

Abstract
We have developed a system for producing tubular multilamellar liposomes that incorporate the protein FtsZ on the inside. We start with a mixture of spherical multilamellar liposomes with FtsZ initially on the outside. Shearing forces generated by applying a coverslip most likely distort some of the spherical liposomes into a tubular shape, and causes some to leak and incorporate FtsZ inside. We describe protocols for liposome preparation, and for preparing membrane-targeted FtsZ that can assemble contractile Z rings inside the tubular liposomes. We also describe the characterization of the multilamellar liposomes in terms of the permeability or leakiness for a small fluorescent dye and larger protein molecules. These liposomes may be useful for reconstitution of other biological systems.
1. Introduction
FtsZ is a bacterial tubulin homologue that forms a ring structure called the “Z ring” at the division plane in bacteria. The Z ring is anchored to the membrane and constricts to divide the bacteria. FtsZ recruits a dozen other essential division proteins, which are mostly involved in remodeling the peptidoglycan cell wall. We recently succeeded in reconstituting Z rings inside tubular liposomes, and found that they generated a constriction force on the liposome wall (Osawa et al., 2008). The assembly of Z rings and the generation of the constriction force were achieved with FtsZ alone, and did not require any other division protein. This was an important discovery itself for understanding the mechanism of bacterial cell division. Now the liposome system we developed should provide a simple in vitro system for studying molecular details of how FtsZ works.
To achieve these results we had to overcome two technical problems. The first problem was to tether FtsZ to the membrane. Pichoff and Lutkenhaus (2005) discovered that the carboxy terminus of FtsZ binds to FtsA, and FtsA has an amphipathic helix at its carboxy terminal that inserts into the membrane. We made an FtsZ that could tether itself to the membrane by fusing an amphipathic helix (membrane targeting sequence: mts) to the carboxy terminus of FtsZ. To visualize the protein, we inserted a yellow fluorescent protein (YFP) before the mts, giving FtsZ-YFP-mts. Here we provide detailed protocols for the purification of FtsZ-YFP-mts.
The second problem was how to get the protein inside liposomes. We have succeeded in getting FtsZ-YFP-mts inside spherical unilamellar liposomes using the emulsion method (Noireaux and Libchaber, 2004; Pautot et al., 2003). However, we have never found Z rings assembled in such spherical unilamellar liposomes.
Eventually, we discovered a procedure that produced tubular multilamellar liposomes, and incorporated FtsZ-YFP-mts inside, where it formed Z rings. Initially this was a fortunate accident, since the cylindrical geometry was not designed, and the FtsZ was initially on the outside. We have since refined the procedures for producing tubular multilamellar liposomes, and we now understand some aspects of the permeability or leakiness that lets FtsZ inside. We describe here our protocols for producing the tubular liposomes and the tests of permeability.
2. Reagents
The following reagents are used in our experiments:
Column buffer: 50 mM Tris/HCl, pH 7.9, 50 mM KCl, 1 mM EDTA, 10% (v/v) glycerol
HMKCG buffer: 50 mM HEPES/KOH, pH 7.7, 5 mM MgAc, 300 mM KAc, 50 mM KCl 10% (v/v) glycerol
HMK50-350 buffer: 50 mM HEPES/KOH, pH 7.7, 5 mM MgAc, 50–350 mM KAc
DOPG:1,2-dioleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (Avanti)
Egg PC: phosphatidylcholine (Avanti)
HccA: 7-Hydroxycoumarin-3-carboxylic acid (Invitrogen)
Teflon disc, 37 mm diameter
3. Bacterial Expression of Membrane Targeting FtsZ
The FtsZ-YFP-mts is expressed from a pET-11b expression vector, with FtsZ366-YFP-mts or FtsZ366-mts genes inserted at NdeI/BamHI sites (366 indicates that the FtsZ was truncated there, removing the FtsA-binding C-terminal peptide). The YFP we use is the variety Venus (Nagai et al.,2002), which gave superior results in FtsZ fusions in E. coli(Osawa and Erickson, 2005). The mts used here is the amphipathic helix from E. coli MinD (Szeto et al., 2003). We have not yet tested the amphipathic helix from FtsA, which has three to five additional extra amino acids that extend the amphipathic helix (Pichoff and Lutkenhaus, 2005). The expression vector is transformed into E. coli strain C41 (Miroux and Walker, 1996), which gives better yields of soluble proteins than BL21.
