

Abstract
Objective
The mechanisms of cancer metastasis have been intensely studied recently and may provide vital therapeutic targets for metastasis prevention. We sought to review the contribution of epithelial-mesenchymal transition and the tumor microenvironment to cancer metastasis.
Summary Background Data
Epithelial-mesenchymal transition is the process by which epithelial cells lose cell-cell junctions and baso-apical polarity and acquire plasticity, mobility, invasive capacity, stemlike characteristics, and resistance to apoptosis. This cell biology program is active in embryology, wound healing, and pathologically in cancer metastasis, and along with the mechanical and cellular components of the tumor microenvironment, provides critical impetus for epithelial malignancies to acquire metastatic capability.
Methods
A literature review was performed using PubMed for “epithelial-mesenchymal transition”, “tumor microenvironment”, “TGF-β and cancer”, “Wnt and epithelial-mesenchymal transition”, “Notch and epithelial-mesenchymal transition”, “Hedgehog and epithelial-mesenchymal transition” and “hypoxia and metastasis”. Relevant primary studies and review articles were assessed.
Results
Major signaling pathways involved in epithelial-mesenchymal transition include TGF-β, Wnt, Notch, Hedgehog, and others. These pathways converge on several transcription factors, including zinc finger proteins Snail and Slug, Twist, ZEB 1/2, and Smads. These factors interact with one another and others to provide crosstalk between the relevant signaling pathways. MicroRNA suppression and epigenetic changes also influence the changes involved in epithelial-mesenchymal transition. Cellular and mechanical components of the tumor microenvironment are also critical in determining metastatic potential.
Conclusions
While the mechanisms promoting metastasis are extremely wide ranging and still under intense investigation, the epithelial-mesenchymal transition program and the tumor microenvironment are both critically involved in the acquisition of metastatic potential. As our understanding of these complexities increases, the ability to target these processes for therapy will offer new promise in the treatment of epithelial malignancy and metastasis.
Keywords: epithelial-mesenchymal transition, tumor microenvironment, metastasis, prevention, therapy
Introduction
The process of metastasis has four significant steps. The metastatic tumor cell must acquire the ability to emigrate from the primary tumor - meaning it must shed cell-cell and cell-matrix interactions, become motile, and acquire plasticity in order to mechanically navigate the tumor stroma. It must then acquire invasive capabilities, allowing it to degrade the surrounding extracellular matrix and allow escape from the primary tumor mass. Next, its invasive capability must be sufficient to allow intravasation by navigation or degradation of the basement membrane and endothelial barrier of its home tissue, vasculature, and/or lymphatic channels. Finally, after migrating through the hematogenous or lymphatic system, it must be able to extravasate, requiring adhesion capability to the distant endothelium and endothelial barrier and basement membrane penetration capability, leading to establishment of a micrometastasis at the distant site [1]. Of these requirements, the first two are directly related to features of and processes active in the primary tumor microenvironment.
This review will provide a broad description of the tumor microenvironment and its contribution to metastatic behavior of carcinomas, with focus on the epithelial-mesenchymal transition and active signaling pathways promoting this phenomenon.
Epithelial-mesenchymal transition
The most comprehensive theory describing how initially quiescent tumor cells acquire metastatic capability is the epithelial-mesenchymal transition (EMT). EMT has become progressively better characterized in the last five to ten years and has emerged as a primary theory of how tumor cells acquire the characteristics necessary to metastasize. First described in corneal epithelial cells in vitro in 1982 [2], the term refers to a program of cell biology that is initially active in embryogenesis and has a physiologic role wound healing. Its molecular hallmark is the downregulation of the cell-cell adhesion molecule E-cadherin, resulting in dissolution of cell-cell tight junctions, and upregulation of a number of mesenchymal markers, including N-cadherin, vimentin, and fibronectin. Phenotypically, cells become more spindle-shaped and lose basal-apical polarity [3]. They become mobile and plastic in shape and acquire apoptosis resistance and stemlike characteristics. In the physiologic setting, these characteristics promote normal cell migration and survival during embryogenesis and the ability of cells to move into denuded areas during wound healing. However, when pathologically activated in the setting of cancer, EMT results in increased metastatic behavior, drug resistance, cancer stem cell transformation, and poorer prognosis for a number of human cancers. The EMT theory as a primary biological program during acquisition of metastatic potential has gained credence with the demonstration that it frequently is observed at the invasive front of primary lesions [4]. (Figure 1)
Figure 1.

Conceptual diagram of molecular, phenotypic, and behavioral transitions of cells undergoing EMT. Reproduced with permission from Micalizzi DS, Farabaugh SM, and Ford HL, Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia 2010; 15: 117-134.
Major signaling pathways in EMT
The signaling regulation of EMT is wide and incredibly complex, involving activation of a number of different pathways converging on several prominent transcription factor families and frequently involving crosstalk between them. In this section, we will review some of the prominent signaling pathways active in EMT and recent findings expanding on our understanding of these pathways.
TGF-β
TGF-β signaling is without question one of the most important and sophisticated regulatory networks under investigation in human cancer. It exerts potent control over cell proliferation, differentiation, apoptosis, adhesion, invasion, and interactions with the cellular microenvironment. These effects have been observed in epithelial, endothelial, and hematopoietic cell lineages [5].
The canonical TGF-β signaling pathway proceeds as follows. Three forms of TGF-β are seen in mammals, all homo- or heterodimers secreted into the extracellular matrix as part of a complex known as the large latency complex (LLC). Once secreted and released from this complex, TGF-β is active. The TGF-β receptors are membrane bound receptors with serine-threonine kinase activity. TGF-β binds as a ligand to the type II receptor, TGFβ-RII, with the assistance of the type III receptor, TGFβ-RIII. Once it is bound tightly to TGF-βRII, this forms a heterotetrameric complex with and phosphorylates the type I receptor, TGF-βRI. The type I receptor then recruits and phosphorylates receptor-regulated Smads (R-Smads), of which there are at least eight known variants, the principals of which are Smads 2 and 3 [6]. Indeed, Smad 2/3 dependent signaling is considered a hallmark of canonical TGF-β signaling [7]. The R-Smad then forms a complex with the common Smad, Smad4, and the complex shuttles into the nucleus, there to function as a transcriptional regulator. However, as Smad complexes in general have weak interactions with DNA, they form transcriptional complexes with a number of other transcriptional regulators to improve binding and also allow interaction with other signaling pathways These include p300/CBP, Forkhead, homeobox, zinc-finger, AP1, Ets, and basic helix-loop-helix families of transcription factors [8].
The TGF-β signaling pathway is under tight control from a variety of separate sources. SARA, TMEPAI, Dab2, and Hgs proteins control presentation and sequestration of Smads, and phosphorylation and rapid dephosphorylation of Smad linker regions creates a rapid activation-deactivation cycle of the Smad complexes [9]. Ubiquitination by E3ligases and Smirf family proteins contribute to degradation of TGF-β pathway constituents. Inhibitory Smads, the principal of which is Smad7, also exist. Smad7 in particular turns off TGF-β signaling by recruiting Smurf 1 and 2 to activated TGF-β receptors, leading to their ubiquitination and degradation [6].
There are also a number of noncanonical pathways by which TGF-β exerts its effects. These include the RhoA-Rock1 signaling axis, Erk-MAP kinase signaling, inflammatory mediator signaling via NF-κB and Cox2, and the p38/JNK pathway [7]. Smads have been shown to have independent action in the nucleus outside of TGF-β mediated activation.
TGF-β is physiologically a cytostatic cytokine that serves to arrest cell cycle progression. As such it is physiologically a tumor suppressor. Simultaneously, however, prometastsatic and cell-survival potentiation in human tumors have been observed as consequences of TGF-β signaling. This phenomenon is knkown as the TGF-β paradox. Human cancers frequently manifest mutations or loss of heterozygosity in TGF-β signaling activation, which results in negation of the tumor-suppressive effects of the cytokine - in other words, the tumor-suppressive role of TGF-β needs to be circumvented to progress from a cytostatic to a prometastatic source of signaling [6,10]. Interestingly, increasing evidence suggests that it is an imbalance between canonical and noncanonical TGF-β signaling that results in the TGF-β paradox, such that noncanonical signaling can promote a prometastatic cell program that overrides the more cytostatic and anti-tumorigenic effects of canonical TGF-β signaling present especially prior to primary tumor formation [7]. Cells can have lost sensitivity for some effects of TGF-β while maintaining intact sensitivity to its signaling overall [11]. The EMT program is thought to be an important effector and manifestation of this imbalance and the primary mechanism behind the TGF-β paradox, enabling the metastatic progression of late stage carcinomas in response to TGF-β [7]. Frequently this involves not only activation of canonical TGF-β signaling, but also downstream action of members of other canonical pathways, including Wnt, Notch, Hedgehog, and others. (Figure 2)
Figure 2.

Canonical TGF-β signaling. Reproduced with permission from Meulmeester E and Ten Dijke P, The dynamic roles of TGF-beta in cancer. J Pathol 2011.
The heterogeneity of ligands and downstream effectors that participate with TGF-β signaling, the variety of transcription factors and complexes at play, and the enormous amount of crosstalk between the TGF-β signaling network and other canonical signaling pathways result in a wide variety of effects of TGF-β on cancer growth and metastasis [12]. Some brief discussion of recent discoveries in TGF-β signaling activity in human cancers is here presented, but we refer the reader to extensive recent reviews of this topic for more detailed information [6,7,9].
