10.1161/01.str.19.4.480. [DOI] [PubMed] [Google Scholar]
- Amemiya S, Kamiya T, Nito C, Inaba T, Kato K, Ueda M, Shimazaki K, Katayama Y. Anti-apoptotic and neuroprotective effects of edaravone following transient focal ischemia in rats. Eur J Pharmacol. 2005;516:125–30. doi: 10.1016/j.ejphar.2005.04.036. [DOI] [PubMed] [Google Scholar]
- Arai T, Watanabe K, Nakao S, Mori H, Murakawa M, Mori K, Tooyama I, Kimura H, Kojima S. Effects of neopterin on ischemic neuronal damage in gerbils. Neurosci Lett. 1994;173:107–10. doi: 10.1016/0304-3940(94)90160-0. [DOI] [PubMed] [Google Scholar]
- Beeson J, Shelton E, Chan H, Gage F. Differential distribution of amyloid protein precursor immunoreactivity in the rat brain studied by using five different antibodies. J Comp Neurol. 1994;342:78–96. doi: 10.1002/cne.903420109. [DOI] [PubMed] [Google Scholar]
- Block M, Zecca L, Hong J. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Dewar D, Yam P, McCulloch J. Drug development for stroke: importance of protecting cerebral white matter. Eur J Pharmacol. 1999;375:41–50. doi: 10.1016/s0014-2999(99)00280-0. [DOI] [PubMed] [Google Scholar]
- Gladstone D, Black S, Hakim A. Toward wisdom from failure: lessons from neuroprotective stroke trials and new therapeutic directions. Stroke. 2002;33:2123–36. doi: 10.1161/01.str.0000025518.34157.51. [DOI] [PubMed] [Google Scholar]
- Hirko A, Dallasen R, Jomura S, Xu Y. Modulation of Inflammatory Responses after Global Ischemia by Transplanted Umbilical-Cord Matrix Stem Cells. Stem Cells. 2008;26:2893–901. doi: 10.1634/stemcells.2008-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn M, Schlote W. Delayed neuronal death and delayed neuronal recovery in the human brain following global ischemia. Acta Neuropathol. 1992;85:79–87. doi: 10.1007/BF00304636. [DOI] [PubMed] [Google Scholar]
- Imai H, Masayasu H, Dewar D, Graham D, Macrae I. Ebselen protects both gray and white matter in a rodent model of focal cerebral ischemia. Stroke. 2001;32:2149–54. doi: 10.1161/hs0901.095725. [DOI] [PubMed] [Google Scholar]
- Irving E, Yatsushiro K, McCulloch J, Dewar D. Rapid alteration of tau in oligodendrocytes after focal ischemic injury in the rat: involvement of free radicals. J Cereb Blood Flow Metab. 1997;17:612–22. doi: 10.1097/00004647-199706000-00003. [DOI] [PubMed] [Google Scholar]
- Jin Y, Mima T, Raicu V, Park K, Shimizu K. Combined argatroban and edaravone caused additive neuroprotection against 15 min of forebrain ischemia in gerbils. Neurosci Res. 2002;43:75–9. doi: 10.1016/s0168-0102(02)00019-6. [DOI] [PubMed] [Google Scholar]
- Jomura S, Uy M, Mitchell K, Dallasen R, Bode C, Xu Y. Potential treatment of cerebral global ischemia with Oct-4+ umbilical cord matrix cells. Stem Cells. 2007;25:98–106. doi: 10.1634/stemcells.2006-0055. [DOI] [PubMed] [Google Scholar]
- Kawai H, Nakai H, Suga M, Yuki S, Watanabe T, Saito K. Effects of a novel free radical scavenger, MCl-186, on ischemic brain damage in the rat distal middle cerebral artery occlusion model. J Pharmacol Exp Ther. 1997;281:921–7. [PubMed] [Google Scholar]
- Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Niinobe M, Mikoshiba K, Hata R, Ueda H, Handa N, Fukunaga R, Isaka Y, Kimura K. Microtubule-associated protein 2 as a sensitive marker for cerebral ischemic damage--immunohistochemical investigation of dendritic damage. Neuroscience. 1989;31:401–11. doi: 10.1016/0306-4522(89)90383-7. [DOI] [PubMed] [Google Scholar]
- Koo E, Sisodia S, Archer D, Martin L, Weidemann A, Beyreuther K, Fischer P, Masters C, Price D. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A. 1990;87:1561–5. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachenko S, Tang P, Hamilton R, Xu Y. A reproducible model of circulatory arrest and remote resuscitation in rats for NMR investigation. Stroke. 1998;29:1229–38. doi: 10.1161/01.str.29.6.1229. discussion 1238–9. [DOI] [PubMed] [Google Scholar]
- Liachenko S, Tang P, Hamilton R, Xu Y. Regional dependence of cerebral reperfusion after circulatory arrest in rats. J Cereb Blood Flow Metab. 2001;21:1320–9. doi: 10.1097/00004647-200111000-00008. [DOI] [PubMed] [Google Scholar]
- Liachenko S, Tang P, Xu Y. Deferoxamine improves early postresuscitation reperfusion after prolonged cardiac arrest in rats. J Cereb Blood Flow Metab. 2003;23:574–81. doi: 10.1097/01.WCB.0000057742.00152.3F. [DOI] [PubMed] [Google Scholar]