After transforming, colonies are selected on an LB (Luria broth) agar plate containing 100 μg/ml ampicillin. A colony is picked and cultured overnight in 50 ml LB media with 100 μg/ml ampicillin at 37 °C.
Five milliliters of the overnight culture is diluted in 500 ml LB and cultured at 37 °C until the optical density at 600 nm reaches 0.8–1.0. Protein expression is induced by addition of 0.5 mM IPTG and at the same time the temperature of the shaker is set to 20 °C (our shaker takes 1–2 h to reach 20 °C).
The cells are cultured overnight and spun down at 3750 rpm for 45 min in a Beckman GPR rotor.
4. Purification of FtsZ-mts and FtsZ-YFP-mts
Since FtsZ-mts and FtsZ-YFP-mts are expressed as soluble proteins, we purify them using the same protocol as for wild-type FtsZ.
The packed cells are resuspended in a final volume of 20 ml column buffer, and 1 mM phenylmethanesulphonylfluoride (PMSF) and 0.1–0.2 mg/ml lysozyme are added. The mixture is then incubated on a rotator at 4 °C for 15 min. They are frozen at 80 °C overnight or longer.
Two cycles of freeze–thaw (fresh 1 mM PMSF is added after each thawing) method are performed. The resultant mixture is sonicated on ice until the viscosity is reduced. We usually sonicate it for three cycles of 20 s, with 1 min cooling intervals.
It is then centrifuged at 32,000 rpm for 20 min at 4 °C (Beckman 42.1 Ti rotor). The supernatant is collected and ammonium sulfate is added to 30% saturation (3.52 g dry ammonium sulfate to the 20 ml volume). This mixture is incubated for 20 min on ice and again centrifuged at 32,000 rpm for 20 min at 4 °C (Beckman 42.1 Ti rotor). The supernatant is discarded and the pellet is resuspended in 10 ml column buffer and passed through a 0.22 μm filter.
The protein is purified on an anion exchange column. A 1 × 10 cm Source Q column (Source 15Q, GE Healthcare) is used. The column is eluted with a 100 ml gradient from 50–500 mM KCl in column buffer.
4.1. For FtsZ-mts
FtsZ has very low UV absorbance, so the peak is located by running each fraction on SDS–PAGE.
The peak fractions are pooled and dialyzed into HMK350.
The protein concentration is determined by the BCA method (Pierce). FtsZ produces 75% as much color as BSA (Lu et al.,1998), so it is necessary to correct for this.
Aliquots are frozen and stored at −80°C.
4.2. For FtsZ-YFP-mts
After elution from the Source 15Q column, the peak fractions are pooled. There are typically two peaks: a large main peak and a following small peak, and both peaks have an indistinguishable activity. These peaks can be identified by yellow fluorescence and confirmed by SDS–PAGE.
They are concentrated using an Amicon Ultra-15 with centrifugation at 5000×g.
We have noted that incomplete boiling of FtsZ-YFP-MTS with SDS sample buffer generates two bands on the gel. The upper band (68 Kd) results from completely denatured protein and the lower band (60 Kd), which still has yellow fluorescence in the gel, is due to FtsZ-YFP-mts where the YFP is not denatured.
The concentration of FtsZ-YFP-mts can be determined from its absorption at 515 nm, using the extinction coefficient for YFP-Venus 92,200 M−1 cm−1.
Our preferred buffer for FtsZ-YFP-mts is now HMKCG because FtsZ-YFP-mts seems to be more stable, as described below; we now use HMKCG for dialysis, dilution, reaction, and storage buffer.