Recent evidence for TGF-β importance in EMT
TGF-β was first identified as a pro-EMT signaling stimulus in 1994 [13], and since that time has come to be regarded as a master regulator in both physiologic and pathologic EMT [9]. The transcriptional activation of Snail, Slug, Zeb1, Twist, and BHLH proteins have been shown to be critical in this process [7,14,15], which begins with dismantling of cell-cell tight junctions and rearrangement of the actin cytoskeleton [7]. Smad4 mutations have been identified in fifty percent of pancreatic cancers and 30% of colorectal cancers [9]. Recently, a novel Smad4 mutation was found to increase homodimerization of Smad4 with the receptor Smads and promote nuclear localization; this resulted in reduction in E-cadherin, increase in N-cadherin, increased fibroblastic phenotype, and ability to grow in anchorage independent conditions of papillary thyroid cancer cells [16]. In a kras-driven animal model of pancreatic cancer, deficiency of Notch2 slowed cancer progression, prolonged survival, and led to a phenotypic switch to anaplastic pancreatic cancer rather than ductal adenocarcinoma [17]. Activated TGF-β signaling constituents were found to be the downstream drivers of this change, along with myc signaling, potentially implicating the Wnt pathway as well as TGF-β and Notch. Fuxe et al described transcriptional crosstalk between TGF-β and the stem-cell promoting pathways, including Wnt, Ras, Hedgehog, and Notch. One mechanism of this crosstalk was found to be the formation of EMT-promoting Smad complexes [18]. From a clinical standpoint, after TGF-β treatment was found to increase cholangiocarcinoma cell migration, invasion, and fibroblastic phenotype, cadherin switch (downregulation of E-cadherin and upregulation of N-cadherin) was observed in tumor samples from cholangiocarcinoma patients [19]. TGF-β has been implicated in resistance to chemotherapy and radiation as well. Radiation treatment has been shown to lead to increased TGF-β levels and increased circulating tumor cells and lung metastatic burden [20], and ionizing radiation was found to promote TGF-β related EMT and associated increases in invasiveness and migration in six different cancer cell types [21]. In this study EMT markers, cell migration and invasiveness, and production of TGF-β were all found to be increased after radiation exposure, and these effects were reduced by treatment with a small molecule inhibitor of TGF-β. TGF-β has been shown to mediate cancer cell protection and recovery after radiation and chemotherapy [9].
Wnt
The Wnt pathway is a known regulator of embryonic cell-fate determination. The canonical Wnt pathway begins with binding of Wnt to its Frizzled receptor. These receptors are seven-transmembrane type receptors with an intracytoplasmic motif known as the disheveled-binding motif. Also spanning the membrane are the LRP5/6 coreceptors for Wnt, which on the cytoplasmic side of the membrane have an Axin binding motif. Wnt’s binding to Frizzled leads to formation of a complex between Frizzled and LRP, resulting in formation of an LRP-Axin-FRAT complex. In the absence of signaling, Wnt’s downstream effector β-catenin is sequestered in the cytoplasm by GSK-3β and its associated destruction complex. Under these conditions, β-catenin is phosphorylated and marked for degradation, preventing accumulation. With Wnt signaling activation, the released LRP-Axin-FRAT complex frees β-catenin from GSK-3β sequestration, thereby preventing its degradation and allowing cytoplasmic accumulation with nuclear translocation. β-catenin then binds to its transcriptional coregulators, forming the TCF/LEF-β-catenin-Legless-PYGO complex in the nucleus to affect gene transcription and repression. This is the ultimate downstream effector of canonical Wnt signaling. Noncanonical signaling also proceeds through Frizzled receptor binding, but utilizes the coreceptors ROR1/2. These are receptor tyrosine kinases with an intracytoplasmic binding domain for casein kinase Iε. The noncanonical pathway has several terminal effectors, including RhoA, Jnk, and Nemo-like kinase [22].
There are at least ten Frizzled receptor variants and a number of variants of Wnt itself, some predisposed towards signaling through one pathway or the other. The multitude of effects of Wnt signaling can therefore be attributed to the various combinations of ligand and receptor that this heterogeny allows. In addition, the secreted frizzled-related protein (SFRP) family of Frizzled-binding proteins, of which there are five members, serves to downregulate Wnt signaling by preventing its interaction with Frizzled, further diversifying Wnt signaling effects [23]. (Figure 3)
Figure 3.

Canonical and noncanonical Wnt signaling. Reproduced with permission from Katoh M, Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol Ther 2006; 5: 1059-1064.
Recent evidence for Wnt activation in EMT
A role for Wnt activation and promotion of the EMT program has been established in a number of human cancers, most recently HCC, prostate, breast, squamous cell, esophageal, renal cell, and ovarian carcinomas.
Yook et al in 2006 demonstrated a Wnt-Axin2-GSK-3β activation axis that regulates Snail1 in breast cancer cells and thereby potentiates EMT. Axin2 was found to act as a nucleocytoplasmic chaperone for GKS-3β, resulting in its inhibition and release of Snail1 from suppression. Snail1 accumulated, leading to increased invasive activity and tissue dedifferentiation [24]. Knockdown of secreted frizzled-related protein 1 (SFRP1), one of the previously described family of competitive antagonists of the Wnt receptor, resulted in changes consistent with EMT including upregulation of ZEB1 and acquisition of characteristics of breast tumor-initiating cells [25]. The same group had previously demonstrated that SFRP1 knockdown led to acquisition of invasive, migratory, stemlike, and EMT characteristics in an immortalized breast cancer cell line [26]. Similar effects of SFRP1 knockdown were observed in cervical cancer cells, in which knockdown resulted in increased invasion and decreased E-cadherin expression with concomitant demonstration of upregulation of Wnt signaling by increased expression of c-myc and cyclin-D1; restoration of SFRP1/2 activity reversed these effects [27]. Stabilization of ubiquitination of β-catenin by Rad6B, a ubiquitin-conjugating enzyme), and subsequent stabilization of cytoplasmic β-catenin levels was observed in early breast cancers and carcinomas but not in normal breast tissue [28]. High Rad6B-expressing cell lines exhibited the EMT phenotype, and when Rad6B was silenced, EMT characteristics were reduced in breast cancer cell lines, including reduction of vimentin and Snail1 levels. This effect required intact Wnt signaling. Oral squamous cell carcinoma cell lines generated with a lack of the GSK-3β phosphorylation site displayed constitutive Wnt signaling activation. Cell morphology changed from polygonal to spindle-shaped, and cells displayed enhanced migration and invasion as well as MMP7 upregulation. A cytoskeletal transition also took place, with redistribution of E-cadherin and rearrangement of actin filaments consistent with EMT [29]. Wnt1 has been shown to induce EMT in murine mammary epithelial cells, with translocation of β-catenin from the cytoplasm to the nucleus, increased cell motility and proliferation, and upregulation of MMP3 (stromelysin-1). These effects were ameliorated by MMP inhibitors and siRNA interference [30]. Interestingly, MMP3 did not appear to be solely a downstream effector of Wnt-EMT cell biology programs, but also cooperated with Wnt3a in regulation of β-catenin activity, suggesting that it functions as both a regulator and an effector of Wnt-induced EMT. Dysregulated Wnt signaling associated with increased MSX2 expression, a downstream β-catenin target, resulted in increased neoplastic potential of ovarian endometrioid adenocarcinoma cells, although in this case EMT changes appeared to be context dependent (manifested primarily by increase in vimentin expression) [31]. From a clinical standpoint, analysis of primary breast cancer biopsy samples revealed accumulation of GSK-3β and Slug in invasive ductal carcinomas and statistical correlation of Slug activation with loss of membranous E-cadherin and nuclear and cytoplasmic β-catenin [32].
If Wnt activation appears to activate EMT programming, then disruption of Wnt signaling leading to its downregulation has revealed opposite effects. Serum response factor (SRF), a known activator of Wnt activity, was overexpressed in HeLa cells and shown to result in increased expression of mesenchymal markers vimentin, N-cadherin, and RhoA. After stable knockdown of SRF via shRNA interference, migration and invasiveness were decreased and mesenchymal marker expression reduced in conjunction with downregulation of c-myc and cyclin D1, indicating Wnt pathway downregulation [33]. The Wnt inhibitory factor 1 (WIF1) promoter is normally methylated and thereby downregulated in prostate cancer. When expression was restored in prostate cancer cells in vitro, epithelial marker expression increased and mesenchymal markers decreased. Cell motility and invasion were also decreased and expression of Slug and Twist, transcription factors active in EMT, were reduced. Tumor growth was also reduced 63% in a xenograft model generated with cells abnormally expressing WIF1 [34]. ShRNA-mediated knockdown of β-catenin in prostate cancer cells resulted in higher expression of E-cadherin and significantly decreased expression of vimentin, N-cadherin, and MMP-2, indicating a reversal of EMT in the setting of low cellular β-catenin and inactive Wnt signaling [35]. In an examination of EMT-positive versus EMT-negative prostate cancer cell lines, EMT-positive cells exhibited higher ratio of phosphorylated GSK-3B and higher β-catenin expression; these changes were correlated with more aggressive invasion and higher proliferative activity [36]. Interestingly, the only EMT-negative cell line to display these changes was a HIF-1α overexpressing cell line. ShRNA mediated knockdown of Wnt5a in canine kidney cells after TGF-β induced EMT resulted in reduced cell migration and invasion [37]. Interestingly, in this study Wnt5a was found to suppress Wnt3a-mediated canonical Wnt pathway activation, and therefore may exert its effects through a noncanonical pathway such as the planar cell polarity pathway. Inhibition of Wnt signaling using low-density lipoprotein receptor-related protein 6 (LRP6), a known Wnt inhibitor, resulted in reduced activation of EMT-related transcription factors including Slug and Twist, increased epithelial markers, and reduced ability of poor-prognosis basal cell breast cancer to metastasize to the lungs in xenografts [38]. Changes at the epigenetic level also have been shown to affect Wnt-related EMT. Mesenchymal-specific DNA hypermethylation in oral squamous cell carcinoma results in silencing of Wnt7A and Wnt10A and is associated with increased T-status, disease stage, and nodal status [39].
