

Abstract
A series of six mono-, di-, and trivalent N,N’-diacetylchitobiose derivatives was conveniently prepared by employing a one-pot procedure for Cu(II)-catalyzed diazo transfer and Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) starting from commercially available amines. These glycoclusters were probed for their binding potencies to the plant lectin wheat germ agglutinin (WGA) from Triticum vulgaris by an enzyme-linked lectin assay (ELLA) employing covalently immobilized N-acetylglucosamine (GlcNAc) as a reference ligand. IC50 values were in the low micromolar/high nanomolar range, depending on the linker between the two disaccharides. Binding enhancements β up to 1000 for the divalent ligands and 2800 for a trivalent WGA ligand, compared to N,N’-diacetylchitobiose as the corresponding monovalent ligand, were observed. Molecular modeling studies, in which the chitobiose moieties were fitted into crystallographically determined binding sites of WGA, correlate the binding enhancements of the multivalent ligands with their ability to bind to the protein in a chelating mode. The best WGA ligand is a trivalent cluster with an IC50 value of 220 nM. Calculated per mol of contained chitobiose, this is the best WGA ligand known so far.
Keywords: carbohydrates, click chemistry, cluster effect, lectins, multivalency
Introduction
The recognition of carbohydrate structures by carbohydrate binding proteins (lectins) plays a fundamental role in numerous intra- and intercellular events during development, inflammation, immune response, cancer metastasis, and pathogen–host interactions [1–2]. Inhibition of such interactions by high-affinity ligands is of high medicinal interest for the treatment of many human diseases. However, carbohydrate–protein interactions are often characterized by low binding affinities. A possible solution to compensate for these weak individual receptor–ligand interactions is the multivalent presentation of sugar epitopes on suitable scaffolds. This principle is not only used in nature but is also a valid strategy for the construction of artificial lectin ligands [3–13]. Prime examples are the recently described ligands for the Shiga-like [14–15] and cholera toxins [16–17] both belonging to the AB5 family of bacterial toxins.
The frequent observation that the binding affinity of a multivalent ligand increases exponentially with the number of binding sites has been termed the glycoside cluster effect [18–19]. Due to the exponential increase of binding affinities, the cluster effect often leads to the amplification of the binding selectivity. This was experimentally demonstrated, for example, by Mortell et al. while investigating glycopolymer ligands of concanavalin A (Con A) [20]. Whereas two diastereomeric (monovalent) C-glycosidic Con A ligands displayed only a small difference in the free energies of binding to Con A, a sizable difference was measured between the corresponding multivalent C-glycosides (calculated per monovalent ligand within the glycopolymer). Such effects can be analyzed in the context of the chelate effect [21], and a number of theoretical models to treat multivalent receptor–ligand interactions have been developed [22–27]. A simple conclusion following from these analyses is that multimerization of monovalent ligands with enhanced binding affinity can lead to multivalent ligands with disproportionally enhanced avidity. A prerequisite for an effective multivalency effect, however, is that the linking spacer between the individual epitopes has the correct geometry to allow a simultaneous multipoint association, i.e., a chelating binding mode.
Wheat germ agglutinin (WGA), besides other plant lectins such as Con A, has been intensively employed as a model lectin to study the influence of the structure of multivalent ligands on the binding affinity. WGA ligands of defined structure containing two to twelve GlcNAc residues obtained either by individual synthesis [28–36] or from screening of combinatorial libraries [37–38] have been reported. WGA is a 36 kDa plant lectin composed of two identical glycine- and cysteine-rich subunits [39] and is enriched in the seeds of Triticum vulgaris. It is specific for terminal N-acetylneuraminic acid and N-acetylglucosamine (GlcNAc) and has been shown to inhibit fungal growth through interaction with fungal cell-wall components [40–42] and to agglutinate transformed cells in vitro [43–44].