- Lin B, Schmidt-Kastner R, Busto R, Ginsberg M. Progressive parenchymal deposition of beta-amyloid precursor protein in rat brain following global cerebral ischemia. Acta Neuropathol. 1999;97:359–68. doi: 10.1007/s004010050999. [DOI] [PubMed] [Google Scholar]
- Lin S, Cox H, Rhodes P, Cai Z. Neuroprotection of alpha-phenyl-n-tert-butyl-nitrone on the neonatal white matter is associated with anti-inflammation. Neurosci Lett. 2006;405:52–6. doi: 10.1016/j.neulet.2006.06.063. [DOI] [PubMed] [Google Scholar]
- Marcoux F, Morawetz R, Crowell R, DeGirolami U, Halsey JJ. Differential regional vulnerability in transient focal cerebral ischemia. Stroke. 1982;13:339–46. doi: 10.1161/01.str.13.3.339. [DOI] [PubMed] [Google Scholar]
- Mattson M. Modification of ion homeostasis by lipid peroxidation: roles in neuronal degeneration and adaptive plasticity. Trends Neurosci. 1998;21:53–7. doi: 10.1016/s0166-2236(97)01188-0. [DOI] [PubMed] [Google Scholar]
- Matus A. Microtubule-associated proteins: their potential role in determining neuronal morphology. Annu Rev Neurosci. 1988;11:29–44. doi: 10.1146/annurev.ne.11.030188.000333. [DOI] [PubMed] [Google Scholar]
- Miyamoto E, Tomimoto H, Nakao S, Wakita H, Akiguchi I, Miyamoto K, Shingu K. Caudoputamen is damaged by hypocapnia during mechanical ventilation in a rat model of chronic cerebral hypoperfusion. Stroke. 2001;32:2920–5. doi: 10.1161/hs1201.100216. [DOI] [PubMed] [Google Scholar]
- Neumar R, Bircher N, Sim K, Xiao F, Zadach K, Radovsky A, Katz L, Ebmeyer E, Safar P. Epinephrine and sodium bicarbonate during CPR following asphyxial cardiac arrest in rats. Resuscitation. 1995;29:249–63. doi: 10.1016/0300-9572(94)00827-3. [DOI] [PubMed] [Google Scholar]
- Otani H, Togashi H, Jesmin S, Sakuma I, Yamaguchi T, Matsumoto M, Kakehata H, Yoshioka M. Temporal effects of edaravone, a free radical scavenger, on transient ischemia-induced neuronal dysfunction in the rat hippocampus. Eur J Pharmacol. 2005;512:129–37. doi: 10.1016/j.ejphar.2005.01.050. [DOI] [PubMed] [Google Scholar]
- Pantoni L, Garcia J, Gutierrez J. Cerebral white matter is highly vulnerable to ischemia. Stroke. 1996;27:1641–6. doi: 10.1161/01.str.27.9.1641. discussion 1647. [DOI] [PubMed] [Google Scholar]
- Pantoni L, Garcia J. Pathogenesis of leukoaraiosis: a review. Stroke. 1997;28:652–9. doi: 10.1161/01.str.28.3.652. [DOI] [PubMed] [Google Scholar]
- Petito C, Feldmann E, Pulsinelli W, Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology. 1987;37:1281–6. doi: 10.1212/wnl.37.8.1281. [DOI] [PubMed] [Google Scholar]
- Pulsinelli W, Brierley J, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–8. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- van Groen T, Puurunen K, Mäki H, Sivenius J, Jolkkonen J. Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke. 2005;36:1551–6. doi: 10.1161/01.STR.0000169933.88903.cf. [DOI] [PubMed] [Google Scholar]
- Vargas M, Barres B. Why is Wallerian degeneration in the CNS so slow? Annu Rev Neurosci. 2007;30:153–79. doi: 10.1146/annurev.neuro.30.051606.094354. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Yuki S, Egawa M, Nishi H. Protective effects of MCI-186 on cerebral ischemia: possible involvement of free radical scavenging and antioxidant actions. J Pharmacol Exp Ther. 1994;268:1597–604. [PubMed] [Google Scholar]
- Yam P, Dewar D, McCulloch J. Axonal injury caused by focal cerebral ischemia in the rat. J Neurotrauma. 1998;15:441–50. doi: 10.1089/neu.1998.15.441. [DOI] [PubMed] [Google Scholar]
- Yam P, Dunn L, Graham D, Dewar D, McCulloch J. NMDA receptor blockade fails to alter axonal injury in focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20:772–9. doi: 10.1097/00004647-200005000-00003. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Yuki S, Watanabe T, Mitsuka M, Saito K, Kogure K. Delayed neuronal death prevented by inhibition of increased hydroxyl radical formation in a transient cerebral ischemia. Brain Res. 1997;762:240–2. doi: 10.1016/s0006-8993(97)00490-3. [DOI] [PubMed] [Google Scholar]
- Yoneda Y, Uehara T, Yamasaki H, Kita Y, Tabuchi M, Mori E. Hospital-based study of the care and cost of acute ischemic stroke in Japan. Stroke. 2003;34:718–24. doi: 10.1161/01.STR.0000056171.55342.FF. [DOI] [PubMed] [Google Scholar]