5. Renatured Preparation of FtsZ-YFP-mts
In our previous study (Osawa et al., 2008), we used a renaturing technique to prepare FtsZ366-YFP-mts. We developed this protocol because an early preparation of soluble protein as described above had no activity. We recently found that the soluble protein has full activity, equal to the best fractions of the renatured preparation. Because of the simplicity and much higher yield, we now use the soluble preparation for all work.
One curious observation with the renatured FtsZ-YFP-mts was that it lost activity when dialyzed into HMK350. We found that addition of 10% glycerol and 50 mM chloride ion to HMK350 would preserve the activity of the renatured FtsZ-YFP-mts during dialysis. Although this precaution may only be necessary for the renatured FtsZ-YFP-mts, our preferred buffer is now HMKCG, which contains this glycerol and chloride ion.
6. Tubular Multilamellar Liposome Preparation
PC and DOPG are dissolved separately in methanol at 100 mg/ml and mixed at a 4:1 ratio (20 μl/5 μl) in a 1.5 ml Eppendorf tube. The mixture is dried with an air current.
250 μl of milliQ water is added to the Eppendorf tube, and the dried lipid is suspended with vigorous vortexing. Many drops (∼ 5 μl each, total 250 μl) of the suspension are placed on a 37 mm diameter Teflon disc (Fig. 1.1A) and dried using an air current (Fig. 1.1B).
Figure 1.1.
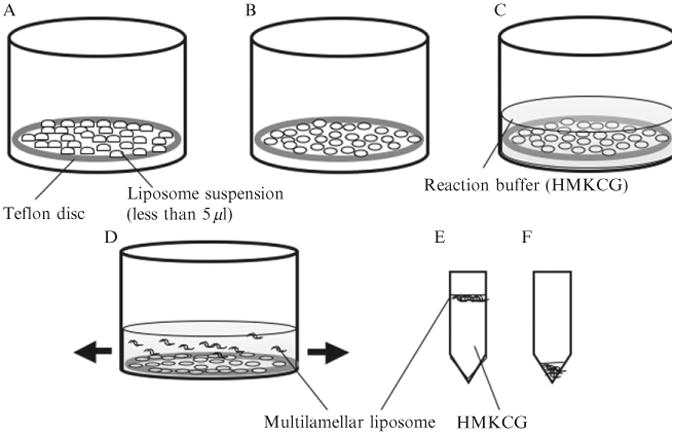
Schematic illustration of the production of multilamellar liposomes. (A) A 37 mm Teflon disc is placed in a small beaker and 250 μl total aqueous suspension of lipid is deposited as many small drops (each drop is less than 5 μl). (B) The drops are dried with an air current and (C) the Teflon disc is covered with 5 ml of HMKCG and incubated overnight at 37 °C. (D) The Teflon disc is gently agitated and multilamellar liposomes floated off. (E) One microliter of the most concentrated suspension, nearest the Teflon, is transferred to an Eppendorf tube and left on the bench for 1 h. The liposomes rise to the surface. (F) HMKCG is carefully removed from the bottom, leaving a concentrated suspension of liposomes.
The Teflon disc is placed in a beaker slightly larger than its diameter, and covered with 5 ml reaction buffer. It is then incubated at 37 °C overnight to hydrate lipid(Fig. 1.1C). High salt buffers such as HMKCG or HMK350 will give aggregated multilamellar liposomes, which are desired to make tubular liposomes for Z-ring formation. The beaker is gently agitated to generate liposomes (Fig. 1.1D). Too vigorous agitation generates many small liposomes which interfere with observation of Z rings.
About 1 ml of the suspension is pipetted out closest to the Teflon, which contains the most concentrated liposomes. This is placed in a 1.5 ml Eppendorf tube and left on a bench for 1 h (Fig. 1.1E). The multilamellar liposomes in HMKCG float to the surface. To obtain concentrated liposomes, buffer from the bottom of the tube is carefully pipetted out, leaving the concentrated top layer (Fig. 1.1F).