Recently, we determined the structural basis of multivalent binding to WGA by X-ray crystallography [36] and EPR spectroscopy [45]. Crystal structure analysis of a complex of WGA and four molecules of a divalent ligand containing two GlcNAc residues showed that each ligand bridged adjacent binding sites with a distance of approx. 13–14 Å between the anomeric oxygen atoms of the GlcNAc residues. This structure confirmed for the first time that all eight sugar binding sites of the WGA dimer [46] are simultaneously functional, and provides the basis for the design of new multivalent ligands with improved binding affinity.
Besides GlcNAc, WGA also binds to chitooligosaccharides with even higher affinity. The association constant for the WGA–N,N’-diacetylchitobiose interaction, for example, has been determined to be K ≈ 5 × 103 to 2 × 104 M–1. The corresponding value for binding to GlcNAc is K ≈ 2 × 102 to 1.3 × 103 M−1 [47]. This prompted us to design a series of multivalent WGA ligands containing two or three N,N’-diacetylchitobiose moieties. To connect the chitobiose moieties we chose several linkers of varying length and flexibility, which were, nevertheless, all expected to allow simultaneous binding to adjacent binding sites in a chelating fashion, thus, leading to especially effective ligands. In this report, we describe the preparation of such a series of multivalent WGA ligands by a one-pot procedure for diazo transfer and azide–alkyne cycloaddition [48] starting from commercially available di- and triamines and the propargyl glycoside of N,N’-diacetylchitobiose. Binding potencies were determined by an enzyme-linked lectin assay (ELLA), resulting in IC50 values in the low-micromolar/high-nanomolar range. A trivalent ligand has a remarkable IC50 value of 220 nM. Molecular dynamics calculations based on published X-ray crystal structures of WGA-ligand complexes provide an explanation for the observed binding affinities.
Results and Discussion
Synthesis of glycoclusters
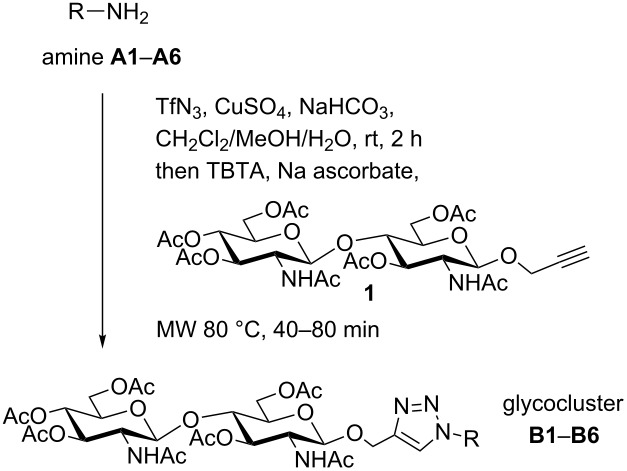
The Cu(I)-catalyzed [49–50] Huisgen [3 + 2] cycloaddition [51] of azides and alkynes (CuAAC) is a frequently used method for the covalent attachment of carbohydrate epitopes to azide- or alkyne-presenting scaffolds [52–54]. Recently, we reported a convenient one-pot procedure for diazo transfer and azide–alkyne cycloaddition [48] giving access to multivalent triazole-linked structures starting from amines. For the synthesis of triazole-linked glycoclusters, commercially available amines A1–A6 (Figure 1) comprising different spacer geometries were selected. These amines were employed in the sequential one-pot procedure [48] for diazo transfer and CuAAC (Table 1). First, the Cu(II)-catalyzed diazo transfer was performed at ambient temperature until complete conversion of the amines to azides. Then, CuAAC was provoked without any workup procedure by the addition of tris(benzyltriazolylmethyl)amine [55] (TBTA), sodium ascorbate, and the propargyl glycoside 1 [56] of N,N’-diacetylchitobiose and heating of the mixture to 80 °C by microwave irradiation, until TLC showed complete consumption of the intermediate azides (see Supporting Information File 1 for full experimental data).
Figure 1.

Amines used for the synthesis of glycoclusters.
Table 1.
