

Abstract
YM155, a novel survivin suppressant, shows potent antitumor activity against various human cancers and is currently in phase II clinical trials. In this study, we investigated whether YM155 selectively inhibits survivin transcription. We hypothesize that inhibition of survivin transcription plays a role in YM155-mediated survivin inhibition. We found that YM155 inhibited survivin promoter activity, while it showed minimal inhibitory effect on four control gene promoters in transfection and luciferase activity assay experiments, indicating its selectivity. Transfection of various survivin promoter-luciferase constructs followed by luciferase assays revealed that the survivin core promoter (269 bp) plays a major role in YM155-mediated inhibitory effects. However, flow cytometry analysis indicated that inhibition of survivin promoter activity by YM155 is cell cycle-independent without G1 cell arrests. Electrophoretic mobility shift assays (EMSA) identified that YM155 abrogates nuclear proteins binding to the region of -149 to -71, in which Sp1 is a major candidate, and that YM155 treatment induces Sp1 re-subcellular localization without inhibiting its expression. Forced expression of Sp1 neutralized YM155-mediated downregulation of survivin promoter activity. Consistently, mutation of the identified Sp1 sites in the oligonucleotide probe diminished DNA-protein interactions in EMSA experiments, and mutation of the Sp1 sites in the survivin promoter-luciferase construct diminished survivin promoter activity. These findings indicate that YM155 inhibition of survivin expression is at least in part through its inhibition of survivin transcription by disruption of Sp1 interaction with the region of -149 to -71 in the survivin core promoter.
Keywords: YM155, the survivin promoter, cancer cells
Introduction
Survivin, a critical member of the inhibitor of apoptosis (IAP1) family proteins [1-3], is highly expressed in human cancer, but undetectable in most normal differentiated adult tissues [4]. Survivin is involved in the regulation of apoptosis and the control of cell cycle and mitosis [5-7], and may also play important roles in other pathophysiological processes [8,9], including its important role in cancer initiation [10] and angiogenesis [11-13], tumor progression, drug/radiation resistance [14], and tumor relapse [7,15]. For example, we recently demonstrated that inhibition of survivin expression by a small chemical molecule SN-38, the active form of irinotecan, enhances effectiveness of a MDR-sensitive compound T138067 [16], suggesting survivin is a drug resistant factor. Given its aberrant expression in cancer, its ability in anti-apoptosis and drug/radiation resistance, promotion of cancer cell proliferation, and its association with various clinicopathological parameters, survivin potentially is one of the most promising targets for cancer therapy. Furthermore, due to its relative absence within normal tissue, it has potential to have very limited toxicity.
YM155, a small chemical molecule survivin suppressant, was discovered via a high throughput screening of compound libraries using a survivin gene promoter-luciferase reporter as a biomarker. YM155 has unique chemical structure (Figure 1A) and shows robust anti-proliferative activities against various human cancer cells regardless of p53 status [17]. Continuous infusion of YM155 induces tumor regression without body weight loss in experimental animal models of human cancer xenografts [17], suggesting a relative low toxicity to normal tissues. Moreover, YM155 induces regression of established human hormone-refractory prostate tumor xenografts [18]. Currently, YM155 is actively studied in various preclinical models [18-26] and clinical trials [27-32]. Recent studies showed that YM155 appears to be a promising compound for drug combination treatment [24,33,34]. For example, Iwasa et. al. found that combination of YM155 with platinum compounds induced synergistic effects including the increase of apoptotic cells and caspase-3 activity, and delayed growth of NSCLC tumor xenografts in animal models greater than either treatment alone [24].
Figure 1.
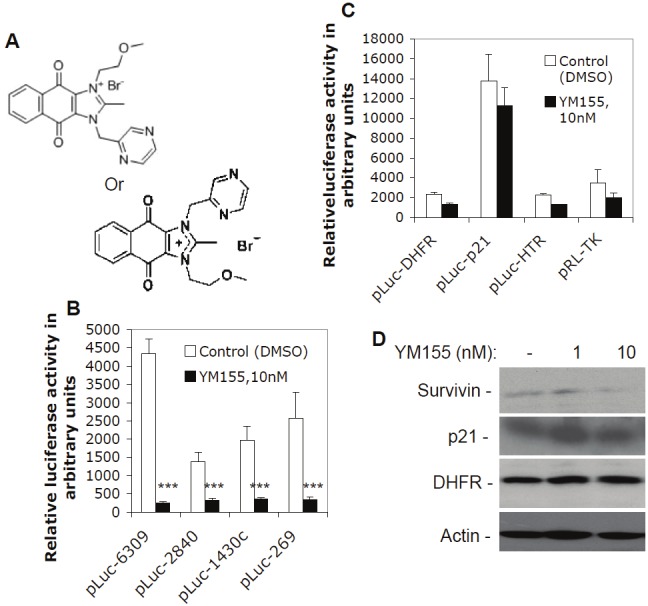
Selective inhibition of survivin promoter activity by YM155: A. Chemical structure of YM155. The two structural formats for YM155 have the same molecular weight (443.3). B. Subconfluent EKVX lung cancer cells were transfected with survivin promoter-luciferase constructs and treated with and without YM155 for 24 hours as shown, followed by a luciferase activity assay. C. Subconfluent EKVX cells were transfected with luciferase reporter constructs driven by promoters from the genes for DHFR, p21, HTR, and TK, followed by either no or YM155 treatment and luciferase activity assays as in B. Data are shown in histograms and each bar is the mean ± SD derived from three independent assays in panels B and C. D. YM155 inhibits endogenous survivin expression, but shows no inhibitory effects on the endogenous expression of p21 and DHFR. Subconfluent EKVX cells were treated with and without YM155 as shown for 24 hours. Cells were then lysed and analyzed by western blots using the corresponding antibodies. Actin was used as an internal control.
Based on the information above, it would be important to define whether inhibition of survivin transcription by YM155 contributes to YM155-mediated inhibition of survivin expression. Here, we report the first study on this topic. We show that inhibition of survivin promoter activity by YM155 is survivin gene-selective, is largely within the 269 bp survivin core promoter, and is cell cycle independent. Our data further shows that abrogation of binding of transcription factor Sp1 to region of -149~-71 in the survivin core promoter by YM155 plays a pivotal role in YM155-mediated suppression of survivin promoter activity.
Materials and methods
Cell culture and reagents
Cancer cell lines from lung (EKVX, A549, H3255, Hop62, H1650), prostate (PC-3, LNCaP-LN3 and PC-3m), and colon (HCT116) were maintained in RPMI1640, supplemented with 10% fetal bovine serum (FBS) (Mediatech Cellgro, Herndon, VA) and 100 units/ml of penicillin, and 100 μg/ml of streptomycin (Invitrogen, Carlsbad, CA). Cancer cell lines from ovarian (2008) and breast (MDA-MB-231) were maintained in DMEM, supplemented with 10% FBS, 100 units/ml of penicillin, and 100 μg/ml of streptomycin. Cells were maintained in a humid atmosphere incubator with 5% CO2 at 37°C and were routinely subcultured twice weekly. YM155 was provided by Astellas Pharma Inc., Japan. For EMSA supershift experiments, Sp1 antibodies (sc-14027x and sc-59) and normal IgG (sc-2027) were purchased from Santa Cruz (Santa Cruz, CA); Ap2 antibody (39304) and Ap2 inhibitory peptide (37504) were purchased from Active Motif (Carlsbad, CA). Propidium iodide (PI) and 4', 6-Diamidino-2-phenylindole (DAPI) were purchased from Sigma (St. Louis, MO). The Dual-Luciferase Reporter Assay System was purchased from Promega (Madison, WI). Restriction enzymes were bought from New England Bio-labs (Beverly, MA). LipofectamineTM 2000 reagents were purchased from Invitrogen (Carlsbad. CA) and Fugene HD transfection reagents were purchased from Roche (Indianapolis, IN).
Human survivin promoter-luciferase constructs
The human survivin promoter-luciferase constructs, pLuc-6270, pLuc-2840, and pLuc-1430c were characterized previously [35]; pLuc-6309 was cloned by addition of the 39 bp DNA sequence (GAA TCG CGG GAC CCG TTG GCA GAG GTG GCG GCG GCG GCA) from the “A’ in the translational start site (ATG) of the survivin gene to the 3’-end of the survivin promoter in pLuc-6270, and pLuc-269 (pLuc-cyc1.2) was characterized previously [36], which contains the 3’-end 269 bp sequence of the survivin promoter in the pLuc-6309 construct. Of note, the number in the pLuc vector is the bp of the promoter length used throughout this report.
YM155 treatment, plasmid transfection and luciferase reporter assay
Cells were treated with various concentrations of YM155, as shown in the corresponding experiments, in the presence of complete cell culture medium containing 10% FBS in all experiments. Cells were seeded in 48-well plates (2.5x104 per well) and grown to approximately 70% cell confluence before transfection. Each of the relevant survivin promoter-luciferase constructs generated previously [35], or in this study, was co-transfected with pRL-TK (TK promoter-Renilla-luciferase construct, internal control) into cells as follows: every 245 ng of a survivin reporter construct and 5 ng of pRL-TK in 100 μl serum-free DMEM was mixed with 100 μl serum-free DMEM containing 0.4 μl LipofectamineTM 2000. After incubation at room temperature for 20-25 minutes, the DNA/Lipofectamine 2000 mixture was added to each well of a 48-well plate 6 hours post-transfection. culture medium in each well containing DNA/Lipofectamine 2000 was substituted by fresh culture medium supplemented with YM155 to designated concentration. Cells were treated with YM155 for up to 24 hours followed by luciferase assays. For the luciferase assay, a Dual-Luciferase Reporter Assay System (Promega) was used. The transfected cells in 48-well plates were washed with PBS and lysed with 150 μl 1x passive lysis buffer on a shaker for up to 1 hour at 4°C. The firefly and Renilla luciferase activity was measured with 20 μl cell lysates and 20 μl reagents in each assay in triplicate using a Synergy 2 microplate reader (Biotek, Winooski, VT). Data were normalized to Renilla luciferase activity (internal control) as arbitrary units and plotted as a histogram from three independent experiments in triplicate.
Western blot analysis
Immuno-blotting analysis of protein expression was performed following our previous protocols [37]. Protein of interest was detected using Western Lightning-ECL (Perkin Elmer, Waltham, MA) and visualized by exposure with an appropriate time (5-60 seconds). Actin was used as an internal control.