Liposomes that have a thinner wall (including unilamellar liposomes) can be easily obtained by using HMK100 with the same methods. If the KAc is reduced to 50 mMor less, many unilamellar liposomes will be obtained. Interestingly, liposomes prepared in HMK100 settle to the bottom of the Eppendorf tube over several hours. This is the opposite of multilamellar liposomes in HMKCG, which float to the top (probably because the 10% glycerol increases the density of the solution).
To produce tubular liposomes, 5 μl of the aggregated multilamellar liposomes in HMKCG or HMK350 is placed on a glass slide and a coverslip is applied. To make liposomes with Z rings, liposomes are mixed first with 1 mM GTP and 4 μMFtsZ-YFP-mts.
In this procedure, tubular liposomes are formed, probably generated by shear force when the suspension is spreading quickly through the narrow space between glasses. The tubular liposomes are always a minority, but with practice they can be found reproducibly. The tubular liposomes are more abundant near the edges of the coverslip, perhaps because that is where shear is maximized. We have noticed that the fluorescence background is higher in the center, where the drop is initially deposited. This may reflect FtsZ-YFP-mts binding to the glass. We have not tested whether the concentration of soluble FtsZ-YFP-mts is substantially reduced by binding to the glass.
7. Permeability of the Multilamellar Liposomes
To check the permeability of the multilamellar tubular liposomes that were produced by the above procedures, a polar fluorescence dye HccA is used as a small molecule indicator (MW 206.15 kDa) and FtsZ fused with YFP-Venus (FtsZ-YFP) as a large molecule indicator (MW 67 kDa). Neither of these probes can cross the lipid bilayer, so they should report on smaller or larger pores in the liposomes. Also, the FtsZ-YFP is missing the mts, and was given no GTP, so it would not make protofilaments or Z rings. It should just report the existence of pores large enough to admit the globular protein. When we mix the multilamellar liposomes with 1 mM HccA and 32 μMFtsZ-YFP and apply a coverslip, we find tubular liposomes with both HccA and YFP fluorescence. More than 80% of tubular liposomes have FtsZ-YFP inside and even more liposomes have HccA inside, indicating that multilamellar liposomes are leaky at least for some time after addition of the fluorescent probes and application of the coverslip.
A simple FRAP (fluorescent recovery after photobleach) assay is used to determine the leakiness of liposomes 30 min after the coverslip is applied. The entire field of view is exposed to UV from the mercury lamp for 20 s, without any filter. This results in complete bleaching of both fluorescent molecules inside and outside the liposomes. HccA fluorescence on the outside recovers over ∼20–60 s as unbleached probe diffuses into the area from outside. At 20 s HccA has entered about half of the liposomes (Fig. 1.2F), and by 5 min almost all the liposomes contain fluorescent HccA. Diffusion of FtsZ-YFP is slower, requiring ∼5 min to recover 70% of original fluorescence on the outside. After 45 min about half of the liposomes have YFP fluorescence inside. We conclude that the multilamellar liposomes are almost all permeable or leaky to the small molecule probe, and about half have pores that will pass the larger protein probe.
Figure 1.2.