Synthesis of glycoclusters B1–B6 using the one-pot procedure for diazo transfer and azide-alkyne cycloaddition.
 | ||
| Amine | Product | Yield (%) |
| A1 | B1 | 45 |
| A2 | B2 | 30 |
| A3 | B3 | 37 |
| A4 | B4 | 28 |
| A5 | B5 | 48a |
| A6 | B6 | 6 |
aIn this case the intermediate diazide 2 was isolated (cf. Scheme 1).
According to TLC all reactions (except for B6) proceeded with complete conversion of the amines to the desired glycoconjugates. However, some loss of material during purification of the acetylated chitobiose derivatives by flash chromatography on silica gel led to the moderate yields indicated in Table 1. Monovalent compounds resulting from partial reactions of the diamines were not observed. To exclude that the observed yields are a result of the one-pot procedure, the preparation of divalent B5 was carried out in two separate steps (Scheme 1). Diazo transfer with A5 gave diazide 2 in a yield of 95%. Subsequent CuAAC of isolated 2 with alkyne 1 delivered B5 after flash chromatography in 51% yield with no observed side products. Severe loss of about 50% of the material during flash chromatography was also experienced when a pure sample of disaccharide 1 was eluted from a silica gel column for a second time. In comparison, the use of the less polar propargyl β-D-glucoside instead of 1 in the one-pot procedure with amine A4 led to the corresponding divalent glycocluster in a yield of 86% [48]. Finally, O-deacetylation of the glycoclusters B1–B6 under Zemplén conditions resulted in WGA ligands C1–C6 (Scheme 2).
Scheme 1.

Synthesis of glycocluster B5 with isolation of the intermediate diazide 2.
Scheme 2.

Deacetylation of glycoconjugates B1–B6. (a) NaOMe, MeOH.
During the synthesis of trivalent compound B6 the formation of a side product with a Rf similar to that of B6 was observed. This is remarkable because all other reactions proceeded without the formation of side-products. The mass of this side product (m/z [M + H]+ = 1500.7) corresponds to a divalent compound in which one arm of the tertiary amine is missing. Since this side product could not be isolated in pure form, we investigated the reaction of A6 with phenylacetylene (3, Scheme 3). Also with this alkyne two main products were obtained which were difficult to separate by chromatography. Apart from the expected tris(triazole) 4, the secondary amine 5 was isolated and characterized. This structure corresponds to the assumed side-product obtained during the synthesis of B6. A contamination of the starting material A6 with secondary amine di(2-aminoethyl)amine was excluded. ESI–MS measurements indicated that side-product formation may already take place during the diazo transfer reaction of A6 because the mass of the corresponding intermediate di(2-azidoethyl)amine (m/z [M + H]+ = 156.2) was found. The mechanism of this side product formation is not clear. We assume that the mechanism is due to the special structure of A6 because comparable side-product formation was not observed with any other amine used.
Scheme 3.

Formation of side-product 5 during the synthesis of 4.
Determination of binding potencies by ELLA
Binding potencies of compounds C1–C6, GlcNAc, and N,N’-diacetylchitobiose were determined by an ELLA employing covalently immobilized GlcNAc as a reference ligand, as described recently [34]. GlcNAc-coated microtiter plates were incubated with mixtures of horseradish-peroxidase-labeled WGA (HRP-WGA) and synthetic WGA ligands in varying concentrations. After incubation, the plates were washed and the remaining HRP-WGA bound to the microtiter plates was quantified by a HRP-catalyzed color reaction. Dose-response curves for inhibition of the binding of HRP–WGA to immobilized GlcNAc are shown in Figure 2. From these curves the concentrations at which the binding of HRP–WGA to GlcNAc is reduced by 50% (IC50 values) were determined as a measure of the potency of the synthesized inhibitors (Table 2). Also shown in Table 2 are the relative inhibitory potencies (β values) referenced to N,N’-diacetylchitobiose (β = 1).
Figure 2.