Flow cytometry analysis of cell cycle distribution
Fluorescence-activated cell sorting (FACS) or flow cytometry was performed for analysis of cell cycle distribution during cell cycle progression following the previous protocol [37]. Subconfluent cells were treated with or without YM155 at various time points, then were harvested by trypsinization, and washed with PBS. Cells (~1x106) were resuspended in 5 ml of 70% ethanol. After the initial fixation, cells were resuspended in 0.5 ml PBS containing 25 μg/ml propidium iodide (PI), 0.2% Triton X-100, and 40 μg/ml RNase A, and incubated for at least 30 minutes at 4°C. Then cells were analyzed for immunofluorescence intensities using FACS (FACScan, Becton Dickinson, San Jose, CA) with 10,000 events per sample. Data from FACS were analyzed using WinList software (Verity Software House Inc., Topsham, ME).
Oligonucleotide-directed mutagenesis of the CDE motifs within the 269 bp survivin promoter-luciferase construct
Oligonucleotide-directed mutagenesis of the CDE motif in the 269 bp survivin promoter was carried out by PCR, followed by cloning mutated PCR fragments into pLuc vector, respectively. All mutated bases are in small letters and follow the role of G/T or C/A exchange at the mutated site, e.g. a “G” will be mutated to “T’ or vice versa. The 5’-end primers used for PCR are hscyc-1 (CGC GGATCC (BamH I) GCG TTC TTT GAA AGC AGT C), hR1m (CG GGATCC GCG TTC TTT tcA AGC AGT CGA GGG Gtg tCT AGG TGT G) and hR23m (CG GGATCC GCG TTC TTT GAA AGC AGT CGA GGG GGC GCT AGG TGT GGG CAG GGA CGA GCT GGa GaG GCG TCG CTG GtTt CAC CGC GAC CAC GGG CAG AGC CAC GCt tat GtA GGA CTA CAA C). The 3’-end primers used for PCR are hscyc-2 (CCC AAG CTT (Hind III) TGC CGC CGC CGC CAC CTC TG), hR4m (CCC AAG CTT TGC CGC CGC CGC CAC CTC TGC CAA CGG GTC CCG CGA TTC AAA TCT GGC GGT TAA TGG CGC GCC GCG GGG CAT GTC GGG AGC Gac aGC CCT CTT AGG CGG TCa cCC CCC ata GGC CTT CTG), hR5m (CCC AAGCTT TGC CGC CGC CGC CAC CTC TGC CAA CGG GTC CCG CGA TTC AAA TCT GGC GGT TAA TGG CGC tCC tCG GGG CAT), hR6m (CCC AAGCTT TGC CGC CGC CGC CAC CTC TGC CAA CGG GTC CCt CGA Tga AcA TCT GGC GG) and hR7m (CCC AAGCTT TGC atC CGC atC CAC CTC TG). PCR amplification with primer pairs of hscyc-1/hR4m, hscyc-1/hR5m, hscyc-1/hR6m, hscyc-1/hR7m, hR1m/hR4m, hR1m/hR5m, hR1m/hR6m, hR1m/hR7m, hR1m/hscyc-2, hR23m/hR4m, hR23m/hR5m, hR23m/hR6m, hR23m/hR7m and hR23m/hscyc-2, respectively, using the survivin pLuc-269 construct as DNA templates, resulted in 269 bp PCR products of 269hR4m, 269hR5m, 269hR6m, 269hR7m, 269hR5m, 269hR5m, 269hR14m, 269hR15m, 269hR16m, 269hR17m, 269hR1m, 269hR234m, 269hR235m, 269hR236m, 269hR237m and 269hR23m in the human survivin promoter, respectively. After digestion with BamH I/Hind III, these mutated PCR products were cloned into the pLuc vector at the BamH I/Hind III sites upstream of the luciferase reporter gene, respectively. The identified new constructs were sequenced and designated as pLuc-269hR4m, pLuc-269hR5m, pLuc-269hR6m, pLuc-269hR7m, pLuc-269hR5m, pLuc-269hR5m, pLuc-269hR14m, pLuc-269hR15m, pLuc-269hR16m, pLuc-269hR17m, pLuc-269hR1m, pLuc-269hR234m, pLuc-269hR235m, pLuc-269hR236m, pLuc-269hR237m and pLuc-269hR23m, respectively. These obtained CDE sites-mutated constructs were used for transfection followed by measuring luciferase activity in the presence and absence of YM155 treatment. The obtained data from YM155-untreated cells were divided by the data from YM155-treated cells as fold decrease. The wild type (pLuc-269) in the same condition was used as a positive control.
Determination of YM155 interaction with DNA using high percentage polyacrylamide gel via electrophoretic gel mobility shift assay (EMSA)
We performed the experiments as we did previously [38,39]. Briefly, to determine the possible direct interaction of SCPO-4 and SCPO-5 DNA elements with YM155, the [γ-32p]ATP-labeled SCPO-4 and SCPO-5 probes were mixed with and without different concentration of YM155 for 20 minutes at 25°C. Other the reactions were then run on a 15% instead of 5% non-denaturing polyacrylamide gel. Gels were peeled off with Waterman paper and covered by Saran wrap, followed by autoradiography to visualize the potential shift resulted from YM155-DNA interactions.
Isolation of cell nuclear extracts
The isolation of cell nuclear extracts was described in detail in our previous publications [38,39]. Briefly, cells treated with or without YM155 were scraped into ice-cold PBS and collected by centrifugation. Cell pellets were then gently resuspended with 3X cpv (cells pellet volume) of the ice cold buffer A, without TritonX-100, to wash the cells. Next, cells were pelleted gently and resuspended well in 3X cpv of ice cold buffer A with 0.1% TritonX-100 (w/v). The resuspended cells were allowed to swell for 12-15 min on ice. Cell nuclei were then collected by centrifugation (3440g) for 15 min at 2°C and resuspended in 1X pnv (packed nuclei volume) of ice cold buffer C. The cell suspension was incubated on ice (~2°C) for 30 min with continuous mixing. The cell nuclear extracts (supernatant) were collected by centrifugation at 25,000g for 30min at 2°C. The nuclear extracts were frozen in liquid nitrogen in aliquots (50-100 μl/1.5 ml tube) and stored at -80°C for gel shift experiments.
Electrophoretic gel mobility shift assay (EMSA)
The EMSA was performed following our previous protocol [38,39]. Briefly, DNA oligonucleotides labeled at the 5’-end with [γ-32P] ATP or by fill-in reaction with [α-32P] dCTP were used as probes. Nuclear extracts (4-10 μg) were pre-incubated in the binding buffer for 20 min at 25°C in a volume of 19 μl with and without unlabeled oligonucleotide competitors in the presence and absence of a relevant antibody (EMSA supershift assay). After addition of 1 μl labeled DNA (1-2x105 cpm), the mixture was incubated for an additional 20 min at 25°C. Each reaction mixture was then run on a 5% non-denaturing polyacrylamide gel and electrophoresed at 200V in 0.5xTBE buffer at 25°C for 1-2 hours. Gels were peeled off with waterman paper and covered by Saran wrap, followed by autoradiography to visualize the protein-DNA interaction complexes. Oligonucleotides used in EMSA experiments are summarized in Table 1.
Table 1.
Summary of oligonucleotides used in EMSA experiments.
| Name | DNA sequence (only forward chains are shown) | Position |
|---|---|---|
| SCPO1 | GCGTTCTTTGAAAGCAGTCGAGGGGGCGCTAGGTGTGGGCAGGGA | -267 to -222 |
| SCPO2 | GGGACGAGCTGGCGCGGCGTCGCTGGGTGCACCGCGACCACGGGCAGAG | -226 to -177 |
| SCPO3 | GGGCAGAGCCACGCGGCGGGAGGACTACAACTCCCGGCACACCC | -185 to -141 |
| SCPO4 | GCACACCCCGCGCCGCCCCGCCTCTACTCCCAGAAGGCCG | -149 to -109 |
| SCPO4m1 | GCACACCaaGCGCCGCCCCGCCTCTACTCCCAGAAGGCCG | |
| SCPO4m2 | GCACACCCCGatCCGCCCCGCCTCTACTCCCAGAAGGCCG | |
| SCPO4m3 | GCACACCCCGCGCCGCCaatCCTCTACTCCCAGAAGGCCG | |
| SCPO4m4 | GCACACCCCGatCCGCCaatCCTCTACTCCCAGAAGGCCG | |
| SCPO5 | GCCGCGGGGGGTGGACCGCCTAAGAGGGCGTGCGCTCCCGACATG | -113 to -67 |
| SCPO5m1 | GCCGatGGGGGTGGACCGCCTAAGAGGGCGTGCGCTCCCGACATG | |
| SCPO5m2 | GCCGCGttGGGTGGACCGCCTAAGAGGGCGTGCGCTCCCGACATG | |
| SCPO5m3 | GCCGCGGGttGTGGACCGCCTAAGAGGGCGTGCGCTCCCGACATG | |
| SCPO5m4 | GCCGCGGGGGGTGGACataaTAAGAGGGCGTGCGCTCCCGACATG | |
| SCPO5m5 | GCCGCGGGGGGTGGACCGCCTccGAtttCGTGCGCTCCCGACATG | |
| SCPO5m6 | GCCGCGGGGGGTGGACCGCCTAAGAGGGatTtaGCTCCCGACATG | |
| SCPO5m7 | GCCGCGGGGGGTGGACCGCCTAAGAGGGCGTGCGCTaaatACATG | |
| SCPO-6 | GACATGCCCCGCGGCGCGCCATTAACCGCCAGATTTGAAT | -74 to -34 |
| SCPO-7 | GAATCGCGGGACCCGTTGGCAGAGGTGGCGGCGGCGGC | -38 to +1 |
| Sp1 | ATTCGATCGGGGCGGGGCGAGC | N/A |
| Ap2 | GATCGAACTGACCGCCCGCGGCCCGT | N/A |
1) SCPO, Survivin Core Promoter Oligonucleotides. 2) Oligonucleotides for Sp1 and Ap2 binding are the consensus DNA sequences for Sp1 and Ap2. 3) SCPO4m1, Ap2 mutant oligo; SCPO4m2, left side Sp1 site mutant oligo; SCPO4m3, right site Sp1 site mutant oligo; and SCPO4m4, mutant oligo in both Sp1 sites. 4) SCPO5m1, Ap2 mutant 1; SCPO5m2, Ap2 mutant 2; and SCPO5m3, Ap2 mutant 3. Names in bold are critical mutations.