β-catenin is known to influence the transcriptional repressors Snail, Slug, and Twist to repress transcription of E-cadherin, which as previously described is the hallmark initiating change of EMT. β-catenin also, as reviewed by Heuberger et al, can be released in response to loss of cadherin mediated adhesion. Under normal circumstances β-catenin binds E-cadherin and α-catenin, providing a direct link between the E-cadherin cell-adhesion complex and the intracellular cytoskeleton. Loss of E-cadherin results in proteolytic cleavage of these connections and release into the cytoplasm of free β-catenin in a manner free from canonical Wnt signaling. Indeed, the Wnt signaling network and the cadherins appear to compete for the same pool of intracellular β-catenin [40].
Finally, Wnt signaling can also result in EMT directly through GSK-3β suppression. As reviewed by Doble et al, the EMT-promoting transcription factor Snail has several consensus sites for GSK-3β, which allow for its phosphorylation. When Wnt signaling is absent, GSK-3β activity is at normal levels and can phosphorylate Snail1, inhibiting its action and preventing its downregulation of E-cadherin. When Wnt signaling is active, GSK-3β is inhibited, Snail remains unphosphorylated, and is free to repress E-cadherin transcription, resulting in EMT [41].
As investigation of the Wnt pathway as relevant in EMT and cancer metastasis has widened, a number of different downstream effectors and regulators of the pathway outside the traditional canonical players have been described. Investigative focus has moved increasingly towards identifying these factors and elucidating their mechanisms of action on the Wnt pathway. A full description of these is outside the scope of this review but includes Bcl9/Bcl9I, DNAJB6, Cripto-1, Sprouty-4, and others.
Notch
Similar to the Wnt pathway, the Notch pathway was initially identified in embryogenesis and directs cell fate decisions. The Notch gene product is a membrane bound receptor with a single transmembrane domain, and large intracellular and extracellular domains for signal transduction and ligand binding respectively. There are four variants of the Notch receptor (1 - 4). The Notch receptors are expressed on the cell surface as heterodimers. Notch ligands include the Delta and Jagged families of membrane-bound ligands. Notch signaling is initiated generally through ligand-receptor binding between adjacent cells. Upon ligand binding, the receptor’s intracellular domain is ubiquitinated, and a conformational change takes place such that the ADAM protease TACE is able to cleave the receptor. An additional cleavage occurs in the transmembrane domain and is performed by gamma-secretase. The culmination of these proteolytic events is the release of the Notch intracellular domain (NICD) into the cytoplasm. It then translocates to the nucleus and binds CSL transcription factors, releasing their targets from inhibition [42]. The Notch-CSL complex can then recruit further transcriptional coactivators and promote transcription of the Notch target genes, which include helix-loop-helix transcription factors, HRT/Herp transcription factors, p21, NRARP, deltex-1, and others [43]. (Figure 4)
Figure 4.
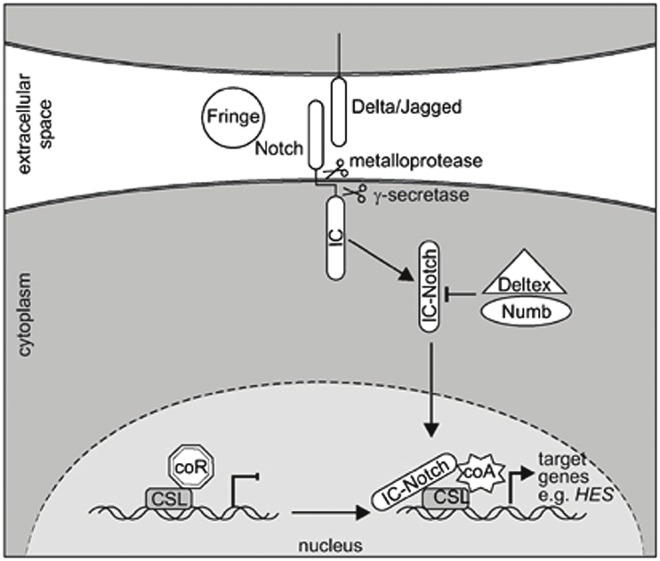
Canonical Notch signaling. Reproduced with permission from Weerkamp F, van Dongen JJ, and Staal FJ, Notch and Wnt signaling in T-lymphocyte development and acute lymphoblastic leukemia. Leukemia 2006; 20: 1197-1205.
Notch signaling in development and cancer has been extensively reviewed [43]. Recent work in Notch pathway activity in cancer cells shows EMT promotion and enhancement of cancer stem cell characteristics, and interestingly, implicates Notch signaling in several cancers displaying resistance to conventional chemotherapy, including cisplatin, gemcitabine, taxotere, taxol, tamoxifen, oxaliplatin, trastuzumab, and others [44]. Notch signaling also appears to be affected significantly by an emerging class of transcriptional inhibitors, the micro RNAs.
Recent evidence for Notch pathway activation in EMT
In work focused primarily on cardiac valve formation, Timmerman in 2003 found that overexpression of activated Notch1 in immortalized cardiac endothelial cells resulted in increased Snail activity and E-cadherin loss, along with oncogenic transformation. Notch pathway mutants were associated with impaired Snail and TGFβ2 expression [45]. In gastric cancer, overexpression of the Notch2 intracellular domain (ICD) was found to promote proliferation and xenograft tumor growth, while knockdown reversed these changes. These changes were shown to be dependent on Notch2’s binding to a Cox2 promoter, and inhibition of Cox2 presence by knockdown or pharmacologic inhibition led to prevention of Notch2 associated tumor progression [46]. Inhibition of Notch1 signaling in MD-MBA-231 breast cancer cells with a monoclonal antibody against Notch1 was shown to result in reduced downstream Notch signaling (decreased Hey1 and HES activation), decreased cell proliferation, and inhibition of mammosphere formation by cancer stem cells; apoptosis was also induced and a reduction of EMT phenotype was observed [47].
Notch interaction with microRNAs has recently been shown to be significant, with implications for its participation in EMT induction. Recently, Jagged2 has been shown to promote metastasis in lung adenocarcinoma cells as a result of Jagged2 promotion of GATA factors, which inhibit the transcription of miR-200 family members. This results in induction of EMT as a result of removal of miR-200 mediated inhibition [48]. Another group has demonstrated specific expression of the Notch ICD to lead to EMT changes via downregulation of the miR-200 family, while showing additionally that miR-200 overexpression led to EMT induction [49]. Brabletz et al showed interactions of transcriptional regulators ZEB1 and miR-200 family members with the Notch pathway. Knockdown of ZEB1 in breast and pancreatic cell lines resulted in reduced Notch reporter activity and downregulation of Jagged1, MamI2/3, and Hey1. Overexpression of miR-200 family members resulted in reduced Notch activity and slightly reduced Jagged1 activity with demonstrated rescue after miR-200 inhibition. Reduction in Notch signaling resulted in reduced proliferation, increased apoptotic susceptibility, and reduced tumorsphere formation. These results were consistent in xexnograft modeling, in which miR-200 family members were increased in xenograft tumors with ZEB1 knockdown, and this correlated with reduced invasiveness and metastatic capacity [50].
Chemoresistance has been observed in relationship to EMT in a number of epithelial cancers, including pancreatic [51], ovarian [52], bladder [53], and breast cancers [54]. While these effects are not exclusively related to Notch signaling, it appears to be an important mediator of chemoresistance in a number of settings, and its contribution to resistance to chemotherapy has recently been explored. Pancreatic cancer cells resistant to gemcitabine were compared to susceptible cells with regard to Notch signaling. Resistant cells were found to have mesenchymal morphology, with downregulation of E-cadherin and β-catenin and upregulation of nestin, α-SMA, vimentin and fibronectin, consistent with EMT induction. Notch2 and Jagged1 were elevated in the gemcitabine resistant cells, and increased activity of MMP9 and uPA/uPAR were observed [55]. In chemoresistant pancreatic ductal adenocarcinoma, a heparin-binding growth factor termed midkine was found to be a downstream target of Notch2. Gemcitabine exposure resulted in a dose-dependent increase in midkine expression, while siRNA mediated knockdown resulted in abrogation of chemoresistance to gemcitabine. The midkine-Notch2 interaction was found to upregulate Notch signaling, EMT markers, and NFκB [56]. Finally, in EGFR-TKI resistant lung cancer, the Notch ICD was found to promote EMT with increased Snail and Vimentin expression, while silencing of NICD reversed this phenotype. This suggested that gefitinib resistance was secondary to Notch-activated EMT [57].
Notch pathway activation has also recently been validated in clinical tissue. An evaluation of prostate cancer specimens and their bony metastases revealed upregulation of Notch1 and expression of EMT markers including E-cadherin, vimentin, ZEB1, PDGF-D, and NFκB in both primary and metastatic sites. Bony metastases revealed increased Notch1 expression relative to their primary sites. In line with interesting recent observations, the EMT phenomenon seemed to be most heavily active at the invasive tumor front [58]. In examination of patient primary breast cancer samples, high expression of Notch1 and Jagged1 were associated with poor prognosis and patient survival [59].
These recent findings indicate that of the described signaling pathways, the Notch pathway may be among the most pivotal in conferring poor clinical prognosis and chemoresistance.