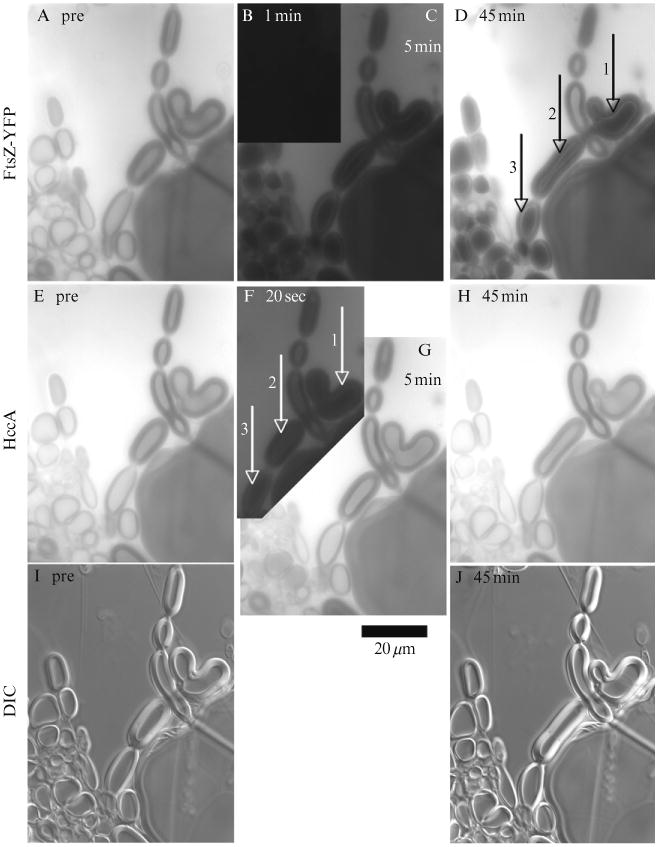
FRAP assay for checking permeability of multilamellar liposomes. The top row shows FtsZ-YFP (lacking the mts and GTP), the middle row shows HccA fluorescence, and the bottom row shows DIC. The first column shows prebleach images, and the numbers on the other panels show time after bleach. In (D), vesicles 1–3 show little or no recovery of FtsZ-YFP inside, but they do show a thin bright layer of internal fluorescence. The vesicle between 1 and 2 shows substantial recovery of FtsZ-YFP. In (F), (G) and (H) vesicles 1–3 show no recovery of HccA at 20 s, but full recovery at 5 min. The exposure of fluorescence images is kept at the same level from prebleach to postbleach. Notice also the change in shape of several vesicles over 45 min. (B) 50% magnification showing that fluorescence is completely bleached.
A curious phenomenon is the appearance of a bright ring on the inner layer of the liposome when the inside of the liposome is dark and the outside is bright (Fig. 1.2D and F). When we repeat the FRAP experiment with a confocal microscope, the bright ring is seen only rarely and is also very dim. We suggest that the bright ring may be an optical artifact seen in the wide field fluorescence microscopy.
8. Z-ring Formation in Liposomes
We have shown previously that FtsZ-YFP-mts can form Z rings inside tubular liposomes when FtsZ-YFP-mts plus GTP was mixed with multilamellar liposomes just before applying the coverslip (Osawa et al.,2008). To examine the leakiness of these liposomes with Z rings inside, we repeated the FRAP assay using 8 μM FtsZ-YFP-mts plus 2 mM GTP instead of 32 μM FtsZ-YFP. Most liposomes that had Z rings before the bleach showed no recovery of fluorescent Z rings, even though HccA fluorescence recovered rapidly (Fig. 1.3). Fluorescent FtsZ-YFP-mts could be seen coating the outside of the liposome but apparently did not enter the liposome, since no fluorescent Z rings were seen.
Figure 1.3.
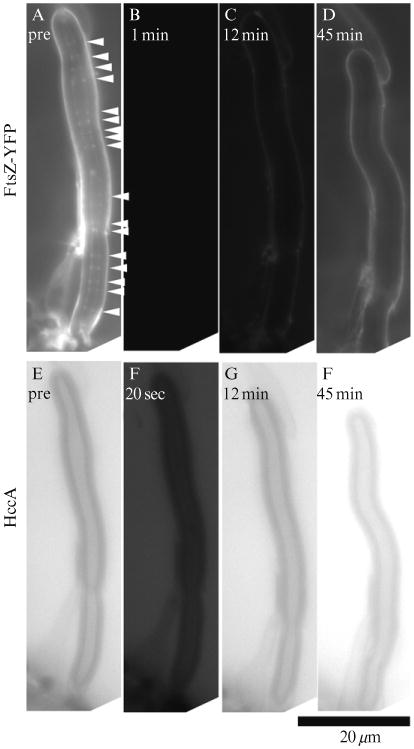
A liposome containing multiple Z rings (arrowheads) before bleach shows recovery of HccA fluorescence both outside and inside. FtsZ-YFP-mts fluorescence is recovered on the outside surface of the liposome, but no fluorescent Z rings recover inside. The exposure of fluorescence images is kept at the same level from prebleach to postbleach. (D) We did not detect any fluorescent Z rings even when we optimized the contrast in this specific panel.