Dose-response curves for the inhibition of binding of HRP-labeled WGA to covalently immobilized GlcNAc by synthetic ligands C1–C6.
Table 2.
Absolute and relative IC50 values of synthetic ligands C1–C6 for inhibition of the binding of HRP-labeled WGA to covalently immobilized GlcNAc from dose-response curves shown in Figure 2.
| Compound | IC50 (µM) | β |
| GlcNAc | 51000 | 1/82 |
| N,N'-diacetylchitobiose | 620 | 1 |
| monovalent C1 | 290 | 2.1 |
| divalent C2 | 1.3 | 480 |
| divalent C3 | 1.9 | 330 |
| divalent C4 | 0.60 | 1000 |
| divalent C5 | 0.72 | 860 |
| trivalent C6 | 0.22 | 2800 |
With an 82 times lower IC50 value, N,N’-diacetylchitobiose is a much better inhibitor than GlcNAc, which is in accordance with the association constants determined by solution binding assays [47]. The benzyl triazolyl appendage of C1 further enhances binding by a factor of two, probably due to additional weak hydrophobic interactions. However, introducing an additional N,N’-diacetylchitobiose epitope leads to dramatically increased affinities. The divalent chitobiose derivatives C2–C5 have IC50 values in the low-micromolar/high-nanomolar range, with some variation due to different spacer properties. Their inhibitory potencies relative to N,N'-diacetylchitobiose are 330–1000. It is interesting to note the differences induced by differing linker geometries. Here, not the most hydrophobic linkers show strongest binding but the ones that apparently promote multivalent binding most efficiently. Whereas the flexible linkers of C4 and C5 lead to β values of 1000 and 860, respectively, the ligands C2 and C3 with the less flexible aromatic linkers have significantly lower binding potencies (β values: 480 and 330). This observation points to the possibility that the aromatic linkers cannot adopt a strain-free conformation if the ligand binds in a chelating mode to WGA. The best divalent WGA ligand is C4 (IC50 0.6 µM), which binds 1000 times stronger to WGA than N,N’-diacetylchitobiose (500 times per chitobiose residue).
As expected, clustering of carbohydrate epitopes with higher WGA binding affinity not only leads to multivalent ligands with higher absolute affinity but also to a higher binding enhancement β relative to the respective monovalent compound. Earlier, we reported WGA binding affinities of β-O-glycosidic divalent GlcNAc derivatives with linker lengths comparable to those of C2–C5 [34]. These GlcNAc derivatives displayed β values of 80–260 relative to GlcNAc, which are much lower than the β values (330–1000, relative to N,N'-diacetylchitobiose) determined for C2–C5 (Table 2). The relative potency of C4 of 500 per chitobiose residue is even higher than that of tetra- to octavalent GlcNAc clusters [28,31–34,37]. We are aware of only one example of a divalent GlcNAc derivative with an exceptional β value of 2350 [36]. In this case, however, the GlcNAc moieties are α-O-glycosidically linked.
For trivalent cluster C6 an IC50 value of 220 nM (660 nM per contained chitobiose) was determined, which is 2800-fold lower than that of N,N'-diacetylchitobiose or 230000-fold lower than the IC50 value of GlcNAc. This is one of the best WGA ligands known. Masaka et al. reported a tetravalent N,N’-diacetylchitobiose derivative with an IC50 value of 180 nM (720 nM per contained chitobiose) determined by a hemagglutination inhibition assay [57]. Calculating the IC50 value per contained chitobiose, trivalent C6 is the better ligand. However, since such numbers are strongly dependent on the employed assay [58–59], they cannot be readily compared. Interestingly, the ligand reported by Masaka et al. led to precipitation of WGA. In this respect it is worth mentioning that we never observed precipitate formation during incubation of WGA with our synthetic ligands. This suggests that in our case intermolecular multivalency (cross-linking) plays a negligible role and that the main mechanism of affinity enhancement is chelating binding to the same WGA dimer.
