

Succinyl-CoA synthetase catalyzes the reaction succinyl-CoA + NDP + Pi ⇌ succinate + CoA + NTP, where N denotes adenosine or guanosine. The enzyme from T. aquaticus was characterized biochemically and its structure was determined in complex with GDP-Mn2+, the preferred nucleotide.
Keywords: thermostability, denaturation, nucleotide specificity, ATP-grasp fold, enzyme kinetics
Abstract
Succinyl-CoA synthetase (SCS) from Thermus aquaticus was characterized biochemically via measurements of the activity of the enzyme and determination of its quaternary structure as well as its stability and refolding properties. The enzyme is most active between pH 8.0 and 8.4 and its activity increases with temperature to about 339 K. Gel-filtration chromatography and sedimentation equilibrium under native conditions demonstrated that the enzyme is a heterotetramer of two α-subunits and two β-subunits. The activity assays showed that the enzyme uses either ADP/ATP or GDP/GTP, but prefers GDP/GTP. This contrasts with Escherichia coli SCS, which uses GDP/GTP but prefers ADP/ATP. To understand the nucleotide preference, T. aquaticus SCS was crystallized in the presence of GDP, leading to the determination of the structure in complex with GDP-Mn2+. A water molecule and Pro20β in T. aquaticus take the place of Gln20β in pig GTP-specific SCS, interacting well with the guanine base and other residues of the nucleotide-binding site. This leads to the preference for GDP/GTP, but does not hinder the binding of ADP/ATP.
1. Introduction
In the citric acid cycle, succinyl-CoA synthetase (SCS) catalyzes the reaction succinyl-CoA + NDP + Pi ⇌ succinate + CoA + NTP, where N denotes adenosine or guanosine. The reaction requires divalent cations, usually Mg2+. The majority of the research investigating SCS has used enzymes from two sources: Escherichia coli and pig heart (reviewed by Bridger, 1974 ▶; Nishimura, 1986 ▶). E. coli SCS can use either ADP/ATP or GDP/GTP, but the adenine base is preferred. The enzyme that has been purified from pig heart is GTP-specific, but pigs are one of the many organisms that have two SCS isozymes, each specific for a single nucleotide. SCSs from different species exhibit either a heterodimeric quaternary structure consisting of one α-subunit and one β-subunit, as observed in pig GTP-specific SCS (Brownie & Bridger, 1972 ▶), or a heterotetrameric quaternary structure (Bridger, 1971 ▶) consisting of a dimer of αβ dimers, as observed in E. coli SCS (Wolodko et al., 1986 ▶, 1994 ▶). The nucleotide specificity is determined by the β-subunit, since the two isozymes contain a common α-subunit but different β-subunits (Johnson, Muhonen et al., 1998 ▶; Johnson, Mehus et al., 1998 ▶). The structures of both E. coli and pig GTP-specific SCS have been determined using X-ray crystallography (Wolodko et al., 1994 ▶; Fraser et al., 1999 ▶, 2000 ▶, 2006 ▶; Joyce et al., 2000 ▶).
Very little is known about the properties of SCS from thermophiles. SCS from Thermus aquaticus has been shown to exist in the ‘large form’ with a relative molecular weight of around 150 000, like E. coli SCS (Weitzman & Kinghorn, 1983 ▶). It was shown to be more thermostable than E. coli SCS in a test that consisted of heating the enzyme for 5 min at each temperature, cooling the solution and assaying the activity at a lower temperature. A thermophilic form of SCS could be a useful reagent in experiments using SCS in a coupled enzyme assay to determine the concentration of succinate in solution (Luo et al., 2006 ▶), since the thermostability of the enzyme could lead to a simplified purification scheme. With the goal of obtaining pure thermophilic enzyme, SCS from T. aquaticus was cloned and overexpressed in E. coli and the purified enzyme was crystallized (Joyce et al., 2007 ▶). Here, we present the biochemical characterization of T. aquaticus SCS via measurements of the activity and quaternary structure of the enzyme as well as its stability and refolding properties. The activity assays showed that T. aquaticus SCS uses either ADP/ATP or GDP/GTP but prefers GDP/GTP. This led to crystallization of the enzyme with GDP and determination of the structure in complex with GDP-Mn2+. The residues that lead to the preference for GDP are outlined on the basis of comparisons with the structures of E. coli SCS in complex with ADP-Mg2+ (Joyce et al., 2000 ▶) and pig GTP-specific SCS in complex with GTP-K+ or GDP-K+ (Fraser et al., 2006 ▶).
2. Materials and methods
T. aquaticus SCS was purified as described previously (Joyce et al., 2007 ▶) and stored either as an ammonium sulfate suspension (20 g ammonium sulfate per 100 ml initial solution) or at 193 K in 20 µl aliquots quick-frozen in liquid nitrogen (Deng et al., 2004 ▶). For all experiments except the crystallization, the ammonium sulfate suspension was collected by centrifugation at 15 000g and 277 K for 30 min and the pellet was dissolved in a minimal volume of 50 mM KCl, 0.1 mM EDTA, 50 mM Tris HCl pH 7.4. The resultant solution was clarified by centrifugation and dialyzed for 16 h with three changes of the same buffer, all at 277 K. For the crystallization experiments, the enzyme solution stored at 193 K was thawed and used directly.
2.1. Enzymatic activity
The ability of T. aquaticus SCS to catalyze the formation of succinyl-CoA from ATP, CoA and succinate was tested over the pH range 6.2–9.6. A 1.9 µg sample of enzyme was diluted in 1 ml buffer consisting of 10 mM MgCl2, 50 mM KCl, 0.1 mM dithiothreitol (DTT), 10 mM succinate, 0.1 mM CoA, 0.4 mM ATP and either 50 mM MOPS (pH 6.2–8.2) or 50 mM Tris–HCl (pH 8.0–9.6) and the formation of the thioester bond in succinyl-CoA was measured spectrophotometrically at 235 nm.
For kinetic analyses, enzymatic activity was measured spectrophotometrically in the direction of succinyl-CoA formation at 295 K. The initial velocity was measured in duplicate at various concentrations of one substrate and constant, saturating concentrations of the other substrates. The concentration ranges for the substrates were 2.50–60.0 µM CoA, 0.125–6.00 mM succinate, 12.5–400 µM ATP and 2.00–100 µM GTP, and the constant concentrations were 114 µM CoA, 10.0 mM succinate or 405 µM ATP or GTP, where appropriate, in a total volume of 1 ml containing 10 mM MgCl2, 50 mM KCl, 50 mM Tris–HCl pH 8.0. The concentrations of the nucleotides and CoA were determined using their standard extinction coefficients for a 1 cm path length: ATP, ∊259 nm = 15.4 mM −1; GTP, ∊252 nm = 13.7 mM −1; CoA, ∊260 nm = 14.6 mM −1 (P-L Biochemicals). K m(app) and V were calculated using the program Enzyme Kinetics (Stanislawski, 1991 ▶). Oxaloacetate, acetoacetate and malic acid were tested for their ability to substitute for succinate as a substrate for T. aquaticus SCS using 20 mM of each and 3.4 µg enzyme.