We did, however, find some liposomes that recovered fluorescent Z rings after the bleach (Fig. 1.4). Figure 1.4C shows an intriguing example where HccA fluorescence invades through a hole at the top side of the tubular liposome (arrow in Fig. 1.4F). FtsZ-YFP-mts followed and fluorescent Z rings assembled as the nonbleached FtsZ-YFP-mts exchanged with the bleached (Fig. 1.4C and D), showing that occasional open liposomes can recover FtsZ from the outside and assemble Z rings. Recovery of Z rings with FtsZ-YFP-mts appeared to be less frequent than recovery of fluorescence of FtsZ-YFP. A potentially important difference is that FtsZ-YFP-mts had GTP, so it should be mostly assembled into protofilaments ∼30 subunits long (Chen and Erickson, 2005). FtsZ-YFP was used without GTP so it should be much smaller protein monomers.
Figure 1.4.

A liposome with Z rings inside shows permeability to both FtsZ-YFP-mts and to HccA. (A) Arrowheads indicate Z rings prebleach. (B) YFP fluorescence is completely eliminated on the outside and inside of the liposome at 1 min after bleach. (C) About 12 min later YFP fluorescence has partially recovered on the outside, and fluorescent Z rings have formed on the inside, showing that unbleached FtsZ-YFP-mts has entered the leaky liposome. (D) At 45 min fluorescent Z rings have spread more toward the bottom. (E, F) About 20 s after bleaching, HccA has begun to enter the liposome, apparently through a hole at the top (note the zone of brighter fluorescence in the top 1/5 of the liposome). (G, H) At 12 min HccA fluorescence has fully recovered inside compared to the prebleached image in (E). Note that the FtsZ rings recover only at the top, near the hole, at 12 min. At 45 min they have spread through the top half of the liposome.
9. A Crude Flow Chamber to Exchange Buffer Outside Liposomes
For various purposes one might want to change the buffer outside the liposomes, especially knowing that most of the liposomes are permeable to small molecules. The best production of tubular liposomes occurs with a small sample volume, which generates an optimal shear when the coverslip is applied. A 5 μl sample spread over a 4.8 cm2 coverslip gives a liquid layer 10 μm thick. If we try to perfuse buffer into this thin layer the liposomes are subject to high shear that results in severe elongation, loss of Z rings, and loss of liposomes. To minimizethese effects, we first apply a 50 μl drop of perfusion buffer to one edge of the coverslip (Fig. 1.5B). We can then drain the liquid by applying a Kimwipe at the opposite edge, while applying a pipette tip or thin rod to prevent the coverslip from slipping. The arrangement shown in Fig. 1.5D, with a thin strip of Kimwipe applied to the coverslip and a large piece farther away, provides an optimal slow flow.
Figure 1.5.