The 2.8-fold increased inhibition potency of C6 over the best divalent ligand C4 indicates that C6 can reach only two WGA binding sites simultaneously due to its geometrical properties, which is fully in accordance with the structural investigations described below. Otherwise, a significantly stronger binding enhancement would have been expected comparable to the several-hundred-fold increase observed when moving from mono- to divalent ligand structures.
Molecular modeling
To provide a structure-based rationalization for the determined binding potencies of C2–C6, we performed molecular modeling studies. Combining information from the crystal structures of WGA3 in complex with N,N’-diacetylchitobiose (PDB ID: 1K7U) [60] and WGA3 binding to a divalent ligand presenting two GlcNAc residues (PDB ID: 2X52) [36], two N,N’-diacetylchitobiose residues were placed in a pair of adjacent primary binding sites of WGA and connected by the respective linker (see Supporting Information File 1 for details). Subsequently, the linkers were energy minimized with the chitobiose moieties fixed in their ideal positions. The models containing the various linker structures were further energy minimized with the terminal GlcNAc residues kept in their optimal positions.
Our molecular modeling studies revealed that the para-disubstituted aromatic linker of C3 cannot adopt a low-energy conformation if the chitobiose residues are kept in their ideal positions in the binding sites of WGA, resulting in significant ring strain of the triazole moieties as well as the central phenyl ring. This ring strain can be reduced by slightly pulling the GlcNAc residues directly attached to the linker out of the binding site, but at the expense of a less efficient multivalent binding of the two chitobiose entities. The chitobiose groups of the divalent ligand C4, on the other hand, can maintain their ideal positions easily with the linker adopting an all-staggered low-energy conformation (Figure 3). Ligand C2 allows for the positioning of its chitobiose moieties ideally in both binding sites when adopting a fully extended conformation. Ligand C2, and even more so ligand C3, possess very limited conformational freedom when adopting a chelating binding mode. Conformational changes within the linkers of C2 and C3 lead to forces that pull one or the other GlcNAc/chitobiose out of its binding site. This situation may increase the entropic costs of chelating binding for these two ligands providing a further explanation for their significantly lower binding affinity.
Figure 3.

Molecular model of divalent ligand C4 with its two chitobiose moieties occupying two adjacent binding sites of WGA. The linker is conformationally largely unrestricted and can adopt several low-energy conformations. WGA chain 1 is colored yellow, chain 2 is blue.
With its linker length and flexibility, trivalent ligand C6 is in an intermediate position between the relatively short-bridged and rigid ligands C2 and C3 and the more flexibly connected ligands C4 and C5. With six chemical bonds between the triazole groups, C6 can present its N,N’-diacetylchitobiose residues in rather similar distances as C2 or C3 having the same linker length. However, C6 has two more rotatable bonds between pairs of chitobiose moieties leading to significantly increased conformational freedom. This is expected to facilitate binding in a chelating fashion at relatively low entropic costs. It is important to note that the third chitobiose unit of C6 cannot reach a third carbohydrate binding site of the WGA dimer. The closest distance to another binding site is approximately 24 Å (measured between the anomeric oxygens of the inner GlcNAc residues). The 2.8-fold increased potency relative to ligand C4 could possibly originate from the facilitated rebinding of monovalently bound trivalent ligand C6 due to the two-fold-higher local concentration of chitobiose compared to the divalent ligands.
Conclusion
In summary, we have presented a series of O-glycosidically linked N,N’-diacetylchitobiose clusters that were conveniently obtained from propargyl glycoside 1 and readily available amine scaffolds by a one-pot procedure for diazo transfer and azide–alkyne cycloaddition. Binding potencies were determined by an ELLA. Divalent ligands were found to have IC50 values in the low-micromolar/high-nanomolar range depending on the linker between the two disaccharides. The observed binding enhancements over the monovalent ligand are significantly higher than those of comparable β-linked GlcNAc clusters. The different binding enhancements can be rationalized by molecular modeling studies that correlate the different linker geometries with their propensities to support chelating binding. The best WGA ligand is trivalent cluster C6 with a remarkable IC50 value of 220 nM. Calculated per mol of contained chitobiose, this is the best WGA ligand published so far.