2.2. Quaternary structure and stability
The stability of SCS was investigated by measuring the activity of T. aquaticus, E. coli and pig GTP-specific SCS at different temperatures and different concentrations of denaturants. For the temperature profile, enzymatic activity was measured in solutions similar to those listed above except that 10 mM potassium phosphate pH 7.4 was used as the buffer because of the small variation of its pK a with temperature. 3.8 µg T. aquaticus SCS, 0.87 µg E. coli SCS and 0.82 µg pig GTP-specific SCS were used in each assay. The stability of SCS was also investigated by measuring the activity of each enzyme after incubation in 60 mM potassium phosphate pH 7.4 with different concentrations of urea (0–10 M) or guanidinium chloride (GdmCl; 0–4.0 M) for 24 h at 295 K.
Gel-filtration chromatography was performed to study the quaternary structure of T. aquaticus SCS in the presence and absence of GdmCl, as well as to separate the subunits. A Varian Vista 5500 liquid chromatograph was used with a Superose 12 HR 10/30 FPLC column. The flow rate was 0.5 ml min−1 and, where needed, one fraction was collected each minute. Firstly, to study the quaternary structure of T. aquaticus SCS with no denaturants, a mixture of 5 mg ml−1 each of T. aquaticus SCS and pig GTP-specific SCS in 5 mM β-mercaptoethanol (β-ME), 0.1 mM EDTA, 60 mM potassium phosphate pH 7.4 was separated by gel-filtration chromatography. Next, to study the quaternary structure just after T. aquaticus SCS had lost activity in GdmCl, 5 mg ml−1 T. aquaticus SCS was incubated for 24 h in 1.8 M GdmCl, 5 mM β-ME, 0.1 mM EDTA, 60 mM potassium phosphate pH 7.4 and chromatography was then performed in the same buffer. Finally, T. aquaticus SCS was incubated in 3.0 M GdmCl, 5 mM β-ME, 0.1 mM EDTA, 60 mM potassium phosphate pH 7.4 and chromatography was performed in the same buffer.
The stability of the secondary structure of T. aquaticus SCS in different concentrations of GdmCl was investigated using far-ultraviolet circular dichroism (far-UV CD). The solution for far-UV CD consisted of between 0 and 7.5 M GdmCl in 5 mM β-ME, 0.1 mM EDTA, 60 mM potassium phosphate pH 7.4. The subunits were separated in 3.0 M GdmCl and then dialyzed in 5 mM β-ME, 0.1 mM EDTA, 60 mM potassium phosphate pH 7.4; far-UV CD was performed at different concentrations of GdmCl. The concentration of whole enzyme was 9.0 or 2.2 µM, the concentration of the α-subunit was 16.2 µM and the concentration of the β-subunit was 10.0 µM. Far-UV CD measurements were made at 298 K using a Jasco J-720 spectropolarimeter (Jasco Inc., Easton, Maryland, USA) and a cuvette with a 0.02 cm path length. The instrument was routinely calibrated with ammonium d-(+)-10-camphor sulfonate at 290.5 and 192 nm and with d-(−)-pantoyllactone at 219 nm. Each sample was scanned ten times from 180 to 255 nm, measuring data every 0.05 nm. Prior to calculating molar ellipticities, noise reduction was applied to the averaged data to remove the high-frequency noise. Molar ellipticities were calculated in deg cm2 dmol−1 using the equation [θ] = θobs × MRW/(10 × l × c), where θobs is the ellipticity measured in millidegrees, MRW is the mean residue weight (molecular weight divided by the number of residues), l is the optical pathlength in cm and c is the protein concentration in mg ml−1. Best-fit lines were fitted to the transitions using the formula y = a + b[1 + exp(c − x)/d] and the program SigmaPlot (Norby et al., 1983 ▶).
Sedimentation-equilibrium experiments were performed to study the quaternary structure of T. aquaticus SCS. The analysis was first performed without GdmCl using three separate loading concentrations of T. aquaticus SCS: 1.77, 1.3 and 0.8 mg ml−1 in 2 mM DTT, 20 mM potassium phosphate pH 7.4. For analysis in the presence of GdmCl, the first GdmCl concentration used was 0.9 M, which is slightly less than that needed to inactivate the enzyme, and the second was 1.8 M GdmCl, both with 5 mM β-ME, 0.1 mM EDTA, 60 mM potassium phosphate pH 7.4. The loading concentrations of T. aquaticus SCS used with GdmCl were 5, 3 and 1 mg ml−1. All sedimentation-equilibrium experiments were carried out at 293 K using a Beckman XL-I ultracentrifuge and interference optics according to procedures outlined by the manufacturer. Sample aliquots of 110 µl were loaded into six-sector charcoal-filled Epon sample cells, allowing the three different concentrations of protein to be run simultaneously. Runs at two different speeds were continued until there was no difference in scans that were taken 3 h apart. The sedimentation-equilibrium data were analyzed using a nonlinear least-squares curve-fitting algorithm (Johnson et al., 1981 ▶) in the NONLIN program (D. A. Yphantis). The equation used to analyze the data assumes a single ideal species, C r = e[ln(C ro) + (1 − νρ)ω/(2RT)M(r 2 − r o 2) − B M(C r − C ro)], where C r is the concentration at radius r, C ro is the concentration at the reference radius r o, ν is the partial specific volume, ρ is the density of the solvent, ω is the angular velocity, R is the universal gas constant, T is the temperature in kelvin, M is the molecular weight and B is the second virial coefficient associated with non-ideality. The program SEDNTERP (Laue et al., 1992 ▶) was used to calculate the partial specific volume of the T. aquaticus SCS using the method of Cohn & Edsall (1943 ▶). For the runs containing GdmCl, the method of Laue was used to apply a correction to ν for the binding of GdmCl to the enzyme and the change in hydration (Laue et al., 1992 ▶).
2.3. Refolding
Refolding studies were conducted by denaturing the enzyme in 6 M GdmCl for 24 h and refolding by rapid dilution [1:20(v:v)] into one of three solutions: benign buffer (50 mM KCl, 60 mM potassium phosphate pH 7.4), arginine buffer at pH 7.4 (0.67 M l-arginine–HCl, 60 mM potassium phosphate pH 7.4) or arginine buffer at pH 8.0 (0.67 M l-arginine–HCl, 60 mM potassium phosphate, 50 mM Tris–HCl pH 8.0). Samples were taken after 5 h and assayed for their ability to catalyze the formation of succinyl-CoA at pH 8.0 and 295 K. The assay solution consisted of 10 mM MgCl2, 50 mM KCl, 0.1 mM DTT, 10 mM succinate, 0.1 mM CoA, 0.4 mM ATP, 50 mM Tris–HCl pH 8.0. The temperature stability of the refolded enzyme was investigated by heating to 348 K for 1 h or 359 K for 20 min and then measuring the activity at pH 8.0 and 295 K. For the enzyme refolded in arginine buffer at pH 7.4, a temperature profile of the enzymatic activity was measured as had been performed for the native T. aquaticus SCS. Enzyme refolded in arginine buffer at pH 7.4 was exchanged into benign buffer using a Millipore centrifugal filter device and its temperature stability was tested by heating to 348 K for 1 h or 359 K for 20 min and then measuring the activity at pH 8.0 and 295 K.
To study the kinetics of refolding and its dependence on temperature, the refolding in arginine buffer at pH 7.4 was performed at 285, 295 and 310 K, taking samples at various times and measuring SCS activity at 295 K and pH 8.0. The data for the refolding were fitted to first-order kinetics and the rate constants were obtained using the program KaleidaGraph (Synergy Software; http://www.synergy.com) and replotted using an Arrhenius plot. The concentration dependence of the kinetics of refolding at 295 K was investigated by the same procedure using final protein concentrations of 0.59, 0.24 and 0.059 mg ml−1 in the refolding buffers.