Hedgehog
Originally identified in flies, the Hedgehog pathway is crucial in embryonic patterning and growth of many human tissues during embryogenesis. Specifically, it has a role in specifying segmental patterns [60]. There are three known Hedgehog variants: sonic hedgehog, desert hedgehog, and Indian hedgehog. Signaling begins with a number of modifications to the protein itself. First, the hedgehog protein autoprocesses itself by proteolytic cleavage to release the N-terminal fragment in addition to a covalently bound cholesterol moiety from the C-terminal fragment. The N-terminal fragment is then palmitoylated, resulting in a lipoprotein N-terminal fragment (HhN) ready for interaction with the other components of the pathway. It binds the Patched receptor, a 12-transmembrane-domain integral membrane protein. In the absence of Hedgehog signaling, Patched acts as an inhibitor of Smoothened (Smo), another transmembrane protein with similarities to G-protein coupled receptors. Upon binding of HhN to Patched, its inhibition of Smo is released, which then accumulates in the plasma membrane. Activated Smo can then activate the Gli (glioma-associated oncogene family zinc finger) family of transcription factors, which are the downstream effectors of Hedgehog signaling. As in the other signaling pathways discussed, the activity of Gli in the nucleus is mediated and chaperoned by a number of coactivators and transcriptional regulators, including Cos2, Fused, and Sufu to name a few [60,61] (Figure 5).
Figure 5.

Canonical Hedgehog signaling. Reproduced with permission from Prasad CP, The Hedgehog Signaling Pathway in Breast Cancer. CML Breast Cancer 2012; 22.
Recent evidence for Hedgehog pathway activation in EMT
Louro in 2002 compared gene expression profiles between cells retrovirally transduced with Gli versus c-myc for downstream signaling activation and found that Gli activation and subsequent Hedgehog signaling resulted in upregulation of Snail mRNA and characteristics of EMT [62]. In non-small cell lung cancer cells chronically exposed to TGF-β, in which EMT was thereby induced, sonic hedgehog signaling activation was observed at protein and mRNA levels with increased clonogenic growth, cell motility, and invasion. Pharmacologic and siRNA-mediated knockdown of sonic hedgehog resulted in reversal of EMT, manifested as upregulation of E-cadherin and downregulation of ZEB1 and fibronectin [63]. In hepatoma cell lines, Chen et all were able to identify a subpopulation of hepatoma cells with high chemoresistance despite being negative for cancer stem cell surface markers. EMT was active in this subpopulation compared to marker positive cells, with downregulation of E-cadherin and upregulation of vimentin and Snail. Hedgehog activation was observed in these cell lines and abrogation of Hedgehog signaling resulted in reduced proliferation [64]. Colon cancer epithelial cells and their stem cells were found to harbor high Hedgehog-Gli activation coincident with the development of metastases. In xenografts, recurrence and metastasis required Hedgehog-Gli function, which led to robust EMT [65].
In human samples, Gli1 was found to be upregulated in HCC tissue and adjacent normal tissue; levels correlated with poor prognosis including increased stage, worsened cirrhosis, presence of metastasis, and portal vein invasion [66]. Gli1 activation positively correlated with sonic hedgehog and S100a4 upregulation, and was negatively correlated with E-cadherin expression. In gastric cancer primary biopsies, sonic hedgehog expression has also been associated with presence of nodal disease and poor prognosis. The same group observed overexpression of sonic hedgehog to result in a higher incidence of metastasis in a mouse xenograft model, and found that the Hedgehog signal was effected through P13K/Akt activation. Inhibition of this axis led to reduced EMT (reduced E-cadherin, and Snail), reduced MMP9 activity, and reduced lymphangiogenesis [67]. In brain tumors, Hedgehog pathway constituents were amplifiable from astrocytomas, gliomas, meningiomas, and metastatic lesions, including sonic hedgehog, Smoothened, Patched, Gli1, Gli2, and N-myc. E-cadherin was seen to be downregulated and Snail concomitantly upregulated in malignant tumors and metastases especially compared to primary tumors. This group concluded that Hedgehog signaling aberrations were detectable and frequent in brain tumors [68].
There have been reports challenging the pro-EMT action of Hedgehog signaling, suggesting that the picture may be more complicated than the literature above suggests. Joost et al demonstrated that in pancreatic ductal adenocarcinoma cells, Hedgehog-dependent Gli1 activity is responsible for epithelial differentiation, and that Gli1 knockdown resulted in EMT-like changes with abolition of characteristics of epithelial differentiation. The EMT conversion was seen to be independent of Snail and Slug, and rather was the result of direct Gli1 regulation of E-cadherin transcription [69]. These results suggest that, as with TGF-β, the Hedgehog contribution to EMT may be context- and cell-type dependent.
Major transcription factors
A primary group of transcription factors appear to effect many of the changes seen in EMT. These include Snail, Slug, Twist, ZEB1 and 2, the Smad proteins, and microRNAs among others. These transcriptional factors can work independently but also appear to be simultaneously activated under the influence of many of the major pathways in EMT activation, as seen in the previous discussion. These transcription factors all directly or indirectly affect E-cadherin transcription. The ZEB proteins and zinc finger proteins Snail and Slug are direct inihibitors of the E-cadherin promoter, while Twist and others repress E-cadherin transcription indirectly [70]. Smads function under the influence of TGF-β signaling as well as independently to form transcriptional complexes with Snail and other EMT-relevant transcription factors. The complexity of these interactions obscures any underlying organization, but there is emerging evidence of a hierarchical relationship between these different transcription factors, with Snail1 being most active at the initiation of EMT, and Snail2, ZEB1/2, and Twist being subsequently involved in maintaining the mesenchymal state [71,72].
Snail/Slug
Snail has been shown structurally to have the capability of interacting with a number of pathways. Yook in 2005 demonstrated β-catenin-like canonical motifs in Snail, which result in GSK-3β dependent phosphorylation, β-TRCP directed ubiquitination, and proteosomal degradation similar to the effects of these Wnt pathway constituents on β-catenin itself. Wnt signaling was found to inhibit Snail phosphorylation, ubiquitination, and subsequent degradation, allowing it to accumulate and drive EMT. These effects were suppressed following Snail knockdown [73].
cDNA microarray analysis comparing Snail-expressing and Snail-knockdown melanoma cell lines has shown downregulation of genes associated with EMT including MMP2, EMMPRIN, SPARC, TIMP-1, t-PA, RhoA, and Notch4 in Snail-deficient cells. E-cadherin expression was also upregulated in these cells, although it was not found to have a regulatory role in the process [74]. Ectopic Snail expression in epidermoid carcinoma cell lines has been shown to induce EMT, manifested by an increase in motility and invasiveness. These changes coincided with increased Wnt5a/Ror2 and MMP2 expression. Interestingly, suppression of Wnt5a/Ror2 interaction did not result in changes in EMT markers, but rather in a reduction of motility and invasiveness at least partially mediated by MMP2 [75]. Yaguchi et al showed that breast cancer cells with high Snail expression undergo EMT and demonstrate increased immunoresistance. These effects are likely mediated by the observed increased production of TGF-β, IL-10, TSP01, induction of suppressive regulatory T-cells, and impairment of dendritic cells [76].
Siemens et al have demonstrated a double negative feedback loop between Snail and microRNAs. P53 expression was found to induce miR-34a/b/c genes, leading to microRNA-mediated downregulation of Snail. When the miRNA activity was suppressed, Snail was released from inhibition and led to upregulation of EMT markers and increased migration and invasiveness. Treating the cells with ectopic miR-34 family members led to downregulation of the EMT program, Snail, Slug, and ZEB1, as well as a multitude of stem cell markers. Interestingly, Snail and ZEB1 were also found to be able to bind to the miR-34 promoter and suppress its transcription, thus defining a double feedback loop. The authors suggested that p53 inactivation or miR34 inactivation might therefore abolish this double negative feedback loop and lock the cells into a metastatic biology program [77].
Crosstalk with the TGF-β signaling pathway may also be made possible by Snail transcription factors. Vincent et al describe the formation of Snail1-Smad3/4 transcriptional repressor complexes that promote TGF-β mediated EMT. These complexes were shown to transcriptionally repress expression of E-cadherin, claudin3, CAR, and occludin during TGF-β induced EMT in breast epithelial cells. Silencing of Snail1 and Smad3/4 by siRNA interference resulted in increased CAR and occludin expression. Interestingly, these effects were observed to be most active at the invasive front of tumors in human invasive breast cancer samples [78].
Twist
Yang et al described a critical role for Twist expression in metastatic breast cancer, reporting that Twist inhibition in highly metastatic breast cancer lines resulted in inhibition of their ability to metastasize to the lungs of xenograft mice. Ectopic expression of Twist resulted in loss of E-cadherin and activation of mesenchymal markers and cell motility, indicating EMT activation. Clinically these results were verified upon finding high levels of Twist expression in invasive lobular carcinoma [79]. Stable expression of Twist in breast and cervical cancer cell lines resulted in a phenotypic change to spindle-shaped morphology consistent with EMT, which correlated with decreased E-cadherin expression and increased N-cadherin and vimentin consistent with EMT. Twist expression also enhanced migration, tumorsphere formation, and expression of stemlike markers. β-catenin and Akt were both found to be activated in Twist over-expressing cells [80].
Smads
Smads are interesting subjects of investigation in EMT because they appear to have functions both dependent and independent of TGF-β signaling. The formation of Smad complexes that interact at the transcriptional level with factors traditionally seen as part of other signaling pathways is a major mechanism of EMT potentiation, and also may explain the highly variable effects of TGF-β signaling in tumorigenesis. Smads alone have a relatively low affinity for DNA, but in interacting with Snail, ZEB1/2, Twist, and other transcription factors, they acquire higher DNA affinity and the ability to affect transcription [18]. This by necessity results in crosstalk between the TGF-β, Wnt, Notch, Hedgehog, and other signaling pathways. The Notch ICD has also been shown to interact with Smad3, 1, and 5 in embryonic settings to affect chromatin organization [81]. In carcinogenesis, direct binding of Notch to Smads or indirect transcriptional inhibition of Smad transcriptional coactivators have been described [82]. Notch action in general seems to inhibit the protumorigenic, prometastatic activities of TGF-β and return the signaling milieu to one in which TGF-β signals result in inhibitory effects on tumorigenesis and metastasis.