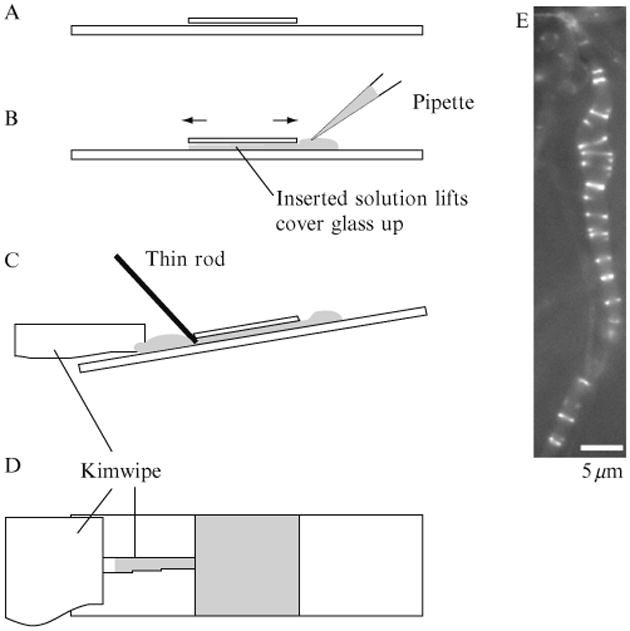
Changing buffer by flow/perfusion. (A) The normal preparation has a very thin layer of liquid between the slide and coverslip. (B) When a drop of the new buffer is placed on the side, the coverslip rises and is able to move back and forth. (C) To adsorb the liquid, the slide is tilted and a small piece of Kimwipe is placed on the edge opposite the drop. To prevent the coverslip from moving we used a thin rod or a pipette. (D) To minimize shear we slow the draining by placing a narrow piece of Kimwipe touching the coverslip and a larger piece farther away. (E) As a demonstration of buffer exchange, we prepared liposomes with FtsZ-YFP-mts but no GTP. FtsZ was trapped inside some tubular liposomes but did not form Z rings. We then flowed through a buffer containing GTP but no FtsZ. The GTP entered the liposome and initiated Z-ring assembly. This liposome was apparently leaky to GTP but not to protein, since the FtsZ did not leak out.
We tested the system by adding FtsZ-YFP-mts without GTP to multilamellar liposomes. Cylindrical liposomes were found with the protein inside but Z rings could not form without GTP. We then perfused HMKCG containing 1 mM GTP. As the GTP entered the liposomes, Z rings assembled (Fig. 1.5E). An additional advantage of this system is that the fluorescent protein outside the liposome is washed away during the perfusion. This removes the fluorescent protein in solution and binding to the outside surface of liposomes and enhances the contrast of the Z rings inside. Because there is no FtsZ outside to hydrolyze the GTP, the Z rings can last a long time.
10. Factors Affecting Z-ring Formation in Liposomes
During months of experimenting with the liposome system, we have made a number of observations that seem to be important for Z-ring formation.
The shape of the liposome is very important, the cylindrical geometry being optimal for Z-ring assembly. We have rarely seen Z rings inside spherical liposomes. When these have internalized FtsZ-YFP-mts, the protein localizes to small patches or forms arcs that move unstably (Fig. 1.6B). One possible interpretation is that when a Z ring forms inside a spherical liposome, the contractile force causes it to collapse to a small patch. Another interpretation is that FtsZ filaments cannot determine the direction to form stable Z rings. We have sometimes observed this situation in which a large round liposome had radially formed FtsZ filaments inside (Fig. 1.6B, liposome on right). With a cylindrical geometry, a contractile force will cause it to form a circle in a plane perpendicular to the axis.
The diameter of the liposome is important. Z-ring assembly is optimal in tubular liposomes less than 2 μm in diameter (bacteria are typically 1 μm in diameter). In larger diameter tubular liposomes, some Z rings are formed, but many structures appear to be helices or spirals rather than closed rings (Fig. 1.6A).
A mixture of neutral (PC) and negatively charged (DOPG) lipids is important. We have obtained Z rings with 2–40% DOPG, but typically use 1:4 DOPG:PC. 100% PC or DOPG, or a 2:3 ratio of DOPG:PC failed to support Z-ring assembly. This is probably because the amphipathic helix has positively charged amino acids on the hydrophilic side, and these need some negatively charged phospholipids in the bilayer to bind to. Interestingly, tubular liposomes produced by E. coli polar lipid (Avanti) did not support Z rings. This particular batch of E. coli lipid that we used may have contained more than 40% of charged lipid.
We have made several attempts to assemble Z rings in unilamellar liposomes, without success. As mentioned above, the spherical shape of unilamellar liposomes may not support Z-ring assembly. Another factor may be the rigidity of the wall. The rigidity of the multilamellar wall may slow constriction and stabilize the Z rings.
Figure 1.6.
