

Abstract
Apoptosis-enabling neuronal potassium efflux is mediated by an enhancement of K+ currents. In cortical neurons, increased currents are triggered by dual phosphorylation of Kv2.1 by Src and p38 at channel residues Y124 and S800. It was recently shown that a K+ current surge is also present in hepatocytes undergoing apoptosis, and that the hepatitis C virus (HCV) nonstructural protein 5A (NS5A) could inhibit Kv2.1-mediated currents and block cell death. Here, we show that NS5A1b (from HCV genotype 1b) expression in rat neurons depresses delayed rectifier potassium currents, limits the magnitude of the K+ current surge following exposure to activated microglia, and is neuroprotective. In a non-neuronal recombinant expression system, cells expressing Kv2.1 mutated at residue Y124, but not S800 mutants, are insensitive to NS5A1b-mediated current inhibition. Accordingly, NS5A1b coexpression prevents phosphorylation of wild-type Kv2.1 by Src at Y124, but is unable to inhibit p38 phosphorylation of the channel at S800. The actions of the viral protein are genotype-selective, as NS5A1a does not depress neuronal potassium currents nor inhibit Src phosphorylation of Kv2.1. Our results indicate that NS5A1b limits K+ currents following injury, leading to increased neuronal viability. NS5A1b may thus serve as a model for a new generation of neuroprotective agents.
Introduction
Neuronal proapoptotic signaling cascades are normally accompanied by a robust enhancement of voltage-dependent K+ currents (Yu, 2003). This phenomenon ensures the completion of cell death programs by providing a venue cytoplasmic K+ loss, establishing a permissive environment for caspase and nuclease activation (Hughes et al., 1997, Yu et al., 1997). Interfering with the processes responsible for the apoptotic K+ current surge effectively blocks neuronal cell death (Yu et al., 1997, McLaughlin et al., 2001, Pal et al., 2003). In mammalian cortical neurons, the current surge is mediated by a SNARE-dependent exocytotic insertion of Kv2.1-encoded K+ channels in the cell membrane (Pal et al., 2003, 2006). This process is triggered by the phosphorylation of Kv2.1 by Src and p38 kinases at two distinct intracellular N- and C-terminal residues of the channel, Y124 and S800 (Redman et al., 2007, 2009).
Liver cells undergoing apoptosis also manifest an apoptotic, Kv2.1-mediated current surge (Mankouri et al., 2009). Remarkably, hepatitis C virus (HCV) infection in hepatocytes blocks cell death by interfering with the functional expression of Kv2.1-mediated currents. A product of the translation and processing of the HCV genomic RNA is the nonstructural protein 5A (NS5A). This protein, among other potential actions (Macdonald and Harris, 2004, He et al., 2006), effectively interferes with Kv2.1 K+ currents in liver cells, limiting the apoptotic current surge, and protecting hepatocytes from oxidant-induced apoptosis (Mankouri et al., 2009). The inhibition of hepatocyte apoptotic cell death by NS5A is thought to permit longer periods of viral replication and increase the likelihood of establishing a chronic viral infection in the liver. Here, we establish that the biological strategy that evolved in the hepatitis C virus to block apoptosis in liver cells can be applied to block injury-mediated neuronal cell death. Importantly, we determine the mechanism of action of NS5A in modulating Kv2.1-mediated potassium currents.
Materials and Methods
Cell culture and transfection.
Cortical neuronal cultures were prepared from embryonic day 16 rat embryos of either sex (Hartnett et al., 1997). Cultures were transfected at 25–29 DIV using Lipofectamine 2000 (Invitrogen; Pal et al., 2003) and maintained for 24–48 h before recording and toxicity assays. Microglial cells (Cheepsunthorn et al., 2001) were activated using 10 U/ml interferon-γ (Millipore Bioscience Research Reagents) as described by Knoch et al. (2008). For electrophysiological studies, trans-well inserts containing microglia were placed on top of neuron-containing coverslips. For toxicity studies, microglia were cultured directly on top of previously transfected neurons. Chinese hamster ovary (CHO) cells were transfected with Lipofectamine.
Electrophysiological measurements.