Immunofluorescence microscopy
The immunofluorescence experiment was performed following our previously established method. [40]. Briefly, EKVX cells were seeded on circular glass coverslips coated with 2% gelatin in 12-well plates. Cells grown to about 50% confluence were fixed with 4% paraformaldehyde in PBS and permeabilized with PBS containing 2% BSA and 0.2% Triton X-100, and then incubated in PBS containing 1% BSA, 0.2% Triton X-100, and anti-Sp1 antibody (1:500) for 60 minutes at 37°C. After washing with PBS, cells were incubated in PBS containing Alexa Fluor 594 goat anti-rabbit IgG antibodies for 45 minutes at room temperature, followed by staining with 4', 6-diamidino-2-phenylindole (DAPI) at a final concentration of 0.5 μg/ml in PBS for 10 min. Coverslips were then mounted on glass slides with Gel/MountTM solution (Biomedia, Foster City, CA). Cell images were digitally captured using a Zeiss Axiovert 100 M Fluorescence Microscopy System.
Systematic deletion of the survivin promoter-luciferase construct pLuc-269
Systematic truncation of the 269 bp survivin core promoter in the pLuc-269 was performed using a PCR approach followed by subcloning steps as follows. The 5’-end set of PCR primers are P257 (CGC GGATCC AAG CAG TCG AGG GGG, the BamH I site is underlined), P237 (CGC GGATCC GGT GTG GGC AGG GAC GA), P217 (CGC GGATCC GGC GCG GCG TCG CT), P197 (CGC GGATCC ACC GCG ACC ACG GG), P177 (CGC GGATCC CAC GCG GCG GGA GGA CTA C), P162 (CGC GGATCC CTA CAA CTC CCG GCA CAC), and P147 (CGC GGATCC CAC CCC GCG CCG CCC CGC CT). The 3’-end PCR primer is hscyc-2 (CCC AAGCTT TGC CGC CGC CGC CAC CTC TG, the Hind III site is underlined). PCR amplification with primer pairs of P257/hscyc-2, P237/hscyc-2, P217/hscyc-2, P197/hscyc-2, P177/hscyc-2, P162/hscyc-2, P147/hscyc-2 using the survivin 269 bp core promoter as a DNA template resulted in PCR products of 257 bp, 237 bp, 217 bp, 197 bp, 177 bp, 162 bp and 147 bp sizes of the human survivin promoter, respectively. After digestion with BamH I/Hind III, these PCR DNA fragments were subcloned into the pLuc vector at the BamH I/Hind III sites upstream of the luciferase reporter gene, respectively. These subclones were sequenced and designated as pLuc-257, pLuc-237, pLuc-217, pLuc-197, pLuc-177, pLuc-162 and pLuc-147, respectively.
Oligonucleotide-directed mutagenesis of the Sp1 sites within the 197 bp survivin promoter-luciferase construct
Generation of mutations of SCPO4m3 and SCPO5m6, alone and in combination in the survivin promoter-luciferase construct, was carried in the pLuc-197 construct via PCR and molecular cloning. This manipulation resulted in three new survivin promoter-luciferase constructs (pLuc197-4m3, pLuc197-5m6 and pLuc197-4m3/5m6) after molecular cloning of the mutated PCR products into pLuc reporter vector. Generation of pLuc197-4m3 used the primers of 197Sp1m (CG GGATCC ACC GCG ACC ACG GGC AGA GCC ACG CGG CGG GAG GAC TAC AAC TCC CGG CAC ACC CCG CGC CGC Caa tCC TCT ACT CCC AG, the BamH I site underlined) and hscyc-2 (CCC AAGCTT TGC CGC CGC CGC CAC CTC TG, the Hind III site underlined) for PCR; generation of pLuc197-5m6 used the primers of P197 (CGC GGATCC ACC GCG ACC ACG GG, the BamH I site underlined), 100PO5m6 (GCC GCC GCC GCC ACC TCT GCC AAC GGG TCC CGC GAT TCA AAT CTG GCG GTT AAT GGC GCG CCG CGG GGC ATG TCG GGA GCt aAa tCC CTC TTA GGC GGT C) to get an intermediate PCR product, which was in turn used as a DNA template for PCR using P197 and hscyc-2 primers to get the final PCR product. Generation of pLuc197-4m3/5m6 used the primers of 197Sp1m and hscyc-2 for PCR with pLuc197-5m6 as DNA templates. Mutated bases in the oligonucleotides above are shown in small letters. The resultant PCR products above were cloned into pLuc luciferase reporter vector at the BamH I and Hind III sites upstream of the luciferase reporter gene, respectively. The generated new survivin promoter-luciferase constructs with the defined mutations were confirmed by sequencing.
Statistical analysis
A t-test was performed for data comparison with corresponding controls shown in Figures 1 and 11. The statistical probability value (p-value) of 0.05 or less was considered to be significant. The p-value at different significance levels was expressed as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
Figure 11.
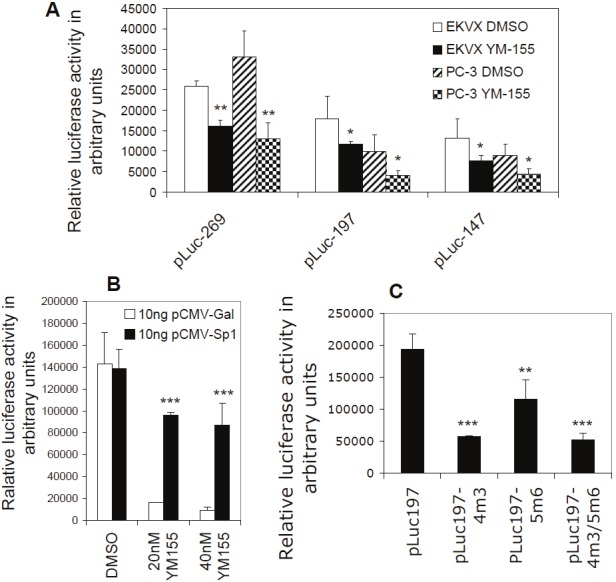
Functional analysis of the Sp1 sites in the SCPO4 and SCPO5 DNA element in YM155-mediated inhibition of survivin promoter activity: A. Luciferase activity assays suggest the most proximate area in the survivin promoter plays a critical role in mediation of YM155’s inhibitory effect on survivin gene activity. Subconfluent EKVX and PC-3 cells were transfected overnight with the survivin core promoter-luciferase constructs with the indicated length as shown. Cells were then treated with YM155 or DMSO (control) for 24 hours, followed by the luciferase activity assay. B. Forced expression of Sp1 neutralized YM155-mediated inhibition of survivin promoter activity. Subconfluent PC-3 cells were cotransfected with both pLuc-269 and the Sp1 expression vectors or control vectors overnight. Cells were then treated with YM155 or DMSO (control) for 24 hours as shown, followed by luciferase activity assay. C. Mutation of the identified Sp1 sites diminished survivin promoter activity. PC-3 cells were transfected with wild type (pLuc197) or Sp1 site-mutated pluc197 alone (4m3, 5m6), or in combination (4m3/5m6) as shown. The transfected cells were grown in complete cell cultural medium for 16 hours, followed by serum starvation treatment for 24 hours. Cells were then lysed for the luciferase activity assay. Data in A, B, and C were normalized with the internal Renilla luciferase activity (internal control). The resultant data are presented in histograms as arbitrary units. Each bar is the mean ± SD derived from three independent assays run in triplicate.
Results
The 269 bp core survivin promoter mediates a major inhibitory effect of YM155 on survivin transcription control, which is a gene-selective event
YM155 was discovered via high throughput screening of in-house chemical libraries using the survivin gene promoter-luciferase reporter system as a biomarker. The chemical structure is shown in Figure 1A. Previous studies indicated that YM155 inhibits both survivin protein and mRNA expression at concentrations as low as 10 nM [18]. To determine the potential inhibitory effect of YM155 on survivin transcription, we explored the effect of YM155 on survivin promoter activity at a low concentration (10 nM) and short time treatment (24 hours), at which there is no significant cell death. We found that YM155 at a 10 nM concentration effectively downregulates survivin promoter activity (Figure 1B). The 269 bp survivin core promoter region in the pLuc-269 construct showed a response as effective as those derived from the constructs with longer survivin promoter sequences (pLuc-6309, 2840 and 1430, Figure 1B). This result suggests that the 269 bp survivin core promoter mediates major (if not all) inhibitory effects of YM155 on survivin gene transcriptional control.
To determine whether downregulation of survivin promoter activity by YM155 is a survivin gene-selective event, we employed four available luciferase constructs driven by promoters for the genes of dihydrofolate reductase (DHFR), human thrombin receptor (HTR), thymidine kinase (TK), and CDK inhibitor p21WAF1/CIP1 (p21), to test YM155’s effect on their promoter activity. Transfection of these constructs, followed by YM155 treatment and luciferase activity assays, revealed that YM155 only exerts minimal inhibitory effects on promoter activity of these genes (Figure 1C), suggesting YM155’s selectivity in nature (see discussion). Consistent with these observations, YM155 inhibited endogenous survivin expression without showing inhibitory effects on the expression of endogenous p21 and DHFR (Figure 1D).
Inhibition of survivin promoter activity by YM155 is not associated with a G1 cell cycle arrest
It is known that survivin expression is very low at the G1 phase, increases in the S phase, and comes to its highest expression at the G2/M phase of the cell cycle [36]. To determine the possibility that inhibition of survivin transcription by YM155 actually resulted from YM155-mediated G1 arrest, we investigated cell cycle distribution after YM155 treatment. Surprisingly, FACS experiments using PC-3 (less sensitive to YM155, Figure 2A) and EKVX (more sensitive to YM155, Figure 2B) indicated that YM155 does not result in a G1 cell arrest (Table 2). Instead, treatment of cancer cells with YM155 over time increased sub-G1 portion (death cells) without G1 arrest (Figure 2 and Table 2), suggesting that inhibition of survivin by YM155 was not due to a G1 cell arrest. Additional experiments from FACS analysis with other cell lines including 2008, Hop62, H1650, A549, H3255, MDA-MB-231, LNCaP-LN3, PC-3m, and HCT116 showed similar results. Accordingly, transfection of cells with various cell cycle-dependent elements (CDE)-mutated survivin promoter-luciferase constructs, followed by luciferase activity assays, showed that CDE motifs could not explain the major inhibitory effect of YM155 on survivin transcription, since after mutation of these motifs in the survivin promoter-luciferase constructs YM155 is still able to significantly inhibit survivin promoter activity (Figure 3). These observations indicated that inhibition of survivin gene transcription by YM155 is relatively a G1 cell cycle arrest-independent event and that induction of cell death by YM155 appears to be associated with G2/M phase cell reduction, rather than with G1 cell cycle arrest.
Figure 2.