Zeb1/2
Similar to the double negative feedback loop between Snail and miR-34 family members described by Siemens, a double negative feedback loop has been described between the ZEB1/2 transcription factors and miR-200 family members. This was first described in 2008 in canine kidney cells [83,84]. The miR-200 family represses ZEB1/2 translation, and conversely the ZEB proteins have the ability to bind to miR-200 family member promoter sites, preventing their transcription. The authors propose that the balance of this feedback loop can be shifted towards ZEB predominance (mesenchymal phenotype) or miR-200 predominance (epithelial phenotype) by the action of exogenous signaling factors, and subsequently identify autocrine TGF-β as a major factor in locking this loop into a pro-mesenchymal state [85]. Interestingly, the mesenchymal phenotype can be perpetuated by methylation of the miR-200 promoter, leading to longer-term mesenchymal changes and supporting the role of epigenetic silencing in EMT and cancer progression. The same feedback loop has also been shown to influence Notch signaling by Brabletz et al [50]. ZEB1 has also been shown to control stemness and survival by inhibiting miR-200 family members [86].
Micro-RNAs
MicroRNAs as regulators of metastatic potential have received a great deal of attention recently. These are short, noncoding RNA molecules that regulate gene expression post-transcriptionally. Base pair complementarity between mature miRNAs and their target mRNAs result in endonuclear cleavage of the mRNA transcript. Imperfect complementarity can also result in interference by forcing transcript degradation or preventing effective translation [87]. In EMT, miRNAs are frequently engaged in feedback loops with previously described signaling pathway constituents such that an imbalance away from miRNA expression results in activation of EMT programming. The miR-200 family has been described to directly target ZEB1 and ZEB2. Korpal et al reported that miR-200 family member expression is repressed in TGF-β induced EMT in murine mammary epithelial cells. Overexpression of the miRNAs individually or as clusters resulted in enhanced E-cadherin expression through direct targeting of ZEB1/2, demonstrated by reduced luciferase reporter activity when miR-200 family members were cotransfected with luciferase-ZEB1/2 plasmid constructs. This effect appeared to be most profound on ZEB2. When a mesenchymal mouse mammary epithelial line was exposed to exogenous mi-R200 miRNAs, the cells changed morphology to an epithelial phenotype and displayed increased E-cadherin expression, consistent with reversal of EMT [88]. In gastric cancer cell lines, miR-200 family members were overexpressed in cell lines also overexpressing Smad3. Luciferase reporter assays showed that Smad3 binds to the miR-200b/a promoter to function as a transcriptional activator. This led to transcription of miR-200 family microRNAs and subsequent downregulation of ZEB1/2, leading to release of E-cadherin from their transcriptional repression. This led to reversal of EMT. Interestingly, although these effects were mediated by Smad3, they were found to ultimately be independent of TGF-β signaling [89].
In general microRNAs have been shown to function as genetic repressors, interfering with transcripts of target genes and preventing their translation and ultimate expression. An interesting study recently described an oncogenic role for miR-27 in gastric cancer. Zhang et al reported increased miR-27 levels in gastric cancer tissues and showed that levels were increased in metastatic compared to nonmetastatic tumors. In vitro, miR-27 overexpression led to increased migration of AGS gastric cancer cells, and when treated with an miR-27 inhibitor, these effects were abrogated. miR-27 was shown to induce an EMT phenotype, including increased ZEB1/2, Slug, and vimentin expression with decreased E-cadherin expression. Finally, the signaling mechanism of miR-27 activity in this study was identified as Wnt/β-catenin based with APC as its target gene [90].
Epigenetics
Epigenetic silencing has been shown in a number of reports to play a role in regulation of the key EMT effectors. Methylation of promoter sites of miR-200 family members [39] and Wnt family members [85] have been shown to affect EMT changes as described above. SFRP5, a Wnt pathway antagonist, was shown to reduce cell proliferation, invasiveness, and tumor formation in mice when expression is allowed. Higher SFRP5 expression was also found to reduce EMT changes and sensitize the ovarian cancer model to chemotherapy with cisplatin. Given evidence for SFRP5 hypermethylation in ovarian cancer patients, the authors proposed that the epigenetic silencing of SFRP5 was a mechanism by which ovarian cancer cells become prometastatic and acquire chemoresistance [91]. Prasad et al demonstrated hypermethylation of CDH1 and APC promoters in invasive ductal breast carcinoma samples, and showed that the hypermethylation status correlated with nuclear localization of β-catenin along with evidence of EMT (E-cadherin loss, increased vimentin expression) [92]. Prolonged induction of EMT has been shown to result in recruitment of DNA methyltransferases and chromatin remodeling enzymes to EMT-related gene promoter regions regulated by TGF-β such as E-cadherin and estrogen receptor α [7,93]. The loss of such epigenetic silencing has been shown sufficient to induce mesenchymal-epithelial transition related to inhibition of Smad2 signaling in breast cancer [94]. Histone modification, which determines repressive versus available chromatin structure, has been shown in a number of cancer models to perform a regulatory role in availability of EMT-related gene products for transcription; many of these effects are under the influence of TGF-β and Notch signaling [82].
Physical components and characteristics of the tumor microenvironment
We are increasingly coming to understand the importance of the specific constituents of the tumor stroma in seeding and metastatic potential of primary tumors. The stroma is generally made up of other cell types, extracellular matrix, and the vasculature and lymphatic systems feeding the tumors. Cell types in the tumor stroma include fibroblasts, myofibroblasts, granulocytes, macrophages, mesenchymal stem cells, and lymphocytes [95]. Work has shown that in the context of the tumor microenvironment, some of these cell types additionally change phenotype and become accessories to tumorigenesis and metastasis, and are therefore referred to as cancer-associated fibroblasts and tumor-associated macrophages. TGF-β signaling has been heavily implicated in the interaction between tumor cell mass and stroma, primarily demonstrated by loss of function studies in which lack of TGF-β signaling results in higher paracrine factor production (such as MMPs, TGF-α, MSP, and HGF) that enhances stromal fibroblast conversion to cancer-associated fibroblasts, and these CAFs in turn can determine to a large extent whether the cytostatic or prometastatic manifestations of TGF-β signaling predominate [70,96]. Reduction of the TGF-β signal has been associated with worsened cancer phenotypes, including increased growth, survival and motility [97]. TGF-β has been shown to stimulate CAFs, resulting in synthesis and deposition of extracellular matrix compounds such as collagen and fibronectin that are associated with tumor invasive potential [98]. CAF are also primary producers of VEGF, which promotes angiogenesis, and facilitates tumor growth and metastasis [99]. Tumor fibroblasts have been shown to promote esophageal tumor progression, and Fu et al have shown that 43% of the known deregulated genes in tumor-associated fibroblasts are associated with proliferation, extracellular matrix remodeling, and immune response, all critical for tumor success [100]. Given the importance of tumor stromal cells in both supporting and enhancing tumor formation, it has been proposed that the signaling impetus driving EMT can be derived from two sources: the genetic and physical changes in the cancer cell population itself resulting in autocrine and paracrine signaling changes leading to EMT, and the input of signaling information from the stromal cells that are recruited to support the tumor mass [95].
In addition, the mechanical characteristics of the extracellular matrix play an important role in the ability of tumor cells to utilize the characteristics acquired during EMT and metastasize. The role of mechanical attributes such as collagen content, fiber thickness, and extent of intrafibrillar crosslinks characterizing the collagen stroma of a given tumor have effects on the degree of plasticity and specific cell-ECM adhesion molecules required by a tumor cell in order to navigate that environment [101,102]. These effects can be mediated by signaling pathways associated with EMT; for example, TGF-β signaling has been studied in the context of 3D-organotype culture systems, in which collagen content, mechanical compliance, and integrin activation can be examined, and these factors have been found to have significant effects on TGF-β signaling both in its tumor-suppressing and tumorigenic roles according to the TGF-β paradox [7]. In addition, matrix rigidity has been shown to change the effect of TGF-β signaling from apoptosis promotion to EMT induction in epithelial cells [103].
An interesting study recently demonstrated Wnt signaling active in tumor-associated fibroblasts at the stromal-tumor interface in oral squamous cell carcinomas. Fu et al demonstrated that in immunohistochemistry of clinical samples of OSCC, Wnt2 signal was only detected in tumor fibroblasts, and primarily at the boundary between stroma and tissue. Wnt2 positivity was associated with lymph node metastasis, higher TNM stage, and worsened disease-specific survival. In vitro studies using conditioned medium from Wnt2 expressing fibroblasts revealed upregulation of cyclin-D1 and c-myc in response to Wnt2 in conditioned medium; this resulted in increased EMT, motility, and invasiveness of OSCC cells [100]. Other authors have reported activity of EMT-associated signaling pathways and transcriptional products primarily at the invasive front as well [4,58,78], recapitulating the idea of EMT as a site-specific phenomenon in the appropriate context.