Whole-cell recordings were conducted with 2 MΩ electrodes (McLaughlin et al., 2001) with a solution containing the following (in mm): 115 NaCl, 2.5 KCl, 2.0 MgCl2, 10 HEPES, 10 d-glucose; pH 7.2; 0.25 μm tetrodotoxin. The electrode contained the following (in mm): 100 K-gluconate, 10 KCl, 1 MgCl2, 1 CaCl2, 2.0 Mg2 ATP, 0.33 GTP, 11 EGTA, 10 HEPES, pH 7.2. Partial compensation (80%) for series resistance was performed; currents were filtered at 2 kHz and digitized at 10 kHz. Delayed-rectifier potassium currents were evoked with a series of incremental 80 ms voltage steps to 50 mV from a holding potential of −80 mV, normalized to cell capacitance. In current clamp, the extracellular solution contained the following (in mm): 146 NaCl, 7.8 glucose, 20 HEPES, 4.7 KCl, 0.6 MgSO4, 1.6 NaHCO3, 0.13 NaH2PO4, 2.5 CaCl2; pH 7.3. The electrode solution contained the following (in mm): 94 K+-gluconate, 30 KCl, 10 phosphocreatine di-tris salt, 10 HEPES, 0.2 EGTA, 4 Mg2ATP, 0.3 Na2GTP; pH 7.3. Measurements were performed with an Axoclamp 2B amplifier and G-clamp 2.2 software (Kullmann et al., 2004). Voltage responses were induced with 1 s current steps ranging from −120 pA to 120 pA. Virtual excitatory synapses were implemented under dynamic clamp (Kullmann et al., 2004).
Toxicity assays.
Neuronal toxicity assays were conducted in luciferase cotransfected cells exposed to activated microglia (AMG) as described earlier (Aras et al., 2008).
Immunoprecipitation.
Protein samples from transfected CHO cells were incubated with mouse anti-Kv2.1 monoclonal antibody (NeuroMab) and immunoprecipitated with protein G plus protein A agarose (Calbiochem). Proteins were probed with either mouse monoclonal antibodies against Kv2.1, phosphotyrosine antibodies (BD Biosciences), or immunoaffinity-purified serum directed at phosphorylated S800 of Kv2.1 (Redman et al., 2007). Blots were quantified by infrared fluorimetry (Li-Cor).
Results
NS5A1b suppresses delayed rectifier K+ currents in cortical neurons and is neuroprotective
We transfected cortical neurons with a plasmid encoding a GFP-tagged, genotype 1b form of HCV NS5A. Control cultures were transfected with the GFP-expressing parent vector. Delayed rectifier K+ currents, which in cortical neurons are primarily mediated by Kv2.1-encoded channels (Pal et al., 2003), were significantly smaller (∼40% of control) in NS5A1b-expressing cells, when compared with vector-expressing cells (Fig. 1A). We next induced an apoptotic current surge by incubating cortical cultures with AMG. This treatment has previously been shown to trigger, within 3 h, a very pronounced, Zn2+-dependent enhancement of K+ currents in neurons (Knoch et al., 2008). Both vector-expressing and NS5A1b-expressing neurons showed a significant enhancement in K+ currents following AMG exposure (Fig. 1A). However, the surge induced by AMG in NS5A1b-transfected neurons resulted in K+ current densities that were essentially identical to vector-expressing cells under basal, resting conditions. That is, although an AMG-induced K+ current surge was present in NS5A1b-expressing neurons, the overall magnitudes of the resulting currents were substantially inhibited when compared with control cells (Fig. 1A).
Figure 1.

NS5A interferes with K+ channel function and is neuroprotective. A, Top, Whole-cell K+ currents in rat cortical neurons in culture, evoked by 10 mV incremental steps (−80 to +50 mV). Neurons were transfected with vector (VEC) or NS5A (genotype 1b) and exposed to vehicle or AMG. Scales are 1 nA and 5 ms. Bottom, Mean (± SEM; n = 7–20 cells per group) current densities (at +10 mV) from cells such as those shown above. ANOVA/Bonferroni, all p < 0.001; ***VEC versus NS5A, †VEC versus VEC/AMG, ‡NS5A versus NS5A/AMG and VEC/AMG versus NS5A/AMG. B, Resting membrane voltage (Vm) and input resistance were measured under current clamp (Rin) and threshold synaptic conductance (Thresh-gsyn) was measured under dynamic clamp in untransfected (UT) and vector-expressing cells (VEC) or NS5A1b-transfected neurons (mean ± SEM; n = 5–8 cells per group). Scales are 20 mV and 500 ms. (C) Repetitive firing was recorded for VEC and NS5A1b transfected neurons using 1 s depolarizing current steps of increased amplitudes. Top, Traces are representative of neurons in each group at three depolarizing currents (n = 8). Bottom, Frequency-current relationships were fit to nonlinear regressions and compared using an F test. NS5A1b transfected neurons have a significantly higher repetitive firing frequency as a function of current when compared with VEC transfected neurons (p < 0.01). D, Rat cortical neurons expressing NS5A1b, but not empty vector (VEC), are protected from exposure to AMG. Shown are mean (±SD) luciferase values (counts per second, CPS) of a representative experiment, performed in quadruplicate; *p < 0.05, ANOVA/Dunnet. Inset, mean ± SEM of viability for a total of 7 independent experiments (each performed in quadruplicate), expressed as a percentage of control; **p < 0.01, paired t test.