Supporting Information
Experimental procedures and analytical data for all new compounds.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (FOR 434) and the University of Konstanz. H.S.G.B. acknowledges a fellowship according to the Landesgraduiertenförderungsgesetz.
This article is part of the Thematic Series "Synthesis in the glycosciences II".
References
- 1.Dwek R A. Chem Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 2.Varki A, Cummings R D, Esko J D, et al., editors. Essentials of Glycobiology. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 3.Mammen M, Choi S-K, Whitesides G M. Angew Chem, Int Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 4.Lee Y C, Lee R T, editors. Neoglycoconjugates: Preparation and Applications. San Diego, CA: Academic Press; 1994. [Google Scholar]
- 5.Kiessling L L, Gestwicki J E, Strong L E. Angew Chem, Int Ed. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins B E, Paulson J C. Curr Opin Chem Biol. 2004;8:617–625. doi: 10.1016/j.cbpa.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Choi S-K. Synthetic Multivalent Molecules. Concepts and Biomedical Applications. Hoboken, NJ: John Wiley & Sons; 2004. [DOI] [Google Scholar]
- 8.Ortiz Mellet C, Defaye J, García Fernández J M. Chem–Eur J. 2002;8:1982–1990. doi: 10.1002/1521-3765(20020503)8:9<1982::AID-CHEM1982>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 9.Lindhorst T K. Top Curr Chem. 2002;218:201–235. doi: 10.1007/3-540-45010-6_7. [DOI] [Google Scholar]
- 10.Chabre Y M, Roy R. Adv Carbohydr Chem Biochem. 2010;63:165–393. doi: 10.1016/S0065-2318(10)63006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dam T K, Brewer C F. Biochemistry. 2008;47:8470–8476. doi: 10.1021/bi801208b. [DOI] [PubMed] [Google Scholar]
- 12.Pieters R J. Org Biomol Chem. 2009;7:2013–2025. doi: 10.1039/b901828j. [DOI] [PubMed] [Google Scholar]
- 13.Wittmann V. Synthetic Approaches to Study Multivalent Carbohydrate-Lectin Interactions. In: Schmuck C, Wennemers H, editors. Highlights in Bioorganic Chemistry: Methods and Applications. Weinheim, Germany: Wiley-VCH; 2004. pp. 203–213. [DOI] [Google Scholar]
- 14.Kitov P I, Sadowska J M, Mulvey G, Armstrong G D, Ling H, Pannu N S, Read R J, Bundle D R. Nature. 2000;403:669–672. doi: 10.1038/35001095. [DOI] [PubMed] [Google Scholar]
- 15.Kitov P I, Mulvey G L, Griener T P, Lipinski T, Solomon D, Paszkiewicz E, Jacobson J M, Sadowska J M, Suzuki M, Yamamura K-i, et al. Proc Natl Acad Sci U S A. 2008;105:16837–16842. doi: 10.1073/pnas.0804919105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan E, Zhang Z, Minke W E, Hou Z, Verlinde C L M J, Hol W G J. J Am Chem Soc. 2000;122:2663–2664. doi: 10.1021/ja993388a. [DOI] [Google Scholar]
- 17.Merritt E A, Zhang Z, Pickens J C, Ahn M, Hol W G J, Fan E. J Am Chem Soc. 2002;124:8818–8824. doi: 10.1021/ja0202560. [DOI] [PubMed] [Google Scholar]
- 18.Lee Y C, Lee R T. Acc Chem Res. 1995;28:321–327. doi: 10.1021/ar00056a001. [DOI] [Google Scholar]
- 19.Lundquist J J, Toone E J. Chem Rev. 2002;102:555–578. doi: 10.1021/cr000418f. [DOI] [PubMed] [Google Scholar]
- 20.Mortell K H, Weatherman R V, Kiessling L L. J