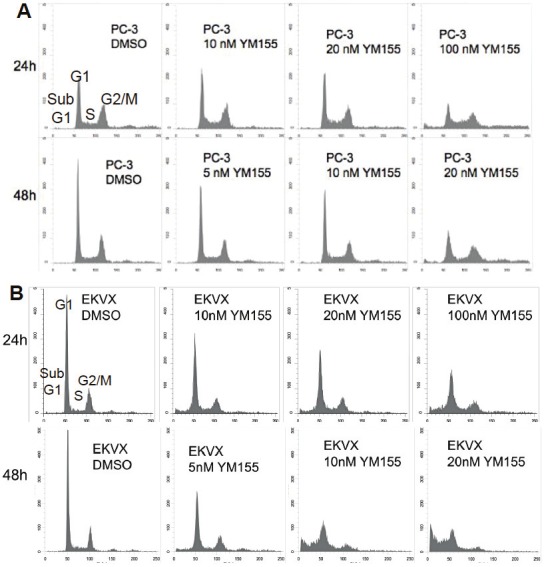
YM155 exerts minimal effects on cell cycle distribution: Subconfluent PC-3 prostate (A) or EKVX lung (B) cancer cells were treated with or without YM155 for 24 and 48 hours as shown. Cells were then harvested for flow cytometry (FACS) analysis as described in the Materials and Methods. Cell phases (sub-G1, G1, S, G2/M) were labeled on the first sub-panel image, and the percentage of each cell phase distribution in each condition was provided in the Table 2. Similar results from 2008, Hop62, H1650, A549, H3255, MDA-MB-231, LNCaP-LN3, PC-3m, and HCT116 were obtained (not shown).
Table 2.
Cell cycle distribution after YM155 treatment.
| Cell lines | Treatment | Cell cycle distribution (%) | |||
|---|---|---|---|---|---|
|
| |||||
| Sub-G1 | G1 | S | G2/M | ||
| PC3 | DMSO(0.1%),24hrs | 1.09 | 29.95 | 14.91 | 54.42 |
| 10nM YM155,24hrs | 0.91 | 31.13 | 21.04 | 47.74 | |
| 20nM YM155,24hrs | 1.54 | 31.99 | 18.09 | 48.66 | |
| 100nM YM155,24hrs | 6.45 | 15.13 | 18.25 | 58.70 | |
| DMSO(0.1%),48hrs | 0.54 | 42.13 | 12.67 | 45.09 | |
| 5nM YM155,48hrs | 0.85 | 43.35 | 13.38 | 42.64 | |
| 10nM YM155,48hrs | 1.18 | 33.92 | 17.57 | 47.81 | |
| 20nM YM155,48hrs | 4.02 | 18.53 | 19.40 | 57.38 | |
|
| |||||
| EKVX | DMSO(0.1%),24hrs | 1.47 | 60.23 | 7.09 | 31.22 |
| 10nM YM155,24hrs | 8.42 | 51.35 | 10.37 | 29.92 | |
| 20nM YM155,24hrs | 7.73 | 49.61 | 10.42 | 32.19 | |
| 100nM YM155,24hrs | 13.57 | 42.01 | 13.32 | 31.23 | |
| DMSO(0.1%),48hrs | 1. 34 | 61.60 | 9.15 | 27.90 | |
| 5nM YM155,48hrs | 8.20 | 45.08 | 9.53 | 37.19 | |
| 10nM YM155,48hrs | 25.63 | 37.81 | 12.85 | 23.87 | |
| 20nM YM155,48hrs | 41.09 | 31.41 | 10.49 | 17.06 | |
Figure 3.
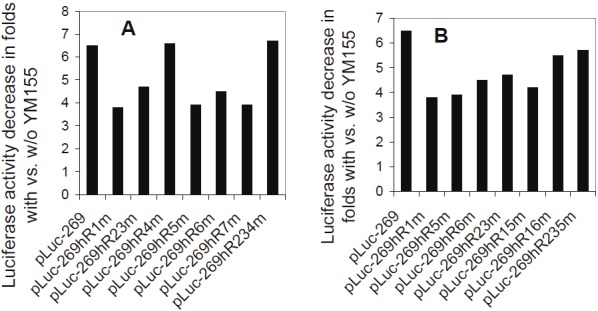
The CDE motifs in the survivin core promoter do not play a major role in YM155-mediated inhibition of survivin promoter activity: Subconfluent EKVX cells were transfected with the wild type pLuc-269 construct (control) or with pLuc-269 constructs containing various mutations in CDE motifs for overnight. Cells were then treated with and with YM155 (10 nM) for 24 hours, followed by cell lysis and luciferase activity assays. The data derived from YM155-untreated cells in triplicate were divided by the data derived from YM155-treated cells in triplicate as relative fold decrease. Each bar is the fold decrease from the mean data of comparable constructs in triplicates shown in A and B. Variations from both YM155 treated and untreated assays before division were within 15%.
YM155 significantly modulates DNA-protein interactions at the region from -185 to -67 in the survivin core promoter
We showed that the 269 bp survivin core promoter plays a major role in YM155-mediated inhibition of survivin promoter activity (Figure 1). However, YM155 did not significantly affect cell cycle distributions (Figure 2 and Table 2) and mutation of CDE motifs could not explain the major inhibitory effect of YM155 inhibition of survivin promoter activity (Figure 3). To identify the DNA elements on which the DNA-protein interactions could be significantly modulated by YM155 treatment, we systematically investigated using electrophoresis mobility gel shift assays (EMSA) DNA-protein interaction profiles of the survivin core promoter region (269 bp) using seven contiguously overlapping oligonucleotide probes (termed SCPO-1, 2, 3, 4, 5, 6 and 7) that cover the entire 269 bp region (Table 1). EMSA analysis revealed that YM155 significantly modulates DNA-protein interactions with the probes of SCPO-4 and 5 (Figure 4A and 4B - lanes 2 versus 3, and 8 versus 9). Using consensus DNA oligonucleotides for Sp1 and Ap2 transcription factors as cold competitors, we demonstrated that while 50x non-specific/scramble DNAs failed to compete the DNA-protein complexes (not shown), 10x Sp1-specific oligonucleotides strongly competed SCPO4-protein complexes (Figure 4B, lanes 2 versus 6), and both 10x Sp1 and Ap2 oligonucleotides appear to compete against SCPO5-protein complexes (Figure 4B, lanes 8 versus 10 and 12). We further determined the Ap2/Sp1-irrelevant proteins in the SCPO4-protein complex using EMSA experiments (Figure 5). Our studies indicated that the unknown protein could be a LyF-1 relevant protein. Specifically, as shown in the Figure 5A, the LyF-1 DNA motif could be a candidate [41]. Our EMSA experiment using SCPO4R21 as a DNA probe indicated that while the murine LyF1 DNA motif appears to slightly inhibit the SCPO4R21 DNA-protein complex, neither SCPO4L19 and the consensus Sp1 DNA motif could inhibit the SCPO4R21 DNA-protein complex (Figure 5B), suggesting that the protein in the Ap2/Sp1-irrelevant SCPO4-protein complex (shown in Figure 4B Lane 6) may be a LyF1-like protein. However, due to unavailability of the EMSA-qualified human LyF1 antibody, we were unable to further confirm it using EMSA gel supershift.
Figure 4.
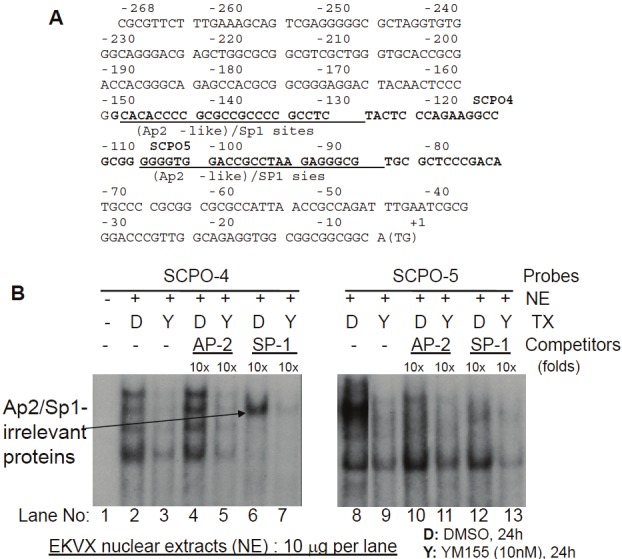
Both consensus Sp1 and Ap2 DNA-binding oligonucleotides can compete the DNA-protein complexes formed on SCPO-4 (-149 to -109) and SCPO-5 (-113 to -67) probes: A. Relevant Ap2-like and Sp1 DNA-binding sites in SCPO-4 and SCPO-5 (bold) in the human survivin 269 bp core promoter are underlined. Of note, the AT rich area appears to be a weak Ap2 binding site. B. EMSA experiments were performed as described in the Methods section. Nuclear extracts (NE) were isolated from EKVX cells treated with and without YM155, and mixed with 32P-labelled SCPO-4 or SCPO-5 probes in the presence and absence of consensus cold Sp1 or Ap2 binding motif oligonucleotides as shown. DNA/protein complexes were separated on a 5% non-denatured PAGE gel and visualized by autoradiography.
Figure 5.
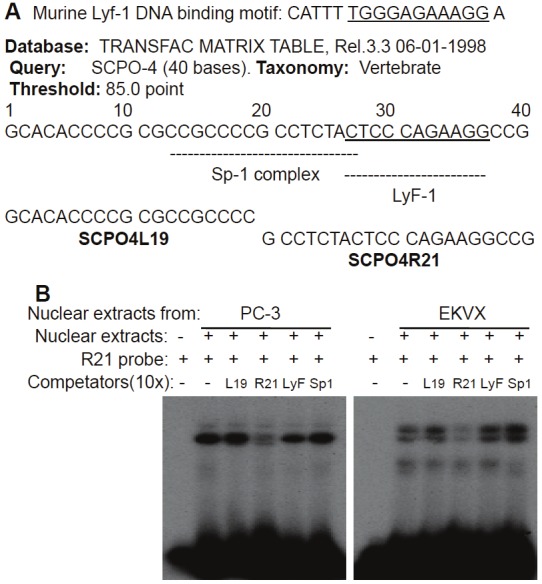
Characterization of the YM155-inhibited but Ap2/Sp1-irrelevant protein: A Diagram of the Sp1 and LyF1 DNA-bindingmotifs in the 40 bp SCPO-4 DNA element. The corresponding DNAoligonucleotides (SCPO4L19 and SCPO4R21) synthesized forEMSA experiments and the synthesized murine LyF1 DNA-bindingmotif are shown. B EMSA results derived from using SCPO4R21(R21) as a probe in the presence and absence of various cold competitors:SCPO4L19 (L19), SCPO4R21 (R21), murine LyF1 DNAelement (LyF) and the consensus Sp1 DNA-binding motif (Sp1) areshown.