2.4. Crystal structure of T. aquaticus SCS in complex with GDP-Mn2+
The experiments undertaken to crystallize T. aquaticus SCS have been described in detail by Joyce et al. (2007 ▶). To crystallize the full-length enzyme in complex with GDP-Mn2+, the protein solution consisted of 7 mg ml−1 protein, 5 mM GDP and 10 mM MnCl2. This solution was mixed with an equal volume of a precipitant solution consisting of 10% polyethylene glycol 3350 (PEG 3350), 100 mM MES pH 6.4 and 200 mM KCl and the droplet was suspended over a reservoir containing the same precipitant solution. Once a crystal grew it was transferred into similar solutions containing an incrementally increasing concentration of PEG 3350 to a maximum of 20% and was then transferred into a solution that also contained 10% glycerol before being vitrified in a stream of cold nitrogen. Crystallographic data were collected on beamline 8.3.1 of the Advanced Light Source, Berkeley, California, USA. The data were processed using the HKL program package (Otwinowski & Minor, 1997 ▶). Subsequent programs used to analyze the data were from the CCP4 package (Winn et al., 2011 ▶).
The structure was solved by molecular replacement using the program AMoRe (Navaza, 1994 ▶). The initial search models were the structures of E. coli SCS (Fraser et al., 2002 ▶; PDB entry 1jkj) and of the α-subunit of T. thermophilus SCS (PDB entry 1oi7; H. Takahashi, Y. Tokunaga, C. Kuroishi, N. Babayeba, S. Kuramitsu, S. Yokoyama, M. Miyano & T. H. Tahirov, unpublished work). The models were refined using the maximum-likelihood target in the programs CNS (Brünger et al., 1998 ▶) and Phenix (Adams et al., 2010 ▶) with noncrystallographic symmetry restraints. The models were visualized and fitted to the electron density using the programs Xfit (McRee, 1999 ▶) and Coot (Emsley et al., 2010 ▶). Stereochemistry was checked using the programs PROCHECK (Laskowski et al., 1993 ▶), WHATCHECK (Hooft et al., 1996 ▶) and MolProbity (Chen et al., 2010 ▶). The programs SPDBV (Guex & Peitsch, 1997 ▶) and O (Jones et al., 1991 ▶) were used to superimpose and visualize the models. The crystallographic data and the model have been deposited in the Protein Data Bank (Berman et al., 2000 ▶) and were assigned the identification code 3ufx.
3. Results and discussion
3.1. Enzymatic activity
The ability of T. aquaticus SCS to catalyze the formation of succinyl-CoA from ATP, CoA and succinate was measured at different pH values and different temperatures. The pH profile shown in Fig. 1 ▶(a) is essentially symmetric, with maximal activity occurring between pH 8.0 and 8.4. This contrasts with the maximal activity at pH 7.4 for E. coli SCS (Wolodko et al., 1994 ▶). The temperature profiles of these two enzymes and pig GTP-specific SCS relative to their activity at 295 K are shown in Fig. 1 ▶(b). Pig GTP-specific SCS ceased to function at 321 K, which is slightly lower than the temperature at which E. coli SCS became inactive (328 K). Both enzymes precipitated in the stock solutions at the temperatures at which they became inactive. In contrast, T. aquaticus SCS remained soluble and active at all temperatures at which the assay could be performed. Its activity reached a plateau at approximately 339 K, with rates comparable with the values observed for E. coli (Wolodko & Bridger, 1987 ▶) and pig GTP-specific SCS (Fraser et al., 2006 ▶) and consistent with the optimal growth temperature of 343–353 K reported for T. aquaticus (Brock & Freeze, 1969 ▶). Although this suggests that the reaction has reached its maximal velocity, the spontaneous hydrolysis of succinyl-CoA becomes significant at these temperatures and this could lead to anomalously low values for the enzymatic activity. The rate of hydrolysis of succinyl-CoA at 348 K was not dependent on its own concentration at 40 µM (data not shown) and if the rate of hydrolysis of succinyl-CoA is not at all dependent on concentration, the maximum rate of synthesis of succinyl-CoA by T. aquaticus SCS in the presence of phosphate is 28.8 U mg−1. This is also an underestimation of the true maximal rate because the phosphate buffer would reduce the specific activity measured for the enzyme, both because its pH did not match the optimal pH of the enzyme and because of product inhibition (Cha & Parks, 1964 ▶). The magnitude of the inhibition can be estimated by comparing the activity of T. aquaticus SCS in 10 mM potassium phosphate with that in 50 mM MOPS. At pH 7.4 and 295 K the activity in phosphate buffer is 87% of that in MOPS.
Figure 1.

Enzymatic activity. (a) The activity of T. aquaticus SCS was measured in 10 mM MgCl2, 50 mM KCl, 0.1 mM dithiothreitol, 10 mM succinate, 0.1 mM CoA, 0.4 mM ATP and 50 mM of either MOPS (squares) or Tris–HCl (circles) to cover the pH range 6.2–9.6. (b) The activities of T. aquaticus (circles), E. coli (triangles) and pig GTP-specific SCS (squares) in 10 mM MgCl2, 50 mM KCl, 0.1 mM dithiothreitol, 10 mM succinate, 0.1 mM CoA, 0.4 mM ATP (for T. aquaticus and E. coli SCS) or GTP (for pig GTP-specific SCS) and 10 mM potassium phosphate pH 7.4 at different temperatures were plotted relative to their activity at 295 K.
The kinetic parameters of T. aquaticus SCS are presented in Table 1 ▶. K m(app) values for all substrates except GTP are within an order of magnitude of those determined for E. coli SCS (Joyce et al., 1999 ▶). Surprisingly, the value of K m(app) for GTP is approximately 16 times less than the comparable value with E. coli SCS. T. aquaticus SCS could not use oxaloacetate, acetoacetate or malic acid to replace succinate, which is consistent with the results using E. coli SCS (Gibson et al., 1967 ▶). As might be expected for a thermophilic enzyme operating well below its optimal temperature, the limiting rate of succinyl-CoA production at room temperature is considerably lower than the values measured for E. coli SCS and pig GTP-specific SCS at room temperature. The specific activities typical for E. coli SCS and pig GTP-specific SCS are 45 and 30 U mg−1, respectively (Wolodko et al., 1986 ▶).
Table 1. Kinetic parameters for T. aquaticus SCS at 295K in 10mM MgCl2, 50mM KCl, 50mM TrisHCl pH 8.0.
| ATP | GTP | CoA | Succinate | |
|---|---|---|---|---|
| K m(app) (M) | 121 5 | 24 5 | 5.5 1.0 | (1.4 0.1) 103 |
| V (Umg1) | 8.8 0.2 | 5.9 0.5 | 5.7 0.5 | 5.0 0.5 |
3.2. Quaternary structure and stability
The quaternary structure of T. aquaticus SCS was evaluated by both gel-filtration chromatography and sedimentation equilibrium. The elution profile of a mixture of purified T. aquaticus and pig GTP-specific SCS revealed two separate peaks (Fig. 2 ▶ a). The first was T. aquaticus SCS and the second was pig GTP-specific SCS, as shown by determining the activities of the protein in each peak with ATP and GTP. This elution profile resembles the elution profile of a mixture of tetrameric E. coli SCS and a dimeric mutant (Bailey et al., 1999 ▶). The results match those of Weitzman and Kinghorn, who used SCS purified from T. aquaticus rather than enzyme overexpressed in E. coli (Weitzman & Kinghorn, 1983 ▶), and indicate that T. aquaticus SCS is a tetramer, since the differences in molecular weight between the subunits of E. coli, T. aquaticus and pig GTP-specific SCS are small on the scale examined by gel filtration. The interpretation is supported by sedimentation equilibrium under native conditions (Fig. 2 ▶ b). These data fitted a model in which T. aquaticus SCS exists as a non-dissociating species with a molecular weight of 1.35 × 105. As calculated from the amino-acid sequence deduced for our expression plasmid (Joyce et al., 2007 ▶), the weight of tetrameric T. aquaticus SCS would be 147 333. The conclusion is that T. aquaticus SCS is a heterotetramer of two α-subunits and two β-subunits.