We evaluated whether NS5A1b expression had a significant effect on the resting membrane potential, input resistance, and threshold synaptic conductance (Fig. 1B). As expected from the voltage dependence of Kv2.1 activation, there were no significant differences between untransfected, vector-expressing, and NS5A1b-expressing neurons in any of these parameters. However, frequency-current relationships (Fig. 1C) revealed that NS5A1b-expressing neurons have a slight, but significantly higher firing frequency as a function of current, when compared with vector-transfected cells, consistent with decreased delayed rectifier current densities in neurons transfected with the viral protein. These results argue that the actions of NS5A1b specifically inhibit Kv2.1 function. They also suggest that the reduction in Kv2.1-mediated currents by NS5A1b may modulate the integrative properties of cortical neurons.
Next, we tested whether NS5A1b expression would render neurons resistant to cell death following AMG exposure. We observed that ∼40% of vector-expressing neurons were lethally injured by AMG within 24 h. In contrast, cells expressing NS5A1b were virtually immune to microglial-induced toxicity (Fig. 1D). These results are consistent with prior observations suggesting that limiting the apoptotic K+ current surge can enhance the viability of injured neurons (Yu et al., 1997, McLaughlin et al., 2001, Pal et al., 2003). Our results, however, do not rule out the possibility that NS5A1b may also interfere with other cell death processes (He et al., 2006), in addition to the functional inhibition of K+ currents. Indeed, as an apoptotic surge is still present in NS5A1b-expressing cells, additional mechanisms may be involved in neuroprotection. These issues are addressed further below.
NS5A1b inhibits Src, but not p38, phosphorylation of Kv2.1-encoded K+ channels
To investigate the molecular mechanism of K+ channel modulation by NS5A1b, we tested whether the viral protein could inhibit functional Kv2.1 channels in CHO cells. These cells have no endogenous voltage-dependent K+ channels (Yu and Kerchner, 1998) but have all the necessary signaling components to generate an apoptotic surge when expressing Kv2.1 (Pal et al., 2003, 2006). Similar to the effects on native neuronal K+ currents, we observed that currents in CHO cells coexpressing Kv2.1 and NS5A1b were substantially decreased when compared with control cells (Fig. 2A).
Figure 2.
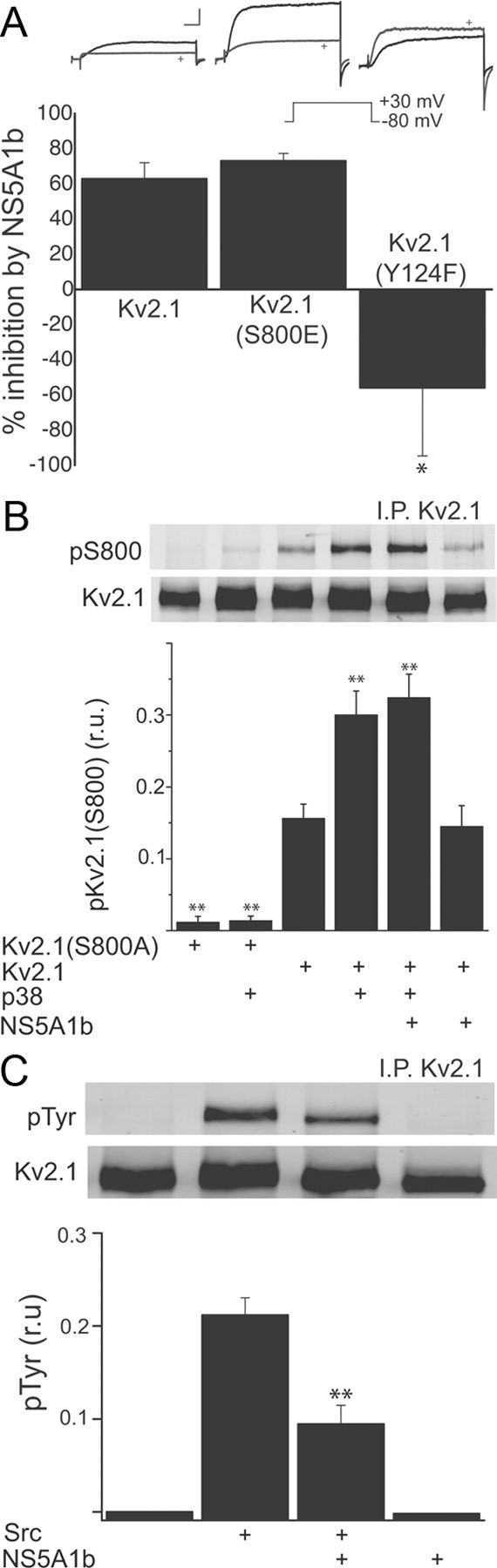
NS5A1b inhibits Src, but not p38, phosphorylation of Kv2.1. A, Top, Whole-cell currents (−80 to +30 mV step) in CHO cells expressing Kv2.1, Kv2.1(S800E), or Kv2.1 (Y124F) together with either VEC or NS5A1b (marked by a +). Scales are 5 nA and 5 ms. Bottom, Average K+ current suppression by NS5A1b coexpression, expressed as a percentage of the average currents present in vector-expressing cells; n = 9–19 cells per group (42 cells total); *p < 0.05, significantly different from the other two groups, ANOVA/Dunnet. B, CHO cells were cotransfected with Kv2.1(S800A), Kv2.1, p38, NS5A1b or vector. Kv2.1 immunoprecipitates were run by SDS-PAGE and probed with a phospho-specific antibody recognizing phosphorylated residue S800. Top, Representative immunoblot; Bottom, summary and quantification of results in three independent experiments, normalized to total Kv2.1 protein; **p < 0.01, ANOVA/Dunnet versus Kv2.1 control (third bar). C, CHO cells were cotransfected with Kv2.1, Src, NS5A1b or vector. Kv2.1 immunoprecipitates were run by SDS-PAGE and probed with a phosphotyrosine antibody. Top, Representative immunoblot; Bottom, summary and quantification of results in three independent experiments; **p < 0.01, ANOVA/Bonferroni versus Src/vector group (second vs third bar).