The Sp1 transcription factor is a major component in the DNA-protein complexes formed on SCPO4 and SCPO5
To confirm the presence of Sp1 and Ap2 transcription factors in the DNA-protein complexes on SCPO-4 and 5 probes, EMSA supershift experiments with Sp1 and Ap2 antibodies were performed. The results indicated that the Sp1 antibody induces a strong supershift band in both SCPO-4 and 5 (Figure 6, lanes 3 and 7). However, to our surprise, while the Ap2 antibody could supershift a band with the consensus Ap2 oligo-protein complex (Figure 7A, lanes 6 and 10), Ap2 antibodies failed to result in a supershifted band with the DNA-protein complex from the SCPO-5 probe (Figure 7B, lanes 5, 6 and 7), even under further optimized supershift conditions (Figure 7C, lanes 3 and 9), although Ap2 consensus oligonucleotides could compete the band on SCPO-5 (Figure 4B, lanes 8 versus 10). In contrast, experiments with the Sp1 antibody consistently showed supershift bands with SCPO-4 or SCPO-5 in nuclear extracts isolated either from PC-3 or EKVX cancer cell nuclear extracts (Figure 6, lanes 3 & 7; and Figure 7B, lanes 8, 9 & 11; 7C, lanes 5 & 11).
Figure 6.
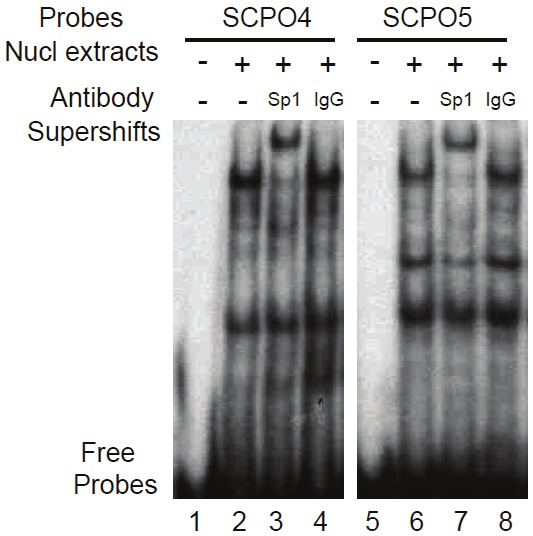
EMSA supershift experiments indicate that Sp1 is a major component in the DNA-protein complex in SCPO-4 and SCPO-5 in the absence of YM155: The presence and absence of relevant antibodies, nuclear extracts, and DNA probes used are indicated. Nuclear extracts were isolated from PC-3 cells without YM155 treatment in order to observe the maximal DNA-protein interaction.
Figure 7.

The Ap2 antibody failed to supershift a band in the SCPO5-protein complex (B and C), while it supershifted astrong band for the consensus Ap2 probe-protein complex (A): EMSA procedures were described in the Methods. A.Positive controls for EMSA supershift experiments. B and C. Two different EMSA supershift experimental conditionsfor the SCPO-5 probe were used. Both conditions produced a strong supershift band by Sp1 antibodies, but not Ap2antibodies. p: PC-3 cell nuclear extracts; e: EKVX cell nuclear extracts; d: DMSO; a: Ap2 antibody; a/pe: Ap2 antibodywith Ap2 inhibitory peptides; s: Sp1 antibody; Ab: antibody; nucl: nuclear; and IgG: normal IgG. D. YM155 does notdirectly bind to the SCPO-4 and SCPO-5 DNA elements. The probes, the presence and absence of YM155 in mM areindicated. YM155 interaction with SCPO-4 and SCPO-5 DNA were determined using 15% gel instead of 5% for DNA-druginteraction. D: DMSO solution (control for YM155).
We previously demonstrated that modulation of survivin expression by hedamycin and hoechst33342 is associated with ligand directly binding to the involved DNA elements identified [38,39]. To determine whether YM155 directly binds SCPO-4 and/or SCPO-5 DNA, we determined the direct interaction of YM155 with SCPO-4 and SCPO-5 using a high percentage gel shift assay established previously [38,39]. Our results revealed that YM155 does not induce a gel shift for SCPO-4 or SCPO-5 DNA probes (Figure 7D), indicating that YM155 does not bind to these two DNA elements.
To further determine which of the two overlapped Sp1 sites in SCPO-4 and SCPO-5 (refer to Figure 4A) is involved in the DNA-protein interaction, we designed a series of mutant SCPO-4 and 5 oligonucleotides on Sp1 (refer to Table 1). Application of these mutant oligonucleotides as competitors in EMSA experiments revealed that in the SCPO-4 element, mutation of the right Sp1 site (Figure 8A, lanes 11-14) resulted in the failure of effective competition of the DNA-protein complexes, while mutation of the Ap2-like or Sp1 sites on the left side does not change its wild-type nature (Figure 8A, lanes 7-10). Thus, we identified SCPO4m3 is a critical base mutation for control of DNA-Sp1 protein interaction (Table 1). Similarly, mutation of the Ap2-like site on the left site using three different ways (refer to Table 1) in the SCPO-5 element showed that their competition ability is as effective as the wild-type SCPO-5 oligonucleotides (Table 1 and Figure 8B, lanes 6-11). These results are consistent with the observation that Ap2 antibodies failed to supershift the DNA-protein complexes formed on SCPO-4 and/or SCPO-5 (Figure 7B and 7C).
Figure 8.
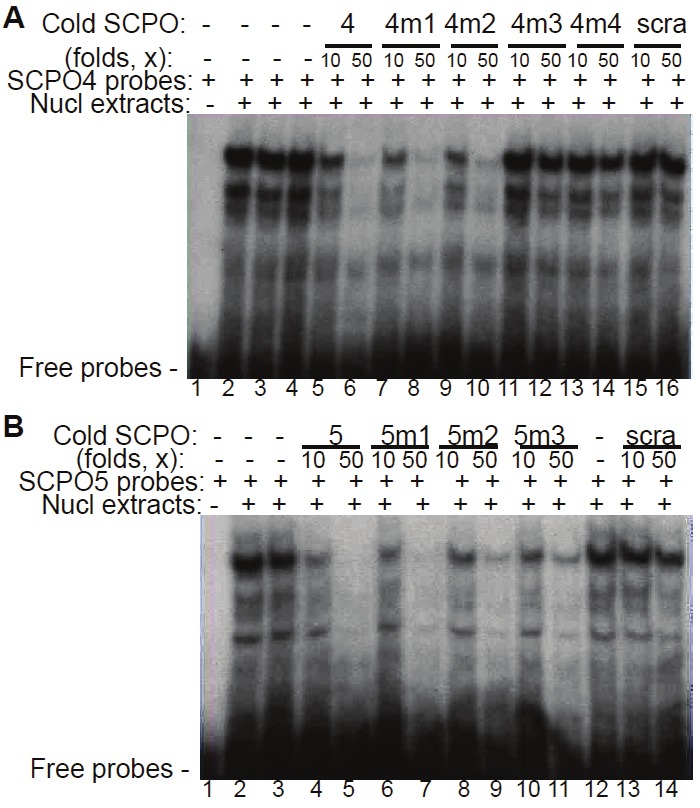
Identification of critical bases in the SCPO4 DNA probe: Oligonucleotides with bp mutations in SCPO-4 and 5 were used as cold competitors in EMSA experiments A. Comparison of the competition ability between wild-type (wt) SCPO-4 and its mutants (SCPO-4m1, m2, m3 and m4) in competitive EMSA using nuclear extracts isolated from PC-3 cells. B. Comparison of the competition ability between wt SCPO-5 and its mutants (SCPO-5m1, m2 and m3) in competitive EMSA using nuclear extracts isolated from PC-3 cells. scra: scramble DNA; 4: SCPO4; 4m1: SCPO4m1; 4m2: SCPO4m2; 4m3: SCPO4m3; 4m4: SCPO4m1; 5: SCPO5; 5m1: SCPO5m1; 5m2: SCPO5m2; 5m3: SCPO5m3.
Given that Ap2 showed inhibitory effects on survivin expression [42], we hypothesized that Ap2 might bind to the Ap2 binding sites in SCPO-4 and 5 only after YM155 treatment. Therefore, we did similar competitive EMSA by using nuclear extracts isolated from YM155-treated cells. However, the results obtained from these EMSA experiments did not support the above hypothesis, instead it is consistent with the conclusion derived from the data shown in the Figure 8A and 8B. Specifically, SCPO-4 oligonucleotides with mutations in the Ap2-like site acted like a wild-type SCPO-4 using nuclear extracts isolated either from EKVX or from PC-3 cells. Similarly, SCPO-5 oligonucleotides with mutations in the Ap2-like site acted as wild-type SCPO-5 in YM155-treated nuclear extracts isolated either from EKVX or from PC-3 cells (data not known).
To further identify critical bases in the SCPO-5 Sp1 site, additional mutations on SCPO-5 (m4 to m7) were generated (Table 1). Using these mutated SCPO-5 as cold competitors for additional EMSA experiments, the resultant data were not very clear, but it appeared that the bases mutated in the SCPO5m6 weakened the ability to compete the SCPO5-protein complex either from YM155-treated (Figure 9A) or untreated (Figure 9B) nuclear extracts.
Figure 9.

Identification of critical bases in the SCPO-5 DNA probe: EMSA experimental procedures were described in the Methods section. DNA probes, nuclear extracts, YM155-treated status and cold DNA oligonucleotide competitors are indicated. A. Using nuclear extracts isolated from YM155-treated PC-3 cells for EMSA competition experiment. B. Using nuclear extracts isolated from DMSO-treated PC-3 cells for EMSA competition experiment. Nucl: nuclear; d: DMSO; scramb: scramble DNA; 5m4: SCPO5m4; 5m5: SCPO5m5; 5m6: SCPO5m6; and 5m7: SCPO5m7.