Hypoxia
The hypoxic tumor microenvironment contributes to its reactive, inflammatory nature and is thought to be relevant in radiation and chemoresistance. Hypoxic stress also promotes angiogenesis, lymphangiogenesis, and inflammation, leading to recruitment of new nutrient supplies and inflammatory cells that can then further potentiate the metastatic phenotype [104]. Cannito et al showed that under hypoxic conditions, several epithelial carcinoma cell lines, including pancreatic, breast, colon, and hepatoblastoma, showed mesenchymal morphologic changes, decreased E-cadherin expression, increased Snail expression, and nuclear translocation of β-catenin consistent with Wnt pathway activation. The cells then displayed increased invasiveness in vitro. The mechanism demonstrated for these changes involved early reactive oxygen species generation in mitochondria and resultant GSK-3β inhibition [105]. Notch signaling has been shown to be activated by hypoxia as well, either directly or synergistically with increased Notch ligand activation. In breast and ovarian cancer cell lines, cells exposed to hypoxia showed E-cadherin and β-catenin downregulation with upregulation of N-cadherin, vimentin, and fibronectin. These changes were ameliorated with specific Notch pathway inhibitor treatment. Further study showed that upregulation of Snail1 directly or by Notch-dependent LOX upregulation was the mechanism of these changes [106]. One mechanism of survival of cancer stem cells and/or EMT-transformed tumor cells may be an increased ability to scavenge free radicals and therefore survive the hypoxia of the tumor microenvironment, as described by Kim et al [107].
Conclusions
As our surgical capability has improved, much of the persistent mortality suffered by cancer patients continues to arise from the presence of distant metastases, converting what seems to begin as a localized process into a systemic disease. Hence, much of our current effort in understanding cancer biology focuses on mechanisms of metastasis with the ultimate goal of targeting those mechanisms for treatment. The tumor microenvironment and the EMT program both contribute in sweeping and complex ways to a metastatic cancer cell’s acquisition of the four properties of metastasis - emigration, invasion, intravasation, and extravasation and micrometastasis establishment. While the challenges of migration from the primary to the distant site and the mechanisms allowing establishment of a micrometastasis are still nebulous, our understanding of EMT and the acquisition of mobile and invasive phenotypes by cancer cells has undergone an explosion in the last decade. The elaboration of the numerous signaling pathways involved in EMT and their interactions, as well as the downstream effectors actually driving the EMT process, has provided us with an extensive number of potential therapeutic targets. While it is unlikely that targeting any specific component of these regulatory networks will result in significant clinical response given the redundancy of these signaling networks, it may be possible to develop multi-agent targeted therapy regimens that block or reverse EMT. In conjunction with surgical removal of the primary tumor and other modal therapies, these potential therapeutic targets provide some hope that a systematic approach to reducing or reversing EMT changes and targeting the components of the tumor microenvironment may allow us to progress on the path to effective prevention and treatment of cancer metastasis.
Acknowledgement
This study was supported by NIH grants R01-GM065113-05A2, T32-GM069331, and UL1-RR024128.
References
- 1.Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10:445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- 2.Hay ED. Interaction of embryonic surface and cytoskeleton with extracellular matrix. Am J Anat. 1982;165:1–12. doi: 10.1002/aja.1001650102. [DOI] [PubMed] [Google Scholar]
- 3.Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010;15:117–134. doi: 10.1007/s10911-010-9178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang D, Du X. Crosstalk between tumor cells and microenvironment via Wnt pathway in colorectal cancer dissemination. World J Gastroenterol. 2008;14:1823–1827. doi: 10.3748/wjg.14.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 6.Meulmeester E, Ten Dijke P. The dynamic roles of TGF-beta in cancer. J Pathol. 2011;223:205–218. doi: 10.1002/path.2785. [DOI] [PubMed] [Google Scholar]
- 7.Wendt MK, Tian M, Schiemann WP. Deconstructing the mechanisms and consequences of TGF-beta-induced EMT during cancer progression. Cell Tissue Res. 2012;347:85–101. doi: 10.1007/s00441-011-1199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koinuma D, Tsutsumi S, Kamimura N, Imamura T, Aburatani H, Miyazono K. Promoter-wide analysis of Smad4 binding sites in human epithelial cells. Cancer Sci. 2009;100:2133–2142. doi: 10.1111/j.1349-7006.2009.01299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tian M, Neil JR, Schiemann WP. Transforming growth factor-beta and the hallmarks of cancer. Cell Signal. 2011;23:951–962. doi: 10.1016/j.cellsig.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Ten Dijke P, Goumans MJ, Itoh F, Itoh S. Regulation of cell proliferation by Smad proteins. J Cell Physiol. 2002;191:1–16. doi: 10.1002/jcp.10066. [DOI] [PubMed] [Google Scholar]
- 12.Postigo AA, Depp JL, Taylor JJ, Kroll KL. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003;22:2453–2462. doi: 10.1093/emboj/cdg226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127:2021–2036. doi: 10.1083/jcb.127.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leptin M. twist and snail as positive and negative regulators during Drosophila mesoderm development. Genes Dev. 1991;5:1568–1576. doi: 10.1101/gad.5.9.1568. [DOI] [PubMed] [Google Scholar]
- 16.D'Inzeo S, Nicolussi A, Donini CF, Zani M, Mancini P, Nardi F, Coppa A. A novel human Smad4 mutation is involved in papillary thyroid carcinoma progression. Endocr Relat Cancer. 2012;19:39–55. doi: 10.1530/ERC-11-0233. [DOI] [PubMed] [Google Scholar]
- 17.Mazur PK, Einwachter H, Lee M, Sipos B, Nakhai H, Rad R, Zimber-Strobl U, Strobl LJ, Radtke F, Kloppel G, Schmid RM, Siveke JT. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc Natl Acad Sci USA. 2010;107:13438–13443. doi: 10.1073/pnas.1002423107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuxe J, Vincent T, Garcia de Herreros A. Transcriptional crosstalk between TGF-beta and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell Cycle. 2010;9:2363–2374. doi: 10.4161/cc.9.12.12050. [DOI] [PubMed] [Google Scholar]
- 19.Araki K, Shimura T, Suzuki H, Tsutsumi S, Wada W, Yajima T, Kobayahi T, Kubo N, Kuwano H. E/N-cadherin switch mediates cancer progression via TGF-beta-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br J Cancer. 2011;105:1885–1893. doi: 10.1038/bjc.2011.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biswas S, Guix M, Rinehart C, Dugger TC, Chytil A, Moses HL, Freeman ML, Arteaga CL. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J Clin Invest. 2007;117:1305–1313. doi: 10.1172/JCI30740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou YC, Liu JY, Li J, Zhang J, Xu YQ, Zhang HW, Qiu LB, Ding GR, Su XM, Mei S, Guo GZ. Ionizing radiation promotes migration and invasion of cancer cells through transforming growth factor-beta-mediated epithelial-mesenchymal transition. Int J Radiat Oncol Biol Phys. 2011;81:1530–1537. doi: 10.1016/j.ijrobp.2011.06.1956. [DOI] [PubMed] [Google Scholar]
- 22.Katoh M. Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol Ther. 2006;5:1059–1064. doi: 10.4161/cbt.5.9.3151. [DOI] [PubMed] [Google Scholar]
- 23.Bovolenta P, Esteve P, Ruiz JM, Cisneros E, Lopez-Rios J. Beyond Wnt inhibition: new functions of secreted Frizzled-related proteins in development and disease. J Cell Sci. 2008;121:737–746. doi: 10.1242/jcs.026096. [DOI] [PubMed] [Google Scholar]
- 24.Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK, Choi YJ, Kim J, Fearon ER, Weiss SJ. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8:1398–1406. doi: 10.1038/ncb1508. [DOI] [PubMed] [Google Scholar]
- 25.Gauger KJ, Chenausky KL, Murray ME, Schneider SS. SFRP1 reduction results in an increased sensitivity to TGF-beta signaling. BMC Cancer. 2011;11:59. doi: 10.1186/1471-2407-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gauger KJ, Hugh JM, Troester MA, Schneider SS. Down-regulation of sfrp1 in a mammary epithelial cell line promotes the development of a cd44high/cd24low population which is invasive and resistant to anoikis. Cancer Cell Int. 2009;9:11. doi: 10.1186/1475-2867-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung MT, Lai HC, Sytwu HK, Yan MD, Shih YL, Chang CC, Yu MH, Liu HS, Chu DW, Lin YW. SFRP1 and SFRP2 suppress the transformation and invasion abilities of cervical cancer cells through Wnt signal pathway. Gynecol Oncol. 2009;112:646–653. doi: 10.1016/j.ygyno.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 28.Gerard B, Tait L, Nangia-Makker P, Shekhar MP. Rad6B acts downstream of Wnt signaling to stabilize beta-catenin: Implications for a novel Wnt/beta-catenin target. J Mol Signal. 2011;6:6. doi: 10.1186/1750-2187-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iwai S, Yonekawa A, Harada C, Hamada M, Katagiri W, Nakazawa M, Yura Y. Involvement of the Wnt-beta-catenin pathway in invasion and migration of oral squamous carcinoma cells. Int J Oncol. 2010;37:1095–1103. doi: 10.3892/ijo_00000761. [DOI] [PubMed] [Google Scholar]