Figure 2.

Quaternary structure. (a) Elution profile of T. aquaticus and pig GTP-specific SCS (5 mg ml−1 each) from gel-filtration chromatography in 5 mM 2-mercaptoethanol, 0.1 mM EDTA, 60 mM potassium phosphate pH 7.4. (b) Sedimentation equilibrium of T. aquaticus SCS under native conditions using 1.77, 1.31 and 0.8 mg ml−1 in 2 mM dithiothreitol, 20 mM potassium phosphate pH 7.4.
To compare the susceptibilities of T. aquaticus, E. coli and pig GTP-specific SCS to chemical denaturation, the activity of each enzyme was measured after incubation for 24 h in various concentrations of either urea or GdmCl (Fig. 3 ▶). Pig GTP-specific SCS was the most sensitive to denaturants, T. aquaticus SCS was the most resistant and E. coli SCS was intermediate. T. aquaticus SCS lost activity between 4 and 5.5 M ures, but remarkably it regained activity with increasing concentrations of urea, reaching 70% of its original activity in 8.5 M urea. This behavior may have arisen from aggregation at the intermediate concentrations of urea, although no precipitation was observed. Lactate dehydrogenase from the thermophile Thermotoga maritima has also been shown to aggregate at intermediate concentrations of GdmCl (Dams et al., 1996 ▶). When the equilibrium unfolding of E. coli SCS in GdmCl was studied (Khan & Nishimura, 1988 ▶), enzymatic activity was lost prior to denaturation of the protein as monitored by far-UV CD. In addition, there were two transitions in the denaturation of E. coli SCS by GdmCl, with a marked plateau between them. To determine whether the equilibrium unfolding of the thermophilic enzyme from T. aquaticus followed the same pattern, the changes in ellipticity at 220 and 222 nm were measured as a function of the denaturant concentration using either 2.2 or 9.0 µM enzyme (Fig. 4 ▶). Unfolding was reversible at all GdmCl concentrations and was independent of the enzyme concentration. This is consistent with a very small dissociation constant for the dissociation of the whole enzyme into subunits. Reminiscent of the observations with the E. coli enzyme (Khan & Nishimura, 1988 ▶), there were two transitions in the denaturation curve, occurring at concentrations of 2.2 and 3.7 M GdmCl. As would be expected for a more stable enzyme, the transitions occurred at higher GdmCl concentrations than those observed for the E. coli enzyme (Khan & Nishimura, 1988 ▶). Similar to the results with the E. coli enzyme, the activity of T. aquaticus SCS was lost prior to the loss of secondary structure. The T. aquaticus enzyme had lost all SCS activity in 1.8 M GdmCl, while the changes in the molar ellipticity did not even begin until 2.0 M, indicating the presence of multiple intermediates in the unfolding process.
Figure 3.

Chemical denaturation. The activities of T. aquaticus (circles), E. coli (triangles) and pig GTP-specific SCS (squares) were plotted relative to their activity in the absence of denaturant. The conditions for activity measurements were 10 mM MgCl2, 50 mM KCl, 0.1 mM dithiothreitol, 10 mM succinate, 0.1 mM CoA, 0.4 mM ATP (for T. aquaticus and E. coli SCS) or GTP (for pig GTP-specific SCS), 10 mM potassium phosphate pH 7.4. (a) After 24 h incubation in 60 mM potassium phosphate pH 7.4 with urea. (b) After 24 h incubation in 60 mM potassium phosphate pH 7.4 with guanidinium chloride (GdmCl).
Figure 4.

Equilibrium unfolding of T. aquaticus SCS. The changes in ellipticity were measured as a function of the concentration of denaturant using either 2.2 µM (circles) or 9.0 µM (filled circles) enzyme in 5 mM 2-mercaptoethanol, 0.1 mM EDTA, 60 mM potassium phosphate pH 7.4. The two transitions are labelled 1 and 2.
To investigate the nature of the intermediate that exists between the two transitions in the denaturation with GdmCl, gel-filtration chromatography of T. aquaticus SCS was performed in 3 M GdmCl. Two peaks were observed in the elution profile (data not shown) and the proteins in these peaks were analyzed by SDS–PAGE. The first peak consisted primarily of β-subunit and the second consisted of α-subunit alone. This raised the possibility that the first transition observed in the denaturation studies corresponded to the dissociation/unfolding of the α-subunit from the tetramer of T. aquaticus SCS and the second transition reflected the unfolding of the remaining enzyme. To determine whether each subunit retained its native structure in the absence of the other subunit, the α-subunits and β-subunits were separated and far-UV CD was carried out on individual subunits in benign buffer. Summation of the two spectra for the individual subunits resulted in a spectrum that closely resembled the spectrum of the native enzyme in the same buffer (data not shown). This indicated that the subunits can adopt a native fold independently and thus major conformational changes are not required to form the whole enzyme from the subunits. To investigate the possibility that the denaturation of the whole enzyme was simply a sum of the denaturation of the subunits, the denaturation of each of the subunits in GdmCl was monitored by far-UV CD. The changes in molar ellipticity at 222 nm were measured as a function of the denaturant concentration. The unfolding transitions for both α-subunits and β-subunits were cooperative and reversible at all denaturant concentrations (data not shown). When the denaturation curves for the two separate subunits were added together, they did not resemble the denaturation curve for the whole enzyme, showing that the denaturation of the whole enzyme is not simply a sum of the denaturation of the individual subunits. In addition, values of 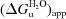 and m for the transitions of the individual subunits and of the whole enzyme were calculated from replots using the molar ellipticity data (Table 2 ▶). is the free energy of unfolding, which represents the stability of the protein in water, and m is proportional to the amount of surface area that is exposed upon denaturation. All of the thermodynamic parameters associated with the transitions for the individual subunits were lower than those for the whole enzyme, indicating that the native enzyme is more stable than the isolated subunits. Thus, the denaturation of the whole enzyme is not simply a sum of the denaturation of the individual subunits, even though the subunits retain most of their native conformation when isolated. It should be noted that for the whole enzyme the calculation of assumed a two-state transition for a monomer, which was obviously not correct. However, the proper calculation for a heterotetramer requires knowledge of the various association constants and was thus not possible. In addition, the prevalent species just prior to the beginning of the first transition in the denaturation of T. aquaticus SCS was observed to be a high-molecular-weight oligomer of uncertain composition with unknown association constants (data not shown).
and m for the transitions of the individual subunits and of the whole enzyme were calculated from replots using the molar ellipticity data (Table 2 ▶). is the free energy of unfolding, which represents the stability of the protein in water, and m is proportional to the amount of surface area that is exposed upon denaturation. All of the thermodynamic parameters associated with the transitions for the individual subunits were lower than those for the whole enzyme, indicating that the native enzyme is more stable than the isolated subunits. Thus, the denaturation of the whole enzyme is not simply a sum of the denaturation of the individual subunits, even though the subunits retain most of their native conformation when isolated. It should be noted that for the whole enzyme the calculation of assumed a two-state transition for a monomer, which was obviously not correct. However, the proper calculation for a heterotetramer requires knowledge of the various association constants and was thus not possible. In addition, the prevalent species just prior to the beginning of the first transition in the denaturation of T. aquaticus SCS was observed to be a high-molecular-weight oligomer of uncertain composition with unknown association constants (data not shown).