Mankouri et al. (2009) suggested that NS5A blocked hepatocyte K+ current function by interfering with p38 kinase activation and subsequent phosphorylation of Kv2.1 by this MAPK. We had previously shown that p38 phosphorylation of residue S800 on Kv2.1 was a critical step necessary for the insertion of new channels into the plasma membrane, underlying the apoptotic K+ current surge (Pal et al., 2006, Redman et al., 2007). We tested this proposed mechanism of NS5A1b by using a recombinant expression system, along with a variant of Kv2.1 in which the serine at position 800 had been mutated to a glutamate, producing a pseudo-phosphorylated K+ channel with enhanced surface expression (Redman et al., 2007). We hypothesized that these mutant channels would be insensitive to NS5A1b expression, as the enhanced currents they generate are independent from p38 activity (Redman et al., 2007). To our surprise, K+ currents in CHO cells expressing Kv2.1(S800E) channels were just as sensitive to inhibition by NS5A1b when compared with wild-type (Fig. 2A).
Since this result suggested a different mechanism of action by the viral protein than that suggested by Mankouri et al. (2009), we investigated the status of p38 phosphorylation of Kv2.1 in CHO cells expressing NS5A1b. We had previously raised a phospho-specific antibody specifically recognizing Kv2.1 when phosphorylated at S800 (Redman et al., 2007), which we used to probe proteins immunoprecipitated from CHO cells using a pan-Kv2.1 antibody. Confirmation of the specificity of our phospho-specific antibody was performed in cells expressing a nonphosphorylatable mutant channel, Kv2.1(S800A) (Redman et al., 2007). Overexpression of p38 led to an expected rise in the levels of S800 phosphorylation in wild-type but not mutant Kv2.1(S800A) (Fig. 1B). Importantly, and as predicted by our electrophysiological data, NS5A1b did not affect the levels of basal or p38-stimulated phosphorylation of residue S800 of Kv2.1. Together, these data indicate that although NS5A1b can depress basal Kv2.1-mediated currents in neurons and in a recombinant expression system, as well as limit the apoptotic surge in neurons, it likely does not interfere with p38 phosphorylation of Kv2.1. Therefore, we searched for an alternative explanation for the observed K+ current inhibition produced by the viral protein.
In addition to the p38 kinase-specific residue, Kv2.1 is phosphorylated by a variety of other kinase systems (Misonou et al., 2005), including Src (Tiran et al., 2003). Indeed, specific phosphorylation and dephosphorylation of residue Y124 by Src and by protein tyrosine phosphatase ε, respectively, can upregulate and downregulate Kv2.1 channel activity (Peretz et al., 2000, Tiran et al., 2003). We also have shown that residue Y124 of Kv2.1, in addition to S800, is critical for the apoptotic current surge mediated by this channel (Redman et al., 2007). Intriguingly, NS5A contains two, very closely spaced class II polyproline motifs that interact with Src-homology 3 (SH3) domains and inhibit the activity of the Src-family kinases Hck, Lck and Fyn (Macdonald et al., 2004). We therefore evaluated the extent of NS5A1b modulation of Kv2.1 in mutants of this channel lacking a phosphorylatable Y124 (n.b., the pseudo phosphorylated mutants Y125E or Y124D are nonfunctional; Redman et al., 2009). Consistent with a putative inhibitory effect of NS5A on Src-mediated processes, we observed that K+ currents in CHO cells expressing Kv2.1(Y124F) mutants were not influenced by the viral protein (Fig. 2A). In fact, CHO cells expressing the mutant channel tended to produce slightly larger currents in NS5A1b-expressing cells, when compared vector-expressing cells (vector = 217.3 ± 39.2 pA/pF, n = 19; NS5A1b = 339.3 ± 84.1 pA/pF, n = 12). This result led us to evaluate whether NS5A1b could prevent Src phosphorylation of Kv2.1.