- 30.Blavier L, Lazaryev A, Shi XH, Dorey FJ, Shackleford GM, DeClerck YA. Stromelysin-1 (MMP-3) is a target and a regulator of Wnt1-induced epithelial-mesenchymal transition (EMT) Cancer Biol Ther. 2010;10:198–208. doi: 10.4161/cbt.10.2.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhai Y, Iura A, Yeasmin S, Wiese AB, Wu R, Feng Y, Fearon ER, Cho KR. MSX2 is an oncogenic downstream target of activated WNT signaling in ovarian endometrioid adenocarcinoma. Oncogene. 2011;30:4152–4162. doi: 10.1038/onc.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prasad CP, Rath G, Mathur S, Bhatnagar D, Parshad R, Ralhan R. Expression analysis of E-cadherin, Slug and GSK3beta in invasive ductal carcinoma of breast. BMC Cancer. 2009;9:325. doi: 10.1186/1471-2407-9-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwon CY, Kim KR, Choi HN, Chung MJ, Noh SJ, Kim DG, Kang MJ, Lee DG, Moon WS. The role of serum response factor in hepatocellular carcinoma: implications for disease progression. Int J Oncol. 2010;37:837–844. doi: 10.3892/ijo_00000734. [DOI] [PubMed] [Google Scholar]
- 34.Yee DS, Tang Y, Li X, Liu Z, Guo Y, Ghaffar S, McQueen P, Atreya D, Xie J, Simoneau AR, Hoang BH, Zi X. The Wnt inhibitory factor 1 restoration in prostate cancer cells was associated with reduced tumor growth, decreased capacity of cell migration and invasion and a reversal of epithelial to mesenchymal transition. Mol Cancer. 2010;9:162. doi: 10.1186/1476-4598-9-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao JH, Luo Y, Jiang YG, He DL, Wu CT. Knockdown of beta-Catenin through shRNA cause a reversal of EMT and metastatic phenotypes induced by HIF-1alpha. Cancer Invest. 2011;29:377–382. doi: 10.3109/07357907.2010.512595. [DOI] [PubMed] [Google Scholar]
- 36.Jiang YG, Luo Y, He DL, Li X, Zhang LL, Peng T, Li MC, Lin YH. Role of Wnt/beta-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1alpha. Int J Urol. 2007;14:1034–1039. doi: 10.1111/j.1442-2042.2007.01866.x. [DOI] [PubMed] [Google Scholar]
- 37.Chen YS, Mathias RA, Mathivanan S, Kapp EA, Moritz RL, Zhu HJ, Simpson RJ. Proteomics profiling of Madin-Darby canine kidney plasma membranes reveals Wnt-5a involvement during oncogenic H-Ras/TGF-beta-mediated epithelial-mesenchymal transition. Mol Cell Proteomics. 2011;10:M110. 001131. doi: 10.1074/mcp.M110.001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DiMeo TA, Anderson K, Phadke P, Fan C, Perou CM, Naber S, Kuperwasser C. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009;69:5364–5373. doi: 10.1158/0008-5472.CAN-08-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurasawa Y, Kozaki K, Pimkhaokham A, Muramatsu T, Ono H, Ishihara T, Uzawa N, Imoto I, Amagasa T, Inazawa J. Stabilization of phenotypic plasticity through mesenchymal-specific DNA hypermethylation in cancer cells. Oncogene. 2012;31:1963–1974. doi: 10.1038/onc.2011.373. [DOI] [PubMed] [Google Scholar]
- 40.Heuberger J, Birchmeier W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb Perspect Biol. 2010;2:a002915. doi: 10.1101/cshperspect.a002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doble BW, Woodgett JR. Role of glycogen synthase kinase-3 in cell fate and epithelial-mesenchymal transitions. Cells Tissues Organs. 2007;185:73–84. doi: 10.1159/000101306. [DOI] [PubMed] [Google Scholar]
- 42.Weerkamp F, van Dongen JJ, Staal FJ. Notch and Wnt signaling in T-lymphocyte development and acute lymphoblastic leukemia. Leukemia. 2006;20:1197–1205. doi: 10.1038/sj.leu.2404255. [DOI] [PubMed] [Google Scholar]
- 43.Bolos V, Grego-Bessa J, de la Pompa JL. Notch signaling in development and cancer. Endocr Rev. 2007;28:339–363. doi: 10.1210/er.2006-0046. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Li Y, Ahmad A, Azmi AS, Banerjee S, Kong D, Sarkar FH. Targeting Notch signaling pathway to overcome drug resistance for cancer therapy. Biochim Biophys Acta. 2010;1806:258–267. doi: 10.1016/j.bbcan.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua-Belmonte JC, de la Pompa JL. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004;18:99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tseng YC, Tsai YH, Tseng MJ, Hsu KW, Yang MC, Huang KH, Li AF, Chi CW, Hsieh RH, Ku HH, Yeh TS. Notch2-induced COX-2 expression enhancing gastric cancer progression. Mol Carcinog. 2011 doi: 10.1002/mc.20865. [DOI] [PubMed] [Google Scholar]
- 47.Sharma A, Paranjape AN, Rangarajan A, Dighe RR. A Monoclonal Antibody against Human Notch1 Ligand Binding Domain Depletes Subpopulation of Breast Cancer Stem-like Cells. Mol Cancer Ther. 2012;11:77–86. doi: 10.1158/1535-7163.MCT-11-0508. [DOI] [PubMed] [Google Scholar]
- 48.Yang Y, Ahn YH, Gibbons DL, Zang Y, Lin W, Thilaganathan N, Alvarez CA, Moreira DC, Creighton CJ, Gregory PA, Goodall GJ, Kurie JM. The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. J Clin Invest. 2011;121:1373–1385. doi: 10.1172/JCI42579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bao B, Wang Z, Ali S, Kong D, Li Y, Ahmad A, Banerjee S, Azmi AS, Miele L, Sarkar FH. Notch-1 induces epithelial-mesenchymal transition consistent with cancer stem cell phenotype in pancreatic cancer cells. Cancer Lett. 2011;307:26–36. doi: 10.1016/j.canlet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Brabletz S, Bajdak K, Meidhof S, Burk U, Niedermann G, Firat E, Wellner U, Dimmler A, Faller G, Schubert J, Brabletz T. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011;30:770–782. doi: 10.1038/emboj.2010.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey DJ, Choi W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–5828. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kajiyama H, Shibata K, Terauchi M, Yamashita M, Ino K, Nawa A, Kikkawa F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol. 2007;31:277–283. [PubMed] [Google Scholar]
- 53.Sayan AE, Griffiths TR, Pal R, Browne GJ, Ruddick A, Yagci T, Edwards R, Mayer NJ, Qazi H, Goyal S, Fernandez S, Straatman K, Jones GD, Bowman KJ, Colquhoun A, Mellon JK, Kriajevska M, Tulchinsky E. SIP1 protein protects cells from DNA damage-induced apoptosis and has independent prognostic value in bladder cancer. Proc Natl Acad Sci USA. 2009;106:14884–14889. doi: 10.1073/pnas.0902042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iseri OD, Kars MD, Arpaci F, Atalay C, Pak I, Gunduz U. Drug resistant MCF-7 cells exhibit epithelial-mesenchymal transition gene expression pattern. Biomed Pharmacother. 2011;65:40–45. doi: 10.1016/j.biopha.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 55.Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009;69:2400–2407. doi: 10.1158/0008-5472.CAN-08-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gungor C, Zander H, Effenberger KE, Vashist YK, Kalinina T, Izbicki JR, Yekebas E, Bockhorn M. Notch signaling activated by replication stress-induced expression of midkine drives epithelial-mesenchymal transition and chemoresistance in pancreatic cancer. Cancer Res. 2011;71:5009–5019. doi: 10.1158/0008-5472.CAN-11-0036. [DOI] [PubMed] [Google Scholar]
- 57.Xie M, Zhang L, He CS, Xu F, Liu JL, Hu ZH, Zhao LP, Tian Y. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J Cell Biochem. 2012;113:1501–1513. doi: 10.1002/jcb.24019. [DOI] [PubMed] [Google Scholar]
- 58.Sethi S, Macoska J, Chen W, Sarkar FH. Molecular signature of epithelial-mesenchymal transition (EMT) in human prostate cancer bone metastasis. Am J Transl Res. 2010;3:90–99. [PMC free article] [PubMed] [Google Scholar]
- 59.Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, Lockwood G, Egan SE. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005;65:8530–8537. doi: 10.1158/0008-5472.CAN-05-1069. [DOI] [PubMed] [Google Scholar]
- 60.Beachy PA, Hymowitz SG, Lazarus RA, Leahy DJ, Siebold C. Interactions between Hedgehog proteins and their binding partners come into view. Genes Dev. 2010;24:2001–2012. doi: 10.1101/gad.1951710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sarkar FH, Li Y, Wang Z, Kong D. The role of nutraceuticals in the regulation of Wnt and Hedgehog signaling in cancer. Cancer Metastasis Rev. 2010;29:383–394. doi: 10.1007/s10555-010-9233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Louro ID, Bailey EC, Li X, South LS, McKie-Bell PR, Yoder BK, Huang CC, Johnson MR, Hill AE, Johnson RL, Ruppert JM. Comparative gene expression profile analysis of GLI and c-MYC in an epithelial model of malignant transformation. Cancer Res. 2002;62:5867–5873. [PubMed] [Google Scholar]