YM155 treatment induces Sp1 subcellular relocalization without affecting Sp-1 protein expression
We demonstrated that Sp1 is the major transcription activator in the DNA-protein complex formed on SCPO4 and SCPO5 (Figures 6 and 7), and that YM155 treatment abrogates the DNA-protein interaction complex formed on SCPO4 and SCPO5 (Figure 4B). To explore the underlying molecular mechanism for this, we further determined whether YM155 treatment of cancer cells could inhibit Sp1 expression and/or induce the change of its subcellular localization. Our data revealed that while YM155 is unable to inhibit Sp1 protein expression (Figure 10A), YM155 treatment of cancer cells induces Sp1 protein subcellular re-localization (Figure 10B). Thus, the observation provides a molecular mechanism by which YM155 inhibits survivin promoter transcription via dissociating Sp1 proteins from its DNA binding sites and inducing Sp1 protein subcellular re-localization to avoid the survivin promoter-binding Sp1 accessing its survivin DNA binding sites.
Figure 10.
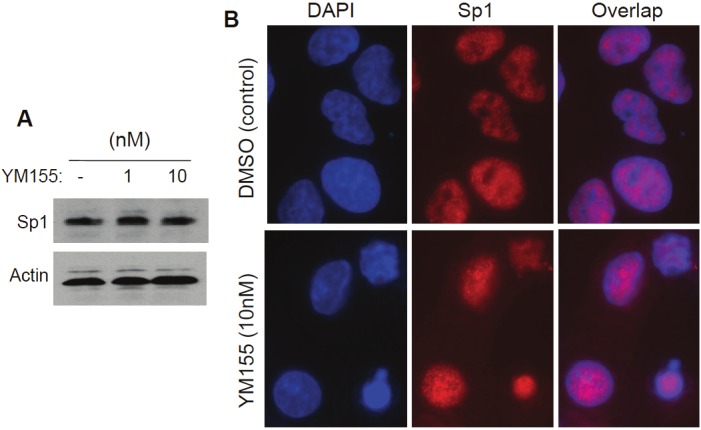
Sp1 protein expression and subcellular localization with and without YM155 treatment: A. YM155 showed no inhibitory effects on Sp1 expression. Subconfluent EKVX lung cancer cells were treated with and without YM155 as shown for 24 hours. Cells were then lysed and analyzed by western blots using Sp1 antibodies. Actin expression is the internal control. B. YM155 treatment induces a re-localization of Sp1 proteins in the subcellular structure of the cell. Sub-confluent EKVX cells grown on circular glass coverslips were treated with YM155 for 12 hours and then processed for immunofluorescence microscopy using DAPI and Sp1 double staining as described in the Methods section. Cell images were digitally captured under a Zeiss Axiovert 100 M Fluorescence Microscopy System.
Functional analysis suggests a role for Sp1 and its binding sites in YM155-mediated inhibition of survivin transcription in the survivin core promoter region
To determine whether the SCPO-4 and SCPO-5 functionally play a role in YM155-induced downregulation of survivin promoter activity, we generated truncated constructs in pLuc-269 and transfected the newly generated survivin-promoter constructs into EKVX and PC-3 cells, followed by treatment with YM155 or DMSO (control) and luciferase activity assays. These experiments indicated that the proximal region (equivalent to SCPO-4 and SCPO-5) of the 269 bp survivin promoter is an essential region for YM155-mediated inhibition of survivin promoter activity (Figure 11A), which is consistent with the results derived from systematic EMSA experiments (Figures 4, 5, 6 and 7). Furthermore, consistent with a role for Sp1 in constitutive expression of survivin [35], forced expression of Sp1 abrogated YM155-mediated inhibition of survivin promoter activity (Figure 11B). Accordingly, mutation of the two identified Sp1 sites in the survivin promoter-luciferase construct, either alone or in combination, significantly diminished survivin promoter activity (Figure 11C). However, YM155 treatment could further decrease the promoter activity in the Sp1-mutated survivin promoter-luciferase constructs (not shown, see discussion), suggesting additional sites may be involved. Nevertheless, in consideration of the fact that inhibition of survivin promoter activity by YM155 is a survivin gene-selective event (Figure 1), the selective role of Sp1 on survivin gene transcription upon YM155 treatment must involve additional transcription factors or co-factors to realize the survivin gene-selective event involving Sp1, which will be of interest for future research.
Discussion
While it was recognized as one of the most important targets for cancer treatment, survivin is a multifunctional molecule with unique multi-subcellular localizations. Its expression is involved in both inhibition of apoptosis [36,43] and regulation of mitotic cell division [44-47]. Follow up studies reveal new roles for survivin in the promotion of the G1/S transition [48] and the regulation of gene transcription [49], and possible involvement in cancer stem cell biology [50]. Previous studies showed that YM155 downregulates survivin mRNA and protein expression [18]. It is possible that YM155-mediated downregulation of survivin expression is a secondary effect. For example, YM155 may induce cancer cells into a G1 arrest status and accordingly, downregulate survivin expression. This is possible because it is known that survivin expression is very low in the G1 phase, its expression increases in the S phase, and comes to its highest expression in the G2/M phase [36]. To unravel the mechanism by which YM155 inhibits survivin expression, we investigated whether YM155 inhibits survivin promoter-driven luciferase activity and whether inhibition of survivin promoter-driven luciferase activity by YM155 is associated with cancer cell G1 arrest. The data from these experiments indicated that YM155 strongly inhibits survivin promoter activity and endogenous protein expression without association with cancer cell G1 phase arrests (Figures 1 and 2, Table 2). Furthermore, the CDE motifs in the survivin promoter seem only to play a minor role in YM155-mediated inhibition of survivin transcription (Figure 3). Together, our data suggested that downregulation of survivin gene expression by YM155 was a cell cycle-independent event without G1 cell arrest. Furthermore, YM155-induced cancer cell death was also not associated with G1 cell cycle arrest.
To determine whether inhibition of survivin promoter activity by YM155 is survivin gene-selective, we employed four available human promoters for the genes of DHFR, p21, HTR, and TK to determine whether YM155 modulates any of these gene promoter-driven luciferase activities. Our results indicate that none of the promoter activity for these genes was significantly modulated by YM155 (Figure 1C). Consistently, the endogenous protein expression of p21 and DHFR was also not inhibited by YM155 (Figure 1D). These observations suggest that inhibition of survivin promoter activity by YM155 is a survivin gene-selective event but not due to cancer cell G1 arrest. Of note, although the selection of these genes for the experiment is largely dependent on the availability, it should be pointed out that similar to the survivin gene promoter, the HTR gene promoter is known to lack a TATA box with a G/C rich sequence, and there are many Sp1 DNA-binding sites in its core promoter [51]. However, our result revealed that YM155 showed a much weaker inhibitory effect on HTR promoter activity (Figure 1C), as compared with its inhibitory effects on the survivin gene promoter (Figure 1B).
Interestingly, our data showed that YM155-mediated modulation of DNA-protein interactions in the SCPO-4 and SCPO-5 DNA elements was largely attributed to the Sp1 transcription factor rather than Ap2 (Figures 4, 6 and 7). However, it is known that Sp1 is largely a ubiquitous transcription factor involved in the constitutive expression of many important survival genes with a G/C rich promoter. Therefore, the gene-selective regulation of survivin transcription by Sp1 via the SCPO-4 and SCPO-5 region on the Sp1 sites must be implicated in other transcription factors or co-factors to the corresponding survivin promoter after YM155 treatment. Consistent with this speculation, our new data indicated that while YM155 is unable to inhibit Sp1 protein expression (Figure 10A), YM155 treatment redistributes Sp1 protein in the subcellular structure of the cell (Figure 10B). The subcellular re-localization of Sp1 proteins after YM155 treatment could therefore exclude survivin promoter-binding Sp1 proteins to interact with the survivin promoter. Of note, in addition to Sp1, other protein factors may also be involved. This notion comes from the observation that Sp1 antibody appears to supershift part of the specific bands formed on SCPO-4 and SCPO-5 (Figures 6 and 7). However, YM155 treatment abrogated all the specific bands (Figure 4B). A typical example is that there is a band shown in the Figure 4B (Lane 6) that could not be competed by cold Sp1 consensus oligonucleotides. To determine the identity of the unknown protein in this DNA-protein complex band, we re-analyzed the 40-bp SCPO4 sequence using a threshold of 85% instead of 90%; we found that at this threshold setting, in addition to the Sp1 complex, there is a LyF1 (Ikaros) binding site beside the Sp1 complex on the right half of the 40 bp SCPO4 element (Figure 5A). Using the SCPO4R21 as a probe in EMSA experiments, in the presence and absence of various cold DNA competitors (including SCPO4L19, SCPO4R21, murine LyF1 and consensus Sp1 DNA oligonucleotides), we demonstrated that the gel shift band could be competed with cold SCPO4R21, but not SCPO4L19, suggesting its high specificity. Moreover, the band appears to be weakly competed by the murine LyF1 DNA oligonucleotides (Of note, there is no consensus human or mouse LyF1 DNA-binding element available in literature due to the lack of examples), but not by the consensus Sp1 DNA-binding oligonucleotides, suggesting that the unknown band could be the human LyF1 protein or a LyF1-like protein (Figure 5B). Nevertheless, these observations place room for further investigation of the mechanism for Sp1 involvement in YM155-selected inhibition of survivin transcription.
Additionally, we found that forced expression of Sp1 neutralized YM155-mediated inhibition of survivin promoter activity (Figure 11B), and that mutation of the Sp1 sites diminished survivin promoter activity (Figure 11C). These findings strongly indicate that Sp1 plays a pivotal role in YM155-mediated inhibition of survivin promoter activity. These findings are consistent with previous studies from our and several other research groups, which showed that Sp1 plays an important role in the constitutive expression of survivin [35,38,52-55]. However, while Sp1 is involved in survivin gene expression, under full growth conditions (Subconfluent cells with 10% FBS) used in this study, we failed to detect a significant increase in survivin promoter activity after Sp1 overexpression. This is consistent with our previous observation that the optimal condition for Sp1 to increase survivin promoter activity is to starve or growth arrest cells before overexpression of Sp1 (unpublished observations). The potential reason is that the endogenous Sp1 protein may already be saturated for binding to its sites on the survivin core promoter region under full growth conditions with 10% serum. However, in this particular experiment, we did not apply this starvation approach because this would make cells more sensitive to YM155 treatment-induced cell death, which could make the results more difficult for interpretation. Nevertheless, in the defined full cell growth condition, YM155’s inhibitory effects on survivin promoter activity was neutralized by forced expression of Sp1 (Figure 11B), indicating a role for Sp1 in YM155-mediated inhibition of survivin transcription. However, one question is whether mutations on the Sp1 binding sites could partially abolish YM155-mediated inhibition of the survivin promoter activity. We carefully determined the possibility and found that while mutation of the Sp1 sites alone or in combination decreases survivin promoter activity (Figure 11C), the mutated survivin promoter-luciferase constructs did not show significantly weaker ability in folds for the YM155-mediated downregulation of survivin promoter activity, although the promoter activity was much weaker overall. While this would need further studies to elucidate its mechanism, the data shown in Figure 10 revealed a potential novel mechanism for Sp1 being involved in YM155-mediated inhibition of survivin promoter activity. As mentioned above, while YM155 treatment abrogates Sp1 protein binding on the Sp1 site in SCPO-4 (major) and in SCPO-5 (minor) (Figures 4, 5, 6 and 7), the Sp1 protein expression level was not affected by YM155 treatment (Figure 10A). In contrast, YM155 treatment is able to relocalize Sp1 proteins in the subcellular structure of the cells (Figure 10B), and thus, this may prevent the survivin promoter-binding Sp1 proteins to access its DNA binding sites on the survivin core promoter region. Nevertheless, we found that there is an unknown DNA/protein band in the SCPO-4 (Figure 4B, lane 6). Further studies indicated that the protein in the unknown DNA/protein complex appears to be LyF-1 or LyF-1 like proteins (Figure 5). Therefore, it is possible that the Sp1 protein may interact with this protein and/or other proteins on the SCOP-4 element to exert the function of the constitutive expression of survivin before YM155 treatment. However, YM155 treatment resulted in the dissociation of Sp1 protein interaction with other proteins in the SCPO-4 element on the survivin promoter, which enables Sp1 protein release from its DNA binding site, and thus resulted in its re-localization to the subcellular structure of the cell, which results in YM155-mediated inhibition of Sp1-controled survivin gene transcription.