We had previously attempted, but were unsuccessful, in raising a phospho-specific antibody against the Kv2.1 phosphorylated at residue Y124 (Redman et al., 2009). Fortunately, most (∼80%) of the Src phosphorylation of Kv2.1 occurs at this residue (Tiran et al., 2003). As such, immunoprecipitated Kv2.1 protein from CHO overexpressing Src in the presence or absence of NS5A1b could be probed with a phosphotyrosine antibody. Under control conditions, we observed little to no immunoprecipitated Kv2.1 that was tyrosine-phosphorylated. Src overexpression, however, led to a pronounced phosphotyrosine signal, which, in agreement with our electrophysiological results, was significantly diminished in cells coexpressing NS5A1b (Fig. 2C). Together, these results strongly suggest that NS5A1b regulates K+ channel function by modulating the actions of Src kinase on residue Y124 of Kv2.1.
A recent report has suggested that two additional tyrosine residues on Kv2.1 (Y686 and Y810) can also be minor targets of Src-mediated phosphorylation (Song et al., 2012) and are thus the likely source of the small, but consistent, remaining phosphorylation signal induced by Src in Kv2.1(Y124F) channels (Tiran et al., 2003). In agreement with these findings, we observed a small (17% of wild-type), but detectable phosphotyrosine signal in Src-overexpressing CHO cells transfected with Kv2.1(Y124F) (phosphotyrosine levels were 0.037 ± 0.003 r.u., n = 3; compared with 0.212 ± 0.019 r.u., n = 3, from Fig. 2C; p < 0.001, unpaired t test). NS5A1b coexpression also inhibited this more modest Src-induced phosphorylation of Kv2.1(Y124F) (0.015 ± 0.004 r.u., n = 3; significantly different from vector-expressing cells, p < 0.05, t test). This suggests that the effects of the viral protein are not selective for any particular tyrosine residue, but rather, they represent a general repressive mechanism of the actions of Src.
NS5A actions are genotype-specific
Mankouri et al. (2009) demonstrated that K+ currents were normal in a hepatocyte-derived cell line harboring a HCV replicon in which the class II polyproline, SH3-binding domains of NS5A had been disrupted by multiple proline to alanine mutations (repliconPA2). We thus evaluated whether K+ currents would be affected in neurons expressing a plasmid encoding NS5A harboring the same mutations. In apparent agreement with a putative action of the viral protein on Src activity via its SH3-binding motif, K+ currents in NS5A(PA2)-expressing neurons were identical to vector-expressing cells (Fig. 3A). We noted however, that the mutated plasmid was derived from a different HCV genotype, 1a (Street et al., 2004). As there is a 20% difference in the NS5A sequences between the 1a and 1b genotypes, we expressed wild-type NS5A1a in neurons and examined its effects on K+ currents as a control. Surprisingly, the K+ current inhibitory effects were absent when using wild-type NS5A1a (Fig. 3A). In accordance with the electrophysiological results, NS5A1a also did not inhibit Src-mediated tyrosine phosphorylation of Kv2.1 channels expressed in CHO cells (Fig. 3B). Moreover, NS5A1a did not afford similar neuroprotection as its 1b counterpart in cortical neurons exposed to AMG (viability was 69.8 ± 2.3% of control; n = 3; compare to Fig. 1B). Together, these data indicate that the inhibitory effects of NS5A on Kv2.1-mediated channels are genotype-specific. Moreover, the neuroprotective actions of the HCV protein also depend on the genotype used, suggesting that the K+ channel inhibitory effect of NS5A1b is a critical component of its antiapoptotic effects.
Figure 3.