Table 2. Thermodynamic parameters for the unfolding of T. aquaticus SCS.
| Protein |
† (kJmol1) |
m † (kJmol1 M 1) | Correlation coefficient† | [GdmCl]1/2 ‡ (M) |
|---|---|---|---|---|
| Whole enzyme | ||||
| First transition | 29 4 | 13 2 | 0.986 | 2.2 |
| Second transition | 25 1 | 7.1 0.4 | 0.998 | 3.7 |
| -Subunit | 6.7 0.4 | 3.1 0.2 | 0.998 | 2.2 |
| -Subunit | 10.0 0.8 | 5.4 0.4 | 0.997 | 2.0 |
These values were calculated by extrapolation of a replot of the molar ellipticities using the equation G = RTlnK, where K = (N
obs)/(obs
U), N is the molar ellipticity observed at 0M GdmCl, U is the molar ellipticity observed at the maximum concentration of GdmCl and obs is the observed molar ellipticity. In the vicinity of the transition the resulting plot was fitted to the equation G = 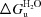 m[GdmCl] (for a review, see Myers et al., 1995 ▶) with the correlation coefficient listed.
m[GdmCl] (for a review, see Myers et al., 1995 ▶) with the correlation coefficient listed.
These values were obtained by interpolation from the graphs and are consistent with the values obtained by the calculation [GdmCl]1/2 = [/m.
3.3. Refolding
Since the T. aquaticus enzyme has the same oligomeric structure as the E. coli enzyme, the question of whether the refolding of T. aquaticus SCS follows the same path as E. coli SCS arises. T. aquaticus SCS did not refold under the conditions used for refolding either E. coli (Wolodko & Bridger, 1987 ▶) or pig GTP-specific SCS (Nishimura et al., 1988 ▶). In the benign buffer, T. aquaticus SCS precipitated and less than 50% of the original activity was recovered. In addition, when refolded T. aquaticus SCS was heated to 348 K for 1 h or to 359 K for 20 min, enzymatic activity was lost. Refolding of T. aquaticus SCS appeared to be successful when carried out by rapid dilution into 0.67 M arginine, 60 mM potassium phosphate pH 7.4 or pH 8.0. No precipitate was visible and 100% of the original activity was recovered, but again this activity was not stable on heating. The thermostability of T. aquaticus SCS was restored by exchanging the arginine buffer for the benign buffer 50 mM KCl, 60 mM potassium phosphate pH 7.4, suggesting that the refolding was not complete in the arginine buffer.
The refolding of T. aquaticus SCS in arginine buffer pH 7.4 was temperature dependent and the rate of refolding followed apparent first-order kinetics. Omission of phosphate from the buffer did not affect the rate. From the Arrhenius plot, the energy of activation (E a) associated with the refolding of T. aquaticus SCS was estimated to be 64.7 kJ mol−1. Consistent with the results of the equilibrium unfolding of T. aquaticus SCS, the kinetics of refolding were also not dependent on the concentration of protein tested.
Despite the high recovery of activity after refolding, the initial experiments indicated that refolded T. aquaticus SCS was not thermostable. To assess the stability of enzyme refolded in arginine buffer pH 7.4, its ability to catalyze the formation of succinyl-CoA was measured at different temperatures. The ‘refolded’ form is much less thermostable than the native form of T. aquaticus SCS (Fig. 5 ▶) and is only slightly more stable than the E. coli enzyme (compare Figs. 1 ▶ a and 5 ▶). Nevertheless, refolded T. aquaticus SCS appeared to have full activity at 295 K when assayed in the standard manner under saturating conditions of substrates. The kinetic parameters of refolded T. aquaticus SCS were evaluated by steady-state analysis of initial rates of succinyl-CoA formation at 295 K, since differences in the apparent kinetic parameters compared with the native enzyme could indicate an area of the enzyme that is perturbed in the refolded enzyme. The apparent kinetic parameters were essentially the same as those of the native enzyme. Remarkably, the instability of refolded T. aquaticus SCS did not disturb substrate binding or catalysis at 295 K.
Figure 5.

Thermostability. The activities of refolded T. aquaticus SCS (squares) and the natively folded enzyme (circles) at different temperatures are plotted. The activities were measured in 10 mM MgCl2, 50 mM KCl, 0.1 mM dithiothreitol, 10 mM succinate, 0.1 mM CoA, 0.4 mM ATP and 10 mM potassium phosphate pH 7.4.
Studies of the conformational stability of dimeric proteins have indicated that dimerization (from monomers) leads to a substantial increase in stability. For a mutant of E. coli SCS that exists as an αβ dimer rather than an α2β2 tetramer, the kinetic parameters remained the same as for the wild-type enzyme but the dimer was less soluble and less stable than the wild-type tetramer (Bailey et al., 1999 ▶). To investigate the possibility that the instability of T. aquaticus SCS was a consequence of the formation of dimeric and not tetrameric enzyme, gel-filtration chromatography of the refolded enzyme was performed. The peak of refolded T. aquaticus SCS eluted essentially where native T. aquaticus SCS eluted from the same column and well before dimeric pig GTP-specific SCS, indicating that the refolded T. aquaticus SCS was tetrameric.
3.4. Crystal structure of T. aquaticus SCS in complex with GDP-Mn2+
Based on the kinetics assays, crystallization trials of T. aquaticus SCS included GDP in the solution with the protein. The best crystal was of T. aquaticus SCS in complex with GDP-Mn2+. Mn2+ can replace Mg2+ in catalysis, although the maximal activity of E. coli SCS with Mn2+ is lower and at a lower concentration of the ion (Gibson et al., 1967 ▶). The asymmetric unit of the crystals in space group C2 includes four α-subunits (chains A, D, F and H) and four β-subunits (chains B, E, G and I). Initial electron-density maps calculated with phases from the protein showed clear electron density for GDP and the ion in each of the domains of the β-subunits possessing the ATP-grasp fold (Fig. 6 ▶). These molecules were added to the model along with water molecules possessing electron density at the 3σ level in the difference Fourier maps and good hydrogen-bonding interactions. After refinement, the temperature factors for the nucleotides were high relative to those of the surrounding residues, suggesting that the sites might not be fully occupied. The occupancy of each GDP-Mn2+ was refined and these values settled near 0.8. All residues of T. aquaticus SCS were fitted, except for residues 353 and 354 of chain G, 374−378 of chain E and 372–378 of chains G and I and any residues added during the cloning (Joyce et al., 2007 ▶). Residues 353 and 354 of chain G are at a crystallographic twofold axis and the electron density suggests that they, like the residues at the carboxy-termini of the molecules, must be disordered. The protein includes one cis-peptide bond in each α-subunit between Gly120α and Gly121α and one in each β-subunit between Asn190β and Pro191β. Both are located at turns near catalytic residues: where the histidine that is phosphorylated during the reaction binds in the α-subunit and where Mn2+ binds in the β-subunit. Both of these peptide bonds are cis in both E. coli and pig GTP-specific SCS, although that in the α-subunit is between Gly and Pro. Table 3 ▶ presents the statistics for the refined model.