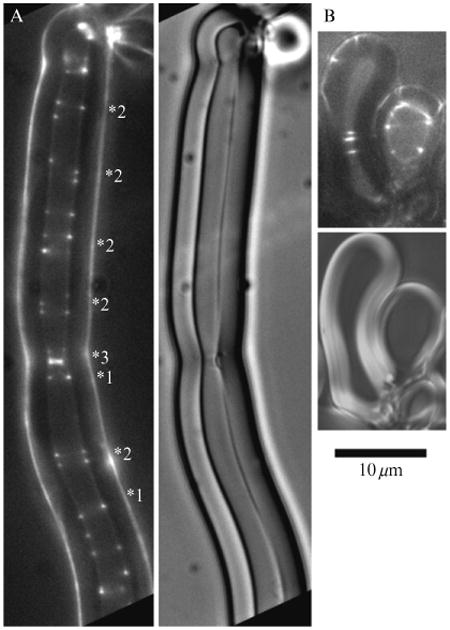
Behavior of FtsZ-YFP-mts in large multilamellar liposomes. (A) In a large tubular liposome (3 μm inside diameter), FtsZ-YFP-mts formed some normal Z rings indicated by two spots of equal intensity directly across from each other (labeled 1⋆). However, many spirals were also formed, indicated by dots spaced irregularly (⋆2). Note the bright ring at (⋆3), which appears to have peeled away the inner layer of the multilamellar wall (DIC image on right). Figure 1.6A is reprinted from Osawa et al. (2008) with permission of the publisher. (B) Several incomplete Z rings, spirals or arc structures, (arrowheads) are formed in a large spherical liposome.
11. Utility of the Liposomes Beyond FtsZ
The liposome system that we have developed may have uses beyond the study of FtsZ. The tubular liposomes are similar in size to a bacterium, and can range up to the size of a yeast cell. Similar to our reconstitution of Z rings, they may eventually be useful in reconstituting the cytokinetic apparatus of yeast or larger animal cells. More generally, we have demonstrated that most of the liposomes are permeable or leaky to small molecules, while about half are leaky to larger proteins. They therefore constitute small femtoliter chambers in which one can enclose proteins and manipulate the small molecule environment.
References
- Chen Y, Erickson HP. Rapid in vitro assembly dynamics and subunit turnover of FtsZ demonstrated by fluorescence resonance energy transfer. J Biol Chem. 2005;280:22549–22554. doi: 10.1074/jbc.M500895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Stricker J, Erickson HP. FtsZ from Escherichia coli, Azotobacter vinelandii, and Thermotoga maritima—Quantitation, GTP hydrolysis, and assembly. Cell Motil Cytoskel. 1998;40:71–86. doi: 10.1002/(SICI)1097-0169(1998)40:1<71::AID-CM7>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Miroux B, Walker JE. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Noireaux V, Libchaber A. A vesicle bioreactor as a step toward an artificial cell assembly. Proc Natl Acad Sci USA. 2004;101:17669–17674. doi: 10.1073/pnas.0408236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa M, Erickson HP. Probing the domain structure of FtsZ by random truncation and insertion of GFP. Microbiology. 2005;151:4033–4043. doi: 10.1099/mic.0.28219-0. [DOI] [PubMed] [Google Scholar]
- Osawa M, Anderson DE, Erickson HP. Reconstitution of contractile FtsZ rings in liposomes. Science. 2008;320:792–794. doi: 10.1126/science.1154520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pautot S, Frisken BJ, Weitz DA. Engineering asymmetric vesicles. Proc Natl Acad Sci USA. 2003;100:10718–10721. doi: 10.1073/pnas.1931005100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichoff S, Lutkenhaus J. Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Mol Microbiol. 2005;55:1722–1734. doi: 10.1111/j.1365-2958.2005.04522.x. [DOI] [PubMed] [Google Scholar]
- Szeto TH, Rowland SL, Habrukowich CL, King GF. The MinD membrane targeting sequence is a transplantable lipid-binding helix. J Biol Chem. 2003;278:40050–40056. doi: 10.1074/jbc.M306876200. [DOI] [PubMed] [Google Scholar]