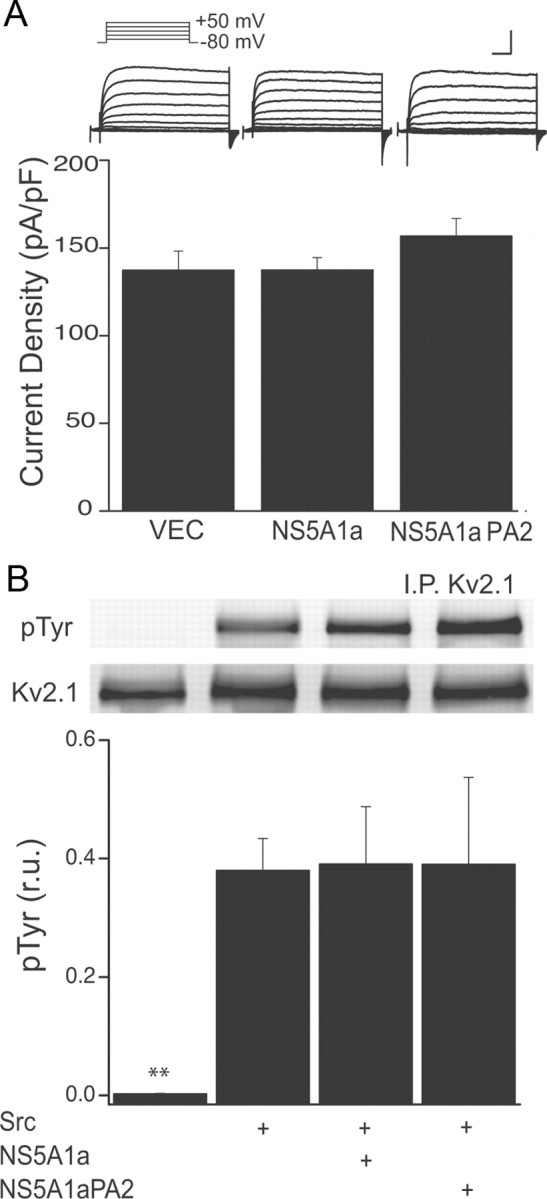
Genotype 1a NS5A does not block K+ currents in neurons, or prevents SRC phosphorylation of Kv2.1. A, Whole-cell K+ currents in rat cortical neurons in culture, evoked by 10 mV incremental steps (−80 to +50 mV). Neurons were transfected with vector (VEC), NS5A1a, or NS5A1a(PA2) plasmids (Top). Scales are 3 nA and 25 ms. Bottom, Mean (±SEM; n = 14–16 cells per group; 44 total cells) current densities (at +10 mV) from neurons transfected with empty vector, NS5A (genotype 1a) or a mutant (PA2) of NS5A1a. B, Experiments similar to those shown in Figure 2C, illustrating the lack of inhibition of Src phosphorylation of Kv2.1 channels in CHO cells expressing NS5A1b (n = 3); **p < 0.01, ANOVA/Dunnet; all groups are significantly different from Kv2.1+vector group (first bar).
Discussion
We report here that the genotype 1b HCV-derived protein NS5A interferes with delayed-rectifier, Kv2.1-mediated K+ currents, and limits the magnitude of the enhancement of these currents that accompanies apoptotic cell death. NS5A1b modulates this process by interfering with the phosphorylation of Kv2.1 by Src. Importantly, the interference of NS5A1b with the functional expression of K+ currents is important for rendering neurons resistant to a lethal, injurious stimulus. Finally, we find that NS5A from genotype 1a HCV is devoid of these K+ channel modulatory properties.
NS5A derived from HCV genotypes 1a and 1b have ∼80% amino acid sequence homology (Reed et al., 1997). Most sequence differences are found near the C-terminal domain of the protein (Kim et al., 2011), the so-called domain 3 (Macdonald and Harris, 2004, He et al., 2006), immediately downstream from the SH3-binding polyproline motifs, which themselves are highly conserved among all HCV genotypes (Tan et al., 1999). It has been suggested that different phosphorylation profiles are present in NS5A1a and NS5A1b, particularly within 70 aa that lie most proximal to the C-terminal (Dal Pero et al., 2007), and that phosphorylation events at NS5A are critical for many of the cellular effects of HCV (Tellinghuisen et al., 2008). Whether the genotype-specific effects of NS5A1b on K+ channel modulation are due to specific amino acid differences, phosphorylation levels, or both, remain to be established. Of interest, however, genotype 1b has been associated with more severe liver disease, more aggressive disease progression, and importantly, increase incidence of hepatocellular carcinoma in patients exhibiting chronic, active HCV infection (Zein, 2000). Although clearly there are many potential reasons for the difference in genotype-dependent disease profile, the anti-apoptotic properties of NS5A1b via its inhibition of Kv2.1 channel function could be important component for its increased persistence and association with liver cancer.