Figure 6.

Complex of T. aquaticus SCS with GDP-Mn2+. In this stereofigure, residues with atoms within 3.2 Å of the GDP-Mn2+ in chain B are shown, as are Glu92β, Ala93β, Pro191β, Ala203β and the water molecules that interact with the guanine base. Atoms are coloured according to type: manganese, magenta; phosphorus, green; oxygen, red; nitrogen, blue; carbon, yellow. Black dashed lines represent ionic interactions with Mn2+ or hydrogen bonds. The electron density, contoured at 2.5σ and shown in green, is from an F o − F c OMIT map calculated after refinement when GDP-Mn2+ had been removed from the model. This figure was drawn using the program PyMOL (DeLano, 2002 ▶).
Table 3. Statistics for the refined model of T. aquaticus SCS in complex with GDP-Mn2+ .
| Resolution limit () | 2.35 |
| Space group | C2 |
| Unit-cell parameters (, ) | a = 261.7, b = 126.8, c = 110.6, = = 90, = 112.8 |
| R factor† (%)/No. of data | 20.4/114027 |
| R free ‡ (%)/No. of data | 24.5/6048 |
| No. of protein atoms§ | 19690 |
| No. of atoms in GDP-Mn2+ | 116 |
| No. of water molecules | 735 |
| R.m.s.d. for bond lengths () | 0.008 |
| R.m.s.d. for bond angles () | 1.0 |
| Ramachandran plot, residues in | |
| Most favoured regions | 2032 (92.4%) |
| Additional allowed regions | 164 (7.5%) |
| Generously allowed regions | 0 |
| Disallowed regions | 4 (0.2%) |
| Average temperature factors (2) | |
| Protein atoms (range) | 43 (12139) |
| GDP-Mn2+ atoms (range) | 48 (3365) |
| Water molecules (range) | 36 (1156) |
| Chains A, D, F, H (-subunits) | 35, 34, 36, 34 |
| Chains B, E, G, I (-subunits) | 48, 49, 49, 49 |
R factor = 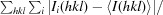
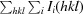 .
.
R factor based on data excluded from the refinement (5%).
The residues modelled are residues 1288 of chains A, D, F and H for the -subunit and residues 178 of chain B, 1373 of chain E, 1352 and 355371 of chain G and 1371 of chain I for the -subunit.
T. aquaticus SCS is an α2β2 heterotetramer, but the asymmetric unit of the crystals contains an octamer, leading to confusion as to which pairs of αβ dimers form the tetramers. For E. coli SCS, the physiologically relevant tetramer was determined by mutating residues at the dimer–dimer interface (Bailey et al., 1999 ▶). By analogy to E. coli SCS, the T. aquaticus tetramers are formed by chains A, B, D and E and chains F, G, H and I.
3.4.1. Nucleotide-binding site
The crystal structure of T. aquaticus SCS in complex with GDP-Mn2+ shows how this form of SCS binds GDP. Figs. 6 ▶ and 7 ▶(a) show the interactions that lead to the binding of the guanine base. O6 and N1 of the base interact with the amide N and carbonyl O atoms of Val94β, one of the residues forming the linker between the two subdomains adopting the ATP-grasp fold. O6 accepts a hydrogen bond from the amide N atom, while N1 donates a hydrogen bond to the carbonyl O atom. A water molecule links N7 of the base to the amine of Lys45β and the carbonyl O atom of Ala203β (Fig. 7 ▶ a) and a second water molecule links the first water molecule, O6 of the base and the carbonyl O atom of Glu92β. This coordination of the guanine base can be compared with the binding of GDP to pig GTP-specific SCS (Fraser et al., 2006 ▶; Fig. 7 ▶ b) and with the binding of ADP to E. coli SCS (Joyce et al., 2000 ▶; Fig. 7 ▶ c). When GDP binds to pig GTP-specific SCS, similar interactions are formed between O6 and N1 of the base and the backbone atoms, but there is also a hydrogen-bonding interaction between NE2 of Gln20β and O6 (Fig. 7 ▶ b). N7 interacts with a water molecule bridging to Lys46β in the complex of pig GTP-specific SCS with GTP (Fraser et al., 2006 ▶). It can be presumed that a similar interaction exists in the complex with GDP (Fig. 7 ▶ b), but the water molecule is not modelled in this crystal structure because of the lower resolution of the data (2.96 versus 2.1 Å for the complex with GTP). Gly20β also accepts a hydrogen bond from this bridging water molecule in the complex with GTP via its side-chain O atom. Essentially, a water molecule and Pro20β in T. aquaticus (Fig. 7 ▶ a) take the place of Gln20β in pig GTP-specific SCS (Fig. 7 ▶ b), interacting well with the guanine base and residues of the nucleotide-binding site. When ADP binds to E. coli SCS (Fig. 7 ▶ c), N6 and N1 of the adenine base interact with the backbone atoms and N6 also donates a weak hydrogen bond (3.2 Å) to Glu99β (Joyce et al., 2000 ▶). In contrast to the guanine base, N6 of the adenine base is protonated and N1 is not, so N6 donates a hydrogen bond to the carbonyl O atom of Ala100β of E. coli SCS and N1 accepts a hydrogen bond from the amide N atom of Thr102β. Relative to GDP (Figs. 7 ▶ a and 7 ▶ b), the adenine base of ADP is pulled into the protein (Fig. 7 ▶ c), allowing the different interactions between the atoms of the base and the backbone atoms of the residues forming the linker between the two subdomains adopting the ATP-grasp fold. In both T. aquaticus and pig GTP-specific SCS (Figs. 7 ▶ a and 7 ▶ b) there is an alanine residue in the position equivalent to Glu99β in E. coli SCS (Fig. 7 ▶ c). These alanine residues do not interact with the base.
Figure 7.

Comparison of the nucleotide-binding sites of T. aquaticus, pig GTP-specific and E. coli SCS. (a) Interactions of T. aquaticus SCS with GDP-Mn2+. (b) Interactions of pig GTP-specific SCS with GDP-K+. (c) Interactions of E. coli SCS with ADP-Mg2+. Atoms are coloured as in Fig. 6 ▶, with the exception that either manganese, potassium or magnesium is shown in magenta. This figure and Fig. 8 ▶ were drawn using MolScript (Kraulis, 1991 ▶) and Raster3D (Merritt & Bacon, 1997 ▶).
T. aquaticus SCS and E. coli SCS will bind either ADP/ATP or GDP/GTP. For E. coli SCS this means that the interaction with Glu99β (Fig. 7 ▶ c) is not sufficient to give specificity for the adenine base. A similar statement can be made about T. aquaticus SCS: the interactions with the water molecules bridging the guanine base to Lys45β, Ala203β and Glu92β (Fig. 7 ▶ a) do not give specificity for the guanine base. When ADP/ATP binds to T. aquaticus SCS, the base would be pulled into the β-subunit as in E. coli SCS (Fig. 7 ▶ c), so that N6 of the base could donate a hydrogen bond to the carbonyl O atom of Glu92β and N1 could accept a hydrogen bond from the amide N atom of Val94β. (Note that this amide N atom is the same atom that donates the hydrogen bond to O6 of the guanine base, so the change of base from guanine to adenine causes a shift along the backbone of the residues forming the linker between the two subdomains adopting the ATP-grasp fold.) A water molecule could occupy the space of the carboxylate O atom of Glu99β of E. coli SCS, accepting the hydrogen bond from N6. Based on the preference of T. aquaticus SCS for GDP/GTP, the interactions with ADP/ATP are known to be weaker than those with GDP/GTP. However, the K m(app) for ATP of T. aquaticus SCS is about the same as the K m(app) for ATP of E. coli SCS (Joyce et al., 1999 ▶). Since the residues in the nucleotide-binding site are otherwise very similar, this suggests that the change from Glu to Ala does not have a significant effect on the binding of the adenine base and that the negative charge is not important for the binding of ADP/ATP.