- 63.Maitah MY, Ali S, Ahmad A, Gadgeel S, Sarkar FH. Up-regulation of sonic hedgehog contributes to TGF-beta1-induced epithelial to mesenchymal transition in NSCLC cells. PLoS One. 2011;6:e16068. doi: 10.1371/journal.pone.0016068. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Chen X, Lingala S, Khoobyari S, Nolta J, Zern MA, Wu J. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J Hepatol. 2011;55:838–845. doi: 10.1016/j.jhep.2010.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, Ruiz i Altaba A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med. 2009;1:338–351. doi: 10.1002/emmm.200900039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng X, Yao Y, Xu Q, Tu K, Liu Q. Evaluation of glioma-associated oncogene 1 expression and its correlation with the expression of sonic hedgehog, E-cadherin and S100a4 in human hepatocellular carcinoma. Mol Med Report. 2010;3:965–970. doi: 10.3892/mmr.2010.375. [DOI] [PubMed] [Google Scholar]
- 67.Yoo YA, Kang MH, Lee HJ, Kim BH, Park JK, Kim HK, Kim JS, Oh SC. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res. 2011;71:7061–7070. doi: 10.1158/0008-5472.CAN-11-1338. [DOI] [PubMed] [Google Scholar]
- 68.Chou CH, Lieu AS, Wu CH, Chang LK, Loh JK, Lin RC, Chen WJ, Liao HD, Fu WS, Chang CS, Lin CC, Hsu CM, Chio CC, Howng SL, Hong YR. Differential expression of hedgehog signaling components and Snail/E-cadherin in human brain tumors. Oncol Rep. 2010;24:1225–1232. doi: 10.3892/or_00000976. [DOI] [PubMed] [Google Scholar]
- 69.Joost S, Almada LL, Rohnalter V, Holz PS, Vrabel AM, Fernandez-Barrena MG, McWilliams RR, Krause M, Fernandez-Zapico ME, Lauth M. GLI1 Inhibition Promotes Epithelial-to-Mesenchymal Transition in Pancreatic Cancer Cells. Cancer Res. 2012;72:88–99. doi: 10.1158/0008-5472.CAN-10-4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 71.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 72.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 73.Yook JI, Li XY, Ota I, Fearon ER, Weiss SJ. Wnt-dependent regulation of the E-cadherin repressor snail. J Biol Chem. 2005;280:11740–11748. doi: 10.1074/jbc.M413878200. [DOI] [PubMed] [Google Scholar]
- 74.Kuphal S, Palm HG, Poser I, Bosserhoff AK. Snail-regulated genes in malignant melanoma. Melanoma Res. 2005;15:305–313. doi: 10.1097/00008390-200508000-00012. [DOI] [PubMed] [Google Scholar]
- 75.Ren D, Minami Y, Nishita M. Critical role of Wnt5a-Ror2 signaling in motility and invasiveness of carcinoma cells following Snail-mediated epithelial-mesenchymal transition. Genes Cells. 2011;16:304–315. doi: 10.1111/j.1365-2443.2011.01487.x. [DOI] [PubMed] [Google Scholar]
- 76.Yaguchi T, Sumimoto H, Kudo-Saito C, Tsukamoto N, Ueda R, Iwata-Kajihara T, Nishio H, Kawamura N, Kawakami Y. The mechanisms of cancer immunoescape and development of overcoming strategies. Int J Hematol. 2011;93:294–300. doi: 10.1007/s12185-011-0799-6. [DOI] [PubMed] [Google Scholar]
- 77.Siemens H, Jackstadt R, Hunten S, Kaller M, Menssen A, Gotz U, Hermeking H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle. 2011;10:4256–4271. doi: 10.4161/cc.10.24.18552. [DOI] [PubMed] [Google Scholar]
- 78.Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL, Crystal RG, de Herreros AG, Moustakas A, Pettersson RF, Fuxe J. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol. 2009;11:943–950. doi: 10.1038/ncb1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 80.Li J, Zhou BP. Activation of beta-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer. 2011;11:49. doi: 10.1186/1471-2407-11-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Blokzijl A, Dahlqvist C, Reissmann E, Falk A, Moliner A, Lendahl U, Ibanez CF. Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J Cell Biol. 2003;163:723–728. doi: 10.1083/jcb.200305112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Blumenberg M, Gao S, Dickman K, Grollman AP, Bottinger EP, Zavadil J. Chromatin structure regulation in transforming growth factor-beta-directed epithelial-mesenchymal transition. Cells Tissues Organs. 2007;185:162–174. doi: 10.1159/000101317. [DOI] [PubMed] [Google Scholar]
- 83.Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68:7846–7854. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 84.Gregory PA, Bracken CP, Bert AG, Goodall GJ. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle. 2008;7:3112–3118. doi: 10.4161/cc.7.20.6851. [DOI] [PubMed] [Google Scholar]
- 85.Gregory PA, Bracken CP, Smith E, Bert AG, Wright JA, Roslan S, Morris M, Wyatt L, Farshid G, Lim YY, Lindeman GJ, Shannon MF, Drew PA, Khew-Goodall Y, Goodall GJ. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 2011;22:1686–1698. doi: 10.1091/mbc.E11-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A, Brunton VG, Morton J, Sansom O, Schuler J, Stemmler MP, Herzberger C, Hopt U, Keck T, Brabletz S, Brabletz T. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 87.Mongroo PS, Rustgi AK. The role of the miR-200 family in epithelial-mesenchymal transition. Cancer Biol Ther. 2010;10:219–222. doi: 10.4161/cbt.10.6312548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ahn SM, Cha JY, Kim J, Kim D, Trang HT, Kim YM, Cho YH, Park D, Hong S. Smad3 regulates E-cadherin via miRNA-200 pathway. Oncogene. 2011 doi: 10.1038/onc.2011.484. [DOI] [PubMed] [Google Scholar]
- 90.Zhang Z, Liu S, Shi R, Zhao G. miR-27 promotes human gastric cancer cell metastasis by inducing epithelial-to-mesenchymal transition. Cancer Genet. 2011;204:486–491. doi: 10.1016/j.cancergen.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 91.Su HY, Lai HC, Lin YW, Liu CY, Chen CK, Chou YC, Lin SP, Lin WC, Lee HY, Yu MH. Epigenetic silencing of SFRP5 is related to malignant phenotype and chemoresistance of ovarian cancer through Wnt signaling pathway. Int J Cancer. 2010;127:555–567. doi: 10.1002/ijc.25083. [DOI] [PubMed] [Google Scholar]
- 92.Prasad CP, Mirza S, Sharma G, Prashad R, DattaGupta S, Rath G, Ralhan R. Epigenetic alterations of CDH1 and APC genes: relationship with activation of Wnt/beta-catenin pathway in invasive ductal carcinoma of breast. Life Sci. 2008;83:318–325. doi: 10.1016/j.lfs.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 93.Dumont N, Wilson MB, Crawford YG, Reynolds PA, Sigaroudinia M, Tlsty TD. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc Natl Acad Sci USA. 2008;105:14867–14872. doi: 10.1073/pnas.0807146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Papageorgis P, Lambert AW, Ozturk S, Gao F, Pan H, Manne U, Alekseyev YO, Thiagalingam A, Abdolmaleky HM, Lenburg M, Thiagalingam S. Smad signaling is required to maintain epigenetic silencing during breast cancer progression. Cancer Res. 2010;70:968–978. doi: 10.1158/0008-5472.CAN-09-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 96.Cheng N, Bhowmick NA, Chytil A, Gorksa AE, Brown KA, Muraoka R, Arteaga CL, Neilson EG, Hayward SW, Moses HL. Loss of TGF-beta type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-alpha-, MSP- and HGF-mediated signaling networks. Oncogene. 2005;24:5053–5068. doi: 10.1038/sj.onc.1208685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fidler IJ. Critical determinants of metastasis. Semin Cancer Biol. 2002;12:89–96. doi: 10.1006/scbi.2001.0416. [DOI] [PubMed] [Google Scholar]
- 98.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 99.Fukumura D, Xavier R, Sugiura T, Chen Y, Park EC, Lu N, Selig M, Nielsen G, Taksir T, Jain RK, Seed B. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–725. doi: 10.1016/s0092-8674(00)81731-6. [DOI] [PubMed] [Google Scholar]
- 100.Fu L, Zhang C, Zhang LY, Dong SS, Lu LH, Chen J, Dai Y, Li Y, Kong KL, Kwong DL, Guan XY. Wnt2 secreted by tumour fibroblasts promotes tumour progression in oesophageal cancer by activation of the Wnt/beta-catenin signalling pathway. Gut. 2011;60:1635–1643. doi: 10.1136/gut.2011.241638. [DOI] [PubMed] [Google Scholar]
- 101.Ungefroren H, Sebens S, Seidl D, Lehnert H, Hass R. Interaction of tumor cells with the microenvironment. Cell Commun Signal. 2011;9:18. doi: 10.1186/1478-811X-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brabek J, Mierke CT, Rosel D, Vesely P, Fabry B. The role of the tissue microenvironment in the regulation of cancer cell motility and invasion. Cell Commun Signal. 2010;8:22. doi: 10.1186/1478-811X-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Leight JL, Wozniak MA, Chen S, Lynch ML, Chen CS. Matrix rigidity regulates a switch between TGF-beta1-induced apoptosis and epithelial-mesenchymal transition. Mol Biol Cell. 2012;23:781–791. doi: 10.1091/mbc.E11-06-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Avni R, Cohen B, Neeman M. Hypoxic stress and cancer: imaging the axis of evil in tumor metastasis. NMR Biomed. 2011;24:569–581. doi: 10.1002/nbm.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cannito S, Novo E, Compagnone A, Valfre di Bonzo L, Busletta C, Zamara E, Paternostro C, Povero D, Bandino A, Bozzo F, Cravanzola C, Bravoco V, Colombatto S, Parola M. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis. 2008;29:2267–2278. doi: 10.1093/carcin/bgn216. [DOI] [PubMed] [Google Scholar]
- 106.Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci USA. 2008;105:6392–6397. doi: 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kim HM, Haraguchi N, Ishii H, Ohkuma M, Okano M, Mimori K, Eguchi H, Yamamoto H, Nagano H, Sekimoto M, Doki Y, Mori M. Increased CD13 Expression Reduces Reactive Oxygen Species, Promoting Survival of Liver Cancer Stem Cells via an Epithelial-Mesenchymal Transition-like Phenomenon. Ann Surg Oncol. 2011 doi: 10.1245/s10434-011-2040-5. [DOI] [PubMed] [Google Scholar]