Distinct to the mechanism proposed by Mankouri et al. (2009), our results suggest that the K+ current modulatory effects of NS5A are not mediated by the SH3-binding, polyproline motifs in this protein, since, as mentioned earlier, both the 1a and 1b genotypes contain these sequences, and NS5A1a lacked function in our assays. In addition, while the SH3-binding domain of NS5A effectively binds and inhibits the action of the Src-family kinases Hck, Lck and Lyn, it does not apparently bind Src itself (Macdonald et al., 2004). However, it was recently reported that Src is required for the interaction between NS5A1b and the HCV RNA-dependent polymerase NS5B (Pfannkuche et al., 2011). NS5A1b can indeed bind Src, not at the kinase SH3 domain, but rather, at a different, SH2 domain (Pfannkuche et al., 2011). Although, the specific NS5A1b SH2-binding interaction region has yet to be defined (Pfannkuche et al., 2011), the likely requirement for phosphorylated tyrosine residues, in combination with the known phosphorylation differences levels between NS5A1b and NS5A1a (Tellinghuisen et al., 2008), could provide important clues at defining the mode of action of the viral protein for modulating K+ channel function. This mechanism, which evolved in a virus to aid in inhibiting cell death, may provide important information for the development of a novel neuroprotective strategy for neurodegenerative disorders associated with an apoptotic K+ current surge following injury.
Footnotes
This work was supported by NIH Grant NS078238 (to E.A.). C.A.N. is supported by NIH Training Grant GM08424 and the Achievement Rewards for College Scientists Foundation, Pittsburgh Chapter. We thank Dr. M. Harris, University of Leeds, for the gift of all NS5A-expressing plasmids, J. Trimmer, University of California, Davis, for Kv2.1, and Ari Elson, Weizmann Institute, for Kv2.1(Y124F).
References
- Aras MA, Hartnett KA, Aizenman E. Assessment of cell viability in primary neuronal cultures. Curr Protoc Neurosci Chapter. 2008;7 doi: 10.1002/0471142301.ns0718s44. Unit 7.18. [DOI] [PubMed] [Google Scholar]
- Cheepsunthorn P, Radov L, Menzies S, Reid J, Connor JR. Characterization of a novel brain-derived microglial cell line isolated from neonatal rat brain. Glia. 2001;35:53–62. doi: 10.1002/glia.1070. [DOI] [PubMed] [Google Scholar]
- Dal Pero F, Di Maira G, Marin O, Bortoletto G, Pinna LA, Alberti A, Ruzzene M, Gerotto M. Heterogeneity of CK2 phosphorylation sites in the NS5A protein of different hepatitis C virus genotypes. J Hepatol. 2007;47:768–776. doi: 10.1016/j.jhep.2007.07.020. [DOI] [PubMed] [Google Scholar]
- Hartnett KA, Stout AK, Rajdev S, Rosenberg PA, Reynolds IJ, Aizenman E. NMDA receptor-mediated neurotoxicity: a paradoxical requirement for extracellular Mg2+ in Na+/Ca2+-free solutions in rat cortical neurons in vitro. J Neurochem. 1997;68:1836–1845. doi: 10.1046/j.1471-4159.1997.68051836.x. [DOI] [PubMed] [Google Scholar]
- He Y, Staschke KA, Tan SL. HCV NS5A: a multifunctional regulator of cellular pathways and virus replication. In: Tan SL, editor. Hepatitis C viruses: genomes and molecular biology. Norfolk, UK: Horizon Bioscience; 2006. pp. 267–292. [PubMed] [Google Scholar]
- Hughes FM, Jr, Bortner CD, Purdy GD, Cidlowski JA. Intracellular K+ suppresses the activation of apoptosis in lymphocytes. J Biol Chem. 1997;272:30567–30576. doi: 10.1074/jbc.272.48.30567. [DOI] [PubMed] [Google Scholar]
- Kim S, Welsch C, Yi M, Lemon SM. Regulation of the production of infectious genotype 1a hepatitis C virus by NS5A domain III. J Virol. 2011;85:6645–6656. doi: 10.1128/JVI.02156-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoch ME, Hartnett KA, Hara H, Kandler K, Aizenman E. Microglia induce neurotoxicity via intraneuronal Zn(2+) release and a K(+) current surge. Glia. 2008;56:89–96. doi: 10.1002/glia.20592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann PH, Wheeler DW, Beacom J, Horn JP. Implementation of a fast 16-Bit dynamic clamp using LabVIEW-RT. J Neurophysiol. 2004;91:542–554. doi: 10.1152/jn.00559.2003. [DOI] [PubMed] [Google Scholar]
- Macdonald A, Harris M. Hepatitis C virus NS5A: tales of a promiscuous protein. J Gen Virol. 2004;85:2485–2502. doi: 10.1099/vir.0.80204-0. [DOI] [PubMed] [Google Scholar]
- Macdonald A, Crowder K, Street A, McCormick C, Harris M. The hepatitis C virus NS5A protein binds to members of the Src family of tyrosine kinases and regulates kinase activity. J Gen Virol. 2004;85:721–729. doi: 10.1099/vir.0.19691-0. [DOI] [PubMed] [Google Scholar]