3.4.2. Active-site histidine residue
In all previous structures of SCS the active-site histidine residue is either phosphorylated or bound to a phosphate or a sulfate ion. As in the other structures of SCS, His246α ND1 of T. aquaticus SCS forms a hydrogen bond to the side chain of Glu208α, so it must be protonated. There is a water molecule interacting with His246α NE2, rather than a phosphate or sulfate ion, and this water molecule bridges to a second water molecule. The second water molecule is tightly bound to residues of the two ‘power helices’, the helices that are donated to the active site by the α- and β-subunits (Wolodko et al., 1994 ▶). This tightly bound water molecule has also been observed in the higher resolution structures of phosphorylated SCSs (Fraser et al., 1999 ▶, 2000 ▶, 2006 ▶).
The residues near the catalytic histidine in T. aquaticus and E. coli SCS are similar and would not indicate a shift in the optimal pH of the enzyme. Analysis of the crystal structure of E. coli SCS, which has an optimal pH of 7.3–7.5, concluded that this pH was consistent with a double charge on the phosphoryl group of the phosphohistidine (Wolodko et al., 1994 ▶). The catalytic mechanism would be expected to be the same for all forms of SCS, so the charge state of the phosphohistidine should be the same. Instead of the optimal pH reflecting the pK a values for specific catalytic residues or substrates, the optimal pH could reflect optimal positioning of the residues and substrate molecules for catalysis. The diversity of optimal pH values for SCS is emphasized by the measurement of an optimal pH of 6 for yeast SCS (Schwartz et al., 1983 ▶).
The only cysteine residue in T. aquaticus SCS is Cys123α, which is located near the catalytic histidine and after the cis-peptide bond that links Gly120α and Gly121α. This cysteine residue is highly conserved in SCSs (Hidber et al., 2007 ▶). Lower proportions of cysteine residues have been observed in thermophilic prokaryotes compared with mesophiles (Singer & Hickey, 2003 ▶) and are thought to diminish the chance of oxidation, which would occur more readily at higher temperatures. In the structure of T. aquaticus SCS this cysteine residue is buried; it is protected in part by the turn formed by Gly120α-Gly121α-Asn122α and Pro124α.
3.4.3. Proposed succinate-binding site
The succinate-binding site in SCSs was proposed based on the binding of citrate to ATP-citrate lyase (Sun et al., 2010 ▶). The binding site is located on a loop of the β-subunit near the two power helices. The carboxylate group of succinate that does not take part in the catalytic reaction is expected to interact with the backbone N atoms of Gly321β and Val323β of E. coli SCS or Gly328β and Val330β of pig GTP-specific SCS. In T. aquaticus SCS the equivalent residues are Gly312β and Thr314β. The loop that includes these residues has a different conformation in each of the three enzymes, providing no further information on the binding site (Fig. 8 ▶). The glycine residues in this loop are likely to provide the flexibility observed in the structures of the different SCSs in the absence of succinate. What is consistent in the structures is that this loop is constrained by hydrogen bonds at the two ends. At the amino-terminal end an asparagine side chain forms hydrogen bonds to the amide N atom and carbonyl O atom of Phe319β, Phe326β or Phe310β in E. coli, pig GTP-specific or T. aquaticus SCS, respectively. At the carboxy-terminal end a hydrogen bond is donated by the amide N atom of residue Cys325β, Cys332β or Ala316β in E. coli, pig GTP-specific or T. aquaticus SCS, respectively, to a carbonyl O atom of a residue on the neighbouring loop: Asn352β, Thr359β or Thr343β in E. coli, pig GTP-specific or T. aquaticus SCS, respectively. In each case, the phenylalanine residue is the penultimate residue of the β-strand preceding the loop and the cysteine or alanine residue is followed by an α-helix (Fig. 8 ▶).
Figure 8.

Comparison of the proposed succinate-binding sites of T. aquaticus, pig GTP-specific and E. coli SCS. The three structures were superposed based on the positions of the backbone atoms of the last five residues in the β-strand preceding the loop and the first six residues in the α-helix following the loop. T. aquaticus SCS is shown in yellow, pig GTP-specific SCS (PDB entry 2fp4; Fraser et al., 2006 ▶) is shown in cyan and E. coli SCS (PDB entry 1cqi; Joyce et al., 2000 ▶) is shown in green. Hydrogen bonds constraining the ends of the loop are shown as dashed lines.
3.4.4. Stability enhancement
The crystal structure suggests some residues that are important in enhancing the thermostability of T. aquaticus SCS and its resistance to chemical denaturation. A simple comparison of the residue content of T. aquaticus, E. coli and pig GTP-specific SCS shows the trend that the more stable the SCS, the lower the content of cysteine, serine, phenylalanine, asparagine and glutamine residues and the higher the content of alanine, valine, methionine, histidine and arginine residues. This is generally consistent with the comparison of protein sequences from mesophilic and thermophilic Methanococcus species, which showed fewer uncharged polar residues and more charged residues (Haney et al., 1999 ▶). Examination of the structures shows 15 salt bridges that are present in T. aquaticus SCS but not in either E. coli or pig GTP-specific SCS. Six are between arginine and glutamate residues, three between arginine and aspartate residues, four between lysine and glutamate residues and two between lysine and aspartate residues. Ionic interactions have been proposed to be a major stabilizing feature of glutamate dehydrogenase from the hyperthermophile Pyrococcus furiosus (Rice et al., 1996 ▶).
4. Conclusions
The structure of T. aquaticus SCS in complex with GDP-Mn2+ shows how changes in specific residues within the ATP-grasp fold can shift the nucleotide preference. Like E. coli SCS, T. aquaticus SCS has a proline residue at position 20 of the β-subunit. T. aquaticus SCS also has an alanine residue in the β-subunit where E. coli SCS has a glutamate residue. The smaller alanine residue allows an additional water molecule to bind between the proline and the guanine base. This gives a preference for GDP/GTP over ADP/ATP by providing similar interactions for the guanine base as observed for the glutamine residue at position 20 of the β-subunit of pig GTP-specific SCS.
Supplementary Material
PDB reference: succinyl-CoA synthetase, 3ufx
Acknowledgments
A Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC) funded the characterization using X-ray crystallography. Crystallographic data were collected on beamline 8.3.1 at the Advanced Light Source (ALS) at Lawrence Berkeley Laboratory under an agreement with the Alberta Synchrotron Institute (ASI). The ALS is operated by the Department of Energy and supported by the National Institutes of Health. Beamline 8.3.1 was funded by the National Science Foundation, the University of California and Henry Wheeler. The ASI synchrotron-access program is supported by grants from the Alberta Science and Research Authority and the Alberta Heritage Foundation for Medical Research (AHFMR). The Canadian Institutes of Health Research supported the research characterizing the enzyme in solution. We thank Kim Oikawa in the laboratory of Dr Cyril Kay for conducting circular-dichroism measurements.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Bailey, D. L., Fraser, M. E., Bridger, W. A., James, M. N. & Wolodko, W. T. (1999). J. Mol. Biol. 285, 1655–1666. [DOI] [PubMed]
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed]
- Bridger, W. A. (1971). Biochem. Biophys. Res. Commun. 42, 948–954. [DOI] [PubMed]
- Bridger, W. A. (1974). The Enzymes, edited by P. D. Boyer, Vol. 10, pp. 581–606. New York: Academic Press.
- Brock, T. D. & Freeze, H. (1969). J. Bacteriol. 98, 289–297. [DOI] [PMC free article] [PubMed]
- Brownie, E. R. & Bridger, W. A. (1972). Can. J. Biochem. 50, 719–724. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Cha, S. & Parks, R. E. (1964). J. Biol. Chem. 239, 1968–1977. [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cohn, E. J. & Edsall, J. T. (1943). Proteins, Amino Acids and Peptides as Ions and Dipolar Ions. New York: Reinhold.
- Dams, T., Ostendorp, R., Ott, M., Rutkat, K. & Jaenicke, R. (1996). Eur. J. Biochem. 240, 274–279. [DOI] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org. [DOI] [PubMed]
- Deng, J., Davies, D. R., Wisedchaisri, G., Wu, M., Hol, W. G. J. & Mehlin, C. (2004). Acta Cryst. D60, 203–204. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Fraser, M. E., Hayakawa, K., Hume, M. S., Ryan, D. G. & Brownie, E. R. (2006). J. Biol. Chem. 281, 11058–11065. [DOI] [PubMed]
- Fraser, M. E., James, M. N., Bridger, W. A. & Wolodko, W. T. (1999). J. Mol. Biol. 285, 1633–1653. [DOI] [PubMed]
- Fraser, M. E., James, M. N., Bridger, W. A. & Wolodko, W. T. (2000). J. Mol. Biol. 299, 1325–1339. [DOI] [PubMed]
- Fraser, M. E., Joyce, M. A., Ryan, D. G. & Wolodko, W. T. (2002). Biochemistry, 41, 537–546. [DOI] [PubMed]
- Gibson, J., Upper, C. D. & Gunsalus, I. C. (1967). J. Biol. Chem. 242, 2474–2477. [PubMed]
- Guex, N. & Peitsch, M. C. (1997). Electrophoresis, 18, 2714–2723. [DOI] [PubMed]
- Haney, P. J., Badger, J. H., Buldak, G. L., Reich, C. I., Woese, C. R. & Olsen, G. J. (1999). Proc. Natl Acad. Sci. USA, 96, 3578–3583. [DOI] [PMC free article] [PubMed]
- Hidber, E., Brownie, E. R., Hayakawa, K. & Fraser, M. E. (2007). Acta Cryst. D63, 876–884. [DOI] [PubMed]
- Hooft, R. W., Vriend, G., Sander, C. & Abola, E. E. (1996). Nature (London), 381, 272. [DOI] [PubMed]
- Johnson, M. L., Correia, J. J., Yphantis, D. A. & Halvorson, H. R. (1981). Biophys. J. 36, 575–588. [DOI] [PMC free article] [PubMed]
- Johnson, J. D., Mehus, J. G., Tews, K., Milavetz, B. I. & Lambeth, D. O. (1998). J. Biol. Chem. 273, 27580–27586. [DOI] [PubMed]
- Johnson, J. D., Muhonen, W. W. & Lambeth, D. O. (1998). J. Biol. Chem. 273, 27573–27579. [DOI] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Joyce, M. A., Brownie, E. R., Hayakawa, K. & Fraser, M. E. (2007). Acta Cryst. F63, 399–402. [DOI] [PMC free article] [PubMed]
- Joyce, M. A., Fraser, M. E., Brownie, E. R., James, M. N., Bridger, W. A. & Wolodko, W. T. (1999). Biochemistry, 38, 7273–7283. [DOI] [PubMed]
- Joyce, M. A., Fraser, M. E., James, M. N., Bridger, W. A. & Wolodko, W. T. (2000). Biochemistry, 39, 17–25. [DOI] [PubMed]
- Khan, I. A. & Nishimura, J. S. (1988). J. Biol. Chem. 263, 2152–2158. [PubMed]
- Kraulis, P. J. (1991). J. Appl. Cryst. 24, 946–950.
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Laue, T. M., Shah, B. D., Ridgeway, T. M. & Pelletier, S. L. (1992). Analytical Ultracentrifugation in Biochemistry and Polymer Science, edited by S. E. Harding, A. J. Rowe & J. C. Horton, pp. 90–125. Cambridge: Royal Society of Chemistry.
- Luo, L., Pappalardi, M. B., Tummino, P. J., Copeland, R. A., Fraser, M. E., Grzyska, P. K. & Hausinger, R. P. (2006). Anal. Biochem. 353, 69–74. [DOI] [PubMed]
- McRee, D. E. (1999). J. Struct. Biol. 125, 156–165. [DOI] [PubMed]
- Merritt, E. A. & Bacon, D. J. (1997). Methods Enzymol. 277, 505–524. [DOI] [PubMed]
- Myers, J. K., Pace, C. N. & Scholtz, J. M. (1995). Protein Sci. 4, 2138–2148. [DOI] [PMC free article] [PubMed]
- Navaza, J. (1994). Acta Cryst. A50, 157–163.
- Nishimura, J. S. (1986). Adv. Enzymol. Relat. Areas Mol. Biol. 58, 141–172. [DOI] [PubMed]
- Nishimura, J. S., Ybarra, J., Mitchell, T. & Horowitz, P. M. (1988). Biochem. J. 250, 429–434. [DOI] [PMC free article] [PubMed]
- Norby, J., Rubenstein, S., Tuerke, T., Schwallie Farmer, C., Forood, R. & Bennington, J. (1983). SigmaPlot: Scientific Graph System, v.4.11. SPSS Inc., Chicago, USA.
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Rice, D. W., Yip, K. S., Stillman, T. J., Britton, K. L., Fuentes, A., Connerton, I., Pasquo, A., Scandura, R. & Engel, P. C. (1996). FEMS Microbiol. Rev. 18, 105–117. [DOI] [PubMed]
- Schwartz, H., Steitz, H. O. & Radler, F. (1983). Antonie van Leeuwenhoek, 49, 69–78. [DOI] [PubMed]
- Singer, G. A. & Hickey, D. A. (2003). Gene, 317, 39–47. [DOI] [PubMed]
- Stanislawski, J. (1991). Enzyme Kinetics, v.1.11. Trinity Software, Campton, New Hampshire, USA.
- Sun, T., Hayakawa, K., Bateman, K. S. & Fraser, M. E. (2010). J. Biol. Chem. 285, 27418–27428. [DOI] [PMC free article] [PubMed]
- Weitzman, P. D. J. & Kinghorn, H. A. (1983). FEBS Lett. 154, 369–372.
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wolodko, W. T. & Bridger, W. A. (1987). Biochem. Cell Biol. 65, 452–457. [DOI] [PubMed]
- Wolodko, W. T., Fraser, M. E., James, M. N. & Bridger, W. A. (1994). J. Biol. Chem. 269, 10883–10890. [DOI] [PubMed]
- Wolodko, W. T., Kay, C. M. & Bridger, W. A. (1986). Biochemistry, 25, 5420–5425. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: succinyl-CoA synthetase, 3ufx