We previously showed that while Ap2 does not significantly downregulate human survivin promoter activity in immortalized ‘normal’ cells, forced expression of Ap2 significantly inhibits survivin promoter activity in cancer cells [42]. Therefore, it is possible that the overlapped mutual competition between Sp1 and Ap2 on the SCPO-4/5 elements plays a role for the constitutive expression of survivin in cancer cells. In other words, in normal cells, the binding of Ap2 at the SCPO-4/5 region may be a major event to inhibit the constitutive expression of survivin, while Sp1 is the major player to replace or override the role of Ap2 binding at the SCPO-4/5 region. Therefore, such a major shift would result in the constitutive expression of survivin in cancer cells. However, although this hypothesis is intriguing, our experiments using cancer cell nuclear extracts could not prove the presence of Ap2 under various conditions (Figure 7), although Ap2-like binding sites were revealed in the SCPO-4 and SCPO-5 DNA elements (Figure 4A). Further studies would be required to unravel its full molecular basis for this interesting hypothesis.
In summary, we demonstrated in this study that inhibition of survivin promoter activity by YM155 is a survivin promoter-selective event without the association of G1 cell cycle arrest. The 269 bp survivin core promoter appears to play a major role in the YM155’s effect on survivin inhibition. We further identified that YM155-mediated modulation of Sp1 protein binding in the region of SCPO-4/5 (-149 to -71) appears to be key mechanism underlying YM155-mediated inhibition of human survivin promoter activity in cancer cells.
Acknowledgements
We would like to thank Dr. Enrico Mihich (Roswell Park Cancer Institute) for helping to coordinate the project between the Li lab and Astellas Pharma Inc, Dr. Suzanne Hess (Roswell Park Cancer Institute) for carefully reading and revising the manuscript, and Dr. Lili Tian (Biostatistical Resource Core of UB/Roswell Park) for statistical consultation for the Li lab.
This work was supported in part by a contract fund from Astellas Pharma Inc., Japan and an R01 Grant [CA109481] from the National Institutes of Health (NIH) to F. Li, as well as a NCI Cancer Center Support Grant (CCSG) [CA16056] to Roswell Park Cancer Institute (RPCI).
References
- 1.Deveraux QL, Reed JC. IAP family proteins--suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 2.Liston P, Fong WG, Korneluk RG. The inhibitors of apoptosis: there is more to life than Bcl2. Oncogene. 2003;22:8568–8580. doi: 10.1038/sj.onc.1207101. [DOI] [PubMed] [Google Scholar]
- 3.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 4.Li F. Survivin Study: What is the next wave? J Cell Physiol. 2003;197:8–29. doi: 10.1002/jcp.10327. [DOI] [PubMed] [Google Scholar]
- 5.Altieri DC. Survivin in apoptosis control and cell cycle regulation in cancer. Prog Cell Cycle Res. 2003;5:447–452. [PubMed] [Google Scholar]
- 6.Wheatley SP, McNeish IA. Survivin: a protein with dual roles in mitosis and apoptosis. Int Rev Cytol. 2005;247:35–88. doi: 10.1016/S0074-7696(05)47002-3. [DOI] [PubMed] [Google Scholar]
- 7.Li F, Ling X. Survivin Study: An update of “What is the next wave? ” J Cell Physiol. 2006;208:476–486. doi: 10.1002/jcp.20634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lechler P, Wu X, Bernhardt W, Campean V, Gastiger S, Hackenbeck T, Klanke B, Weidemann A, Warnecke C, Amann K, Engehausen D, Willam C, Eckardt KU, Rodel F, Wiesener MS. The tumor gene survivin is highly expressed in adult renal tubular cells: implications for a pathophysiological role in the kidney. Am J Pathol. 2007;171:1483–1498. doi: 10.2353/ajpath.2007.070132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li F, Brattain MG. Role of the Survivin Gene in Pathophysiology. Am J Pathol. 2006;169:1–11. doi: 10.2353/ajpath.2006.060121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dasgupta P, Kinkade R, Joshi B, Decook C, Haura E, Chellappan S. Nicotine inhibits apoptosis induced by chemotherapeutic drugs by up-regulating XIAP and survivin. Proc Natl Acad Sci USA. 2006;103:6332–6337. doi: 10.1073/pnas.0509313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iurlaro M, Demontis F, Corada M, Zanetta L, Drake C, Gariboldi M, Peiro S, Cano A, Navarro P, Cattelino A, Tognin S, Marchisio PC, Dejana E. VE-Cadherin Expression and Clustering Maintain Low Levels of Survivin in Endothelial Cells. Am J Pathol. 2004;165:181–189. doi: 10.1016/s0002-9440(10)63287-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conway EM, Zwerts F, Van Eygen V, DeVriese A, Nagai N, Luo W, Collen D. Survivin-dependent angiogenesis in ischemic brain: molecular mechanisms of hypoxia-induced upregulation. Am J Pathol. 2003;163:935–946. doi: 10.1016/S0002-9440(10)63453-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byun SJ, Choi KS, Park SH, Cho NW, Hyun Yoo C, Yun KJ, Koh YJ, Koh GY, So BJ, Yoon KH. Cartilage oligometric matrix protein-angiopoietin-1 promotes revascularization through increased survivin expression in dermal endothelial cells of skin grafts in mice. Am J Pathol. 2007;171:1682–1690. doi: 10.2353/ajpath.2007.070142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Virrey JJ, Guan S, Li W, Schonthal AH, Chen TC, Hofman FM. Increased survivin expression confers chemoresistance to tumor-associated endothelial cells. Am J Pathol. 2008;173:575–585. doi: 10.2353/ajpath.2008.071079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li F. Role of survivin and its splice variants in tumorigenesis. Br J Cancer. 2005;92:212–216. doi: 10.1038/sj.bjc.6602340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ling X, He X, Apontes P, Cao F, Azrak RG, Li F. Enhancing effectiveness of the MDR-sensitive compound T138067 using advanced treatment with negative modulators of the drug-resistant protein survivin. Am J Transl Res. 2009;1:393–405. [PMC free article] [PubMed] [Google Scholar]
- 17.Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, Tominaga F, Kinoyama I, Matsuhisa A, Kudou M, Sasamata M. Broad spectrum and potent antitumor activities of YM155, a novel small-molecule survivin suppressant, in a wide variety of human cancer cell lines and xenograft models. Cancer Sci. 2010;102:614–621. doi: 10.1111/j.1349-7006.2010.01834.x. [DOI] [PubMed] [Google Scholar]
- 18.Nakahara T, Takeuchi M, Kinoyama I, Minematsu T, Shirasuna K, Matsuhisa A, Kita A, Tominaga F, Yamanaka K, Kudoh M, Sasamata M. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007;67:8014–8021. doi: 10.1158/0008-5472.CAN-07-1343. [DOI] [PubMed] [Google Scholar]
- 19.Iwasa T, Okamoto I, Suzuki M, Nakahara T, Yamanaka K, Hatashita E, Yamada Y, Fukuoka M, Ono K, Nakagawa K. Radiosensitizing effect of YM155, a novel small-molecule survivin suppressant, in non-small cell lung cancer cell lines. Clin Cancer Res. 2008;14:6496–6504. doi: 10.1158/1078-0432.CCR-08-0468. [DOI] [PubMed] [Google Scholar]
- 20.Minematsu T, Iwai M, Umehara KI, Usui T, Kamimura H. Characterization of human organic cation transporter 1 (OCT1/SLC22A1)-, and OCT2 (SLC22A2)-mediated transport of YM155 monobromide, a novel survivin suppressant. Drug Metab Dispos. 2010;38:1–4. doi: 10.1124/dmd.109.028142. [DOI] [PubMed] [Google Scholar]
- 21.Minematsu T, Iwai M, Sugimoto K, Shirai N, Nakahara T, Usui T, Kamimura H. Carrier-Mediated Uptake of YM155 Monobromide, 1-(2-Methoxyethyl)-2-methyl-4,9-dioxo-3-(pyrazin-2-ylmethyl)-4,9-dihydro-1 H-naphtho[2,3-d] imidazolium Bromide, a Novel Small-molecule Survivin Suppressant, into Human Solid Tumor and Lymphoma Cells. Drug Metab Dispos. 2009;37:619–628. doi: 10.1124/dmd.108.025254. [DOI] [PubMed] [Google Scholar]
- 22.Minematsu T, Felder L, Oppeneer T, Sakazume M, Oikawa K, Hashimoto T, Usui T, Kamimura H. Liquid chromatography-electrospray tandem mass spectrometric assay suitable for quantitation of YM155, a novel small-molecule survivin suppressant, in dog plasma. Biomed Chromatogr. 2008;22:763–769. doi: 10.1002/bmc.996. [DOI] [PubMed] [Google Scholar]
- 23.Iwai M, Minematsu T, Narikawa S, Usui T, Kamimura H. Involvement of human organic cation transporter 1 in the hepatic uptake of 1-(2-methoxyethyl)-2-methyl-4,9-dioxo-3-(pyrazin-2-ylmethyl)-4,9-dihydro-1 H-naphtho[2,3-d] imidazolium bromide (YM155 monobromide), a novel, small molecule survivin suppressant. Drug Metab Dispos. 2009;37:1856–1863. doi: 10.1124/dmd.109.027359. [DOI] [PubMed] [Google Scholar]