- Mankouri J, Dallas ML, Hughes ME, Griffin SD, Macdonald A, Peers C, Harris M. Suppression of a pro-apoptotic K+ channel as a mechanism for hepatitis C virus persistence. Proc Natl Acad Sci U S A. 2009;106:15903–15908. doi: 10.1073/pnas.0906798106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B, Pal S, Tran MP, Parsons AA, Barone FC, Erhardt JA, Aizenman E. p38 activation is required upstream of potassium current enhancement and caspase cleavage in thiol oxidant-induced neuronal apoptosis. J Neurosci. 2001;21:3303–3311. doi: 10.1523/JNEUROSCI.21-10-03303.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Trimmer JS. Kv2.1: a voltage-gated k+ channel critical to dynamic control of neuronal excitability. Neurotoxicology. 2005;26:743–752. doi: 10.1016/j.neuro.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Pal SK, Takimoto K, Aizenman E, Levitan ES. Apoptotic surface delivery of K+ channels. Cell Death Differ. 2006;13:661–667. doi: 10.1038/sj.cdd.4401792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E. Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci. 2003;23:4798–4802. doi: 10.1523/JNEUROSCI.23-12-04798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz A, Gil-Henn H, Sobko A, Shinder V, Attali B, Elson A. Hypomyelination and increased activity of voltage-gated K(+) channels in mice lacking protein tyrosine phosphatase epsilon. EMBO J. 2000;19:4036–4045. doi: 10.1093/emboj/19.15.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfannkuche A, Büther K, Karthe J, Poenisch M, Bartenschlager R, Trilling M, Hengel H, Willbold D, Häussinger D, Bode JG. c-Src is required for complex formation between the hepatitis C virus-encoded proteins NS5A and NS5B: a prerequisite for replication. Hepatology. 2011;53:1127–1136. doi: 10.1002/hep.24214. [DOI] [PubMed] [Google Scholar]
- Redman PT, He K, Hartnett KA, Jefferson BS, Hu L, Rosenberg PA, Levitan ES, Aizenman E. Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc Natl Acad Sci U S A. 2007;104:3568–3573. doi: 10.1073/pnas.0610159104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman PT, Hartnett KA, Aras MA, Levitan ES, Aizenman E. Regulation of apoptotic potassium currents by coordinated zinc-dependent signalling. J Physiol. 2009;587:4393–4404. doi: 10.1113/jphysiol.2009.176321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed KE, Xu J, Rice CM. Phosphorylation of the hepatitis C virus NS5A protein in vitro and in vivo: properties of the NS5A-associated kinase. J Virol. 1997;71:7187–7197. doi: 10.1128/jvi.71.10.7187-7197.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MY, Hong C, Bae SH, So I, Park KS. Dynamic modulation of the kv2.1 channel by SRC-dependent tyrosine phosphorylation. J Proteome Res. 2012;11:1018–1026. doi: 10.1021/pr200770v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street A, Macdonald A, Crowder K, Harris M. The hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J Biol Chem. 2004;279:12232–12241. doi: 10.1074/jbc.M312245200. [DOI] [PubMed] [Google Scholar]
- Tan SL, Nakao H, He Y, Vijaysri S, Neddermann P, Jacobs BL, Mayer BJ, Katze MG. NS5A, a nonstructural protein of hepatitis C virus, binds growth factor receptor-bound protein 2 adaptor protein in a Src homology 3 domain/ligand-dependent manner and perturbs mitogenic signaling. Proc Natl Acad Sci U S A. 1999;96:5533–5538. doi: 10.1073/pnas.96.10.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellinghuisen TL, Foss KL, Treadaway J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 2008;4:e1000032. doi: 10.1371/journal.ppat.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiran Z, Peretz A, Attali B, Elson A. Phosphorylation-dependent regulation of Kv2.1 Channel activity at tyrosine 124 by Src and by protein-tyrosine phosphatase epsilon. J Biol Chem. 2003;278:17509–17514. doi: 10.1074/jbc.M212766200. [DOI] [PubMed] [Google Scholar]
- Yu SP. Regulation and critical role of potassium homeostasis in apoptosis. Prog Neurobiol. 2003;70:363–386. doi: 10.1016/s0301-0082(03)00090-x. [DOI] [PubMed] [Google Scholar]
- Yu SP, Kerchner GA. Endogenous voltage-gated potassium channels in human embryonic kidney (HEK293) cells. J Neurosci Res. 1998;52:612–617. doi: 10.1002/(SICI)1097-4547(19980601)52:5<612::AID-JNR13>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- Zein NN. Clinical significance of hepatitis C virus genotypes. Clin Microbiol Rev. 2000;13:223–235. doi: 10.1128/cmr.13.2.223-235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]