- 24.Iwasa T, Okamoto I, Takezawa K, Yamanaka K, Nakahara T, Kita A, Koutoku H, Sasamata M, Hatashita E, Yamada Y, Kuwata K, Fukuoka M, Nakagawa K. Marked anti-tumour activity of the combination of YM155, a novel survivin suppressant, and platinum-based drugs. Br J Cancer. 2010;103:36–42. doi: 10.1038/sj.bjc.6605713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kita A, Nakahara T, Yamanaka K, Nakano K, Nakata M, Mori M, Kaneko N, Koutoku H, Izumisawa N, Sasamata M. Antitumor effects of YM155, a novel survivin suppressant, against human aggressive non-Hodgkin lymphoma. Leuk Res. 2011;35:787–792. doi: 10.1016/j.leukres.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 26.Yamanaka K, Nakata M, Kaneko N, Fushiki H, Kita A, Nakahara T, Koutoku H, Sasamata M. YM155, a selective survivin suppressant, inhibits tumor spread and prolongs survival in a spontaneous metastatic model of human triple negative breast cancer. Int J Oncol. 2011;39:569–575. doi: 10.3892/ijo.2011.1077. [DOI] [PubMed] [Google Scholar]
- 27.Tolcher AW, Mita A, Lewis LD, Garrett CR, Till E, Daud AI, Patnaik A, Papadopoulos K, Takimoto C, Bartels P, Keating A, Antonia S. Phase I and Pharmacokinetic Study of YM155, a Small-Molecule Inhibitor of Survivin. J. Clin. Oncol. 2008;26:5198–5203. doi: 10.1200/JCO.2008.17.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Satoh T, Okamoto I, Miyazaki M, Morinaga R, Tsuya A, Hasegawa Y, Terashima M, Ueda S, Fukuoka M, Ariyoshi Y, Saito T, Masuda N, Watanabe H, Taguchi T, Kakihara T, Aoyama Y, Hashimoto Y, Nakagawa K. Phase I study of YM155, a novel survivin suppressant, in patients with advanced solid tumors. Clin Cancer Res. 2009;15:3872–3880. doi: 10.1158/1078-0432.CCR-08-1946. [DOI] [PubMed] [Google Scholar]
- 29.Lewis KD, Samlowski W, Ward J, Catlett J, Cranmer L, Kirkwood J, Lawson D, Whitman E, Gonzalez R. A multi-center phase II evaluation of the small molecule survivin suppressor YM155 in patients with unresectable stage III or IV melanoma. Invest New Drugs. 2009;29:161–166. doi: 10.1007/s10637-009-9333-6. [DOI] [PubMed] [Google Scholar]
- 30.Giaccone G, Zatloukal P, Roubec J, Floor K, Musil J, Kuta M, van Klaveren RJ, Chaudhary S, Gunther A, Shamsili S. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J. Clin. Oncol. 2009;27:4481–4486. doi: 10.1200/JCO.2008.21.1862. [DOI] [PubMed] [Google Scholar]
- 31. NCI Drug Dictionary: Survivin inhibitor YM155 clinical trial: NCI ( http://www.cancer.gov/Templates/drugdictionary.aspx?CdrID=476771) [Google Scholar]
- 32.Tolcher AW, Quinn DI, Ferrari A, Ahmann F, Giaccone G, Drake T, Keating A, de Bono JS. A phase II study of YM155, a novel small-molecule suppressor of survivin, in castration-resistant taxane-pretreated prostate cancer. Ann Oncol. 2012;23:968–973. doi: 10.1093/annonc/mdr353. [DOI] [PubMed] [Google Scholar]
- 33.Yamanaka K, Nakahara T, Yamauchi T, Kita A, Takeuchi M, Kiyonaga F, Kaneko N, Sasamata M. Antitumor activity of YM155, a selective small-molecule survivin suppressant, alone and in combination with docetaxel in human malignant melanoma models. Clin Cancer Res. 2011;17:5423–5431. doi: 10.1158/1078-0432.CCR-10-3410. [DOI] [PubMed] [Google Scholar]
- 34.Nakahara T, Yamanaka K, Hatakeyama S, Kita A, Takeuchi M, Kinoyama I, Matsuhisa A, Nakano K, Shishido T, Koutoku H, Sasamata M. YM155, a novel survivin suppressant, enhances taxane-induced apoptosis and tumor regression in a human Calu 6 lung cancer xenograft model. Anticancer Drugs. 2011;22:454–462. doi: 10.1097/CAD.0b013e328344ac68. [DOI] [PubMed] [Google Scholar]
- 35.Li F, Altieri DC. Transcriptional analysis of human survivin gene expression. Biochem J. 1999;344:305–311. doi: 10.1042/0264-6021:3440305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 37.Ling X, Bernacki RJ, Brattain MG, Li F. Induction of survivin expression by taxol (paclitaxel) is an early event which is independent on taxol-mediated G2/M arrest. J Biol Chem. 2004;279:15196–15203. doi: 10.1074/jbc.M310947200. [DOI] [PubMed] [Google Scholar]
- 38.Wu J, Ling X, Pan D, Apontes P, Song L, Liang P, Altieri DC, Beerman T, Li F. Molecular mechanism of inhibition of survivin transcription by the GC-rich sequence selective DNA-binding antitumor agent, hedamycin: evidence of survivin downregulation associated with drug sensitivity. J Biol Chem. 2005;280:9745–9751. doi: 10.1074/jbc.M409350200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu J, Apontes P, Song L, Liang P, Yang L, Li F. Molecular mechanism of upregulation of survivin transcription by the AT-rich DNA-binding ligand, Hoechst33342: evidence for survivin involvement in drug resistance. Nucleic Acids Res. 2007;35:2390–2402. doi: 10.1093/nar/gkm149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ling X, Cheng Q, Black JD, Li F. Forced Expression of Survivin-2B Abrogates Mitotic Cells and Induces Mitochondria-dependent Apoptosis by Blockade of Tubulin Polymerization and Modulation of Bcl-2, Bax, and Survivin. J Biol Chem. 2007;282:27204–27214. doi: 10.1074/jbc.M705161200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lo K, Landau NR, Smale ST. LyF-1, a transcriptional regulator that interacts with a novel class of promoters for lymphocyte-specific genes. Mol Cell Biol. 1991;11:5229–5243. doi: 10.1128/mcb.11.10.5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spaulding B, Pan D, Ghadersohi A, Nielsen G, Jensen S, Gellert F, Ling X, Zhang M, Black A, Li F. Characterization of the 12C4 survivin monoclonal antibody and insight into the expression of survivin in human adult tissues. Histopathology. 2006;49:622–633. doi: 10.1111/j.1365-2559.2006.02556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dohi T, Beltrami E, Wall NR, Plescia J, Altieri DC. Mitochondrial survivin inhibits apoptosis and promotes tumorigenesis. J Clin Invest. 2004;114:1117–1127. doi: 10.1172/JCI22222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li F, Ackermann EJ, Bennett CF, Rothermel AL, Plescia J, Tognin S, Villa A, Marchisio PC, Altieri DC. Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat Cell Biol. 1999;1:461–466. doi: 10.1038/70242. [DOI] [PubMed] [Google Scholar]
- 45.Reed JC, Reed SI. Survivin' cell-separation anxiety. Nat Cell Biol. 1999;1:E199–200. doi: 10.1038/70227. [DOI] [PubMed] [Google Scholar]
- 46.Skoufias DA, Mollinari C, Lacroix FB, Margolis RL. Human survivin is a kinetochore-associated passenger protein. J Cell Biol. 2000;151:1575–1582. doi: 10.1083/jcb.151.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uren AG, Wong L, Pakusch M, Fowler KJ, Burrows FJ, Vaux DL, Choo KH. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr Biol. 2000;10:1319–1328. doi: 10.1016/s0960-9822(00)00769-7. [DOI] [PubMed] [Google Scholar]
- 48.Li F, Ling X, Huang H, Brattain L, Apontes P, Wu J, Binderup L, Brattain MG. Differential regulation of survivin expression and apoptosis by vitamin D(3) compounds in two isogenic MCF-7 breast cancer cell sublines. Oncogene. 2005;24:1385–1395. doi: 10.1038/sj.onc.1208330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang M, Yang J, Li F. Transcriptional and post-transcriptional controls of survivin in cancer cells: novel approaches for cancer treatment. J Exp Clin Cancer Res. 2006;25:391–402. [PMC free article] [PubMed] [Google Scholar]
- 50.Li F, Cheng Q, Ling X, Stablewski A, Tang L, Foster BA, Johnson CS, Rustum YM, Porter CW. Generation of a Novel Transgenic Mouse Model for Bioluminescent Monitoring of Survivin Gene Activity in Vivo at Various Pathophysiological Processes. Survivin Expression Overlaps with Stem Cell Markers. Am J Pathol. 2010;176:1629–1638. doi: 10.2353/ajpath.2010.090414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li F, Baykal D, Horaist C, Yan CN, Carr BN, Rao GN, Runge MS. Cloning and identification of regulatory sequences of the human thrombin receptor gene. J Biol Chem. 1996;271:26320–26328. doi: 10.1074/jbc.271.42.26320. [DOI] [PubMed] [Google Scholar]
- 52.Li F, Altieri DC. The cancer antiapoptosis mouse survivin gene: characterization of locus and transcriptional requirements of basal and cell cycle-dependent expression. Cancer Res. 1999;59:3143–3151. [PubMed] [Google Scholar]
- 53.Li Y, Xie M, Yang J, Yang D, Deng R, Wan Y, Yan B. The expression of antiapoptotic protein survivin is transcriptionally upregulated by DEC1 primarily through multiple sp1 binding sites in the proximal promoter. Oncogene. 2006;25:3296–3306. doi: 10.1038/sj.onc.1209363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu R, Zhang P, Huang J, Ge S, Lu J, Qian G. Sp1 and Sp3 regulate basal transcription of the survivin gene. Biochem Biophys Res Commun. 2007;356:286–292. doi: 10.1016/j.bbrc.2007.02.140. [DOI] [PubMed] [Google Scholar]
- 55.Chun JY, Hu Y, Pinder E, Wu J, Li F, Gao AC. Selenium inhibition of survivin expression by preventing Sp1 binding to its promoter. Mol Cancer Ther. 2007;6:2572–2580. doi: 10.1158/1535-7163.MCT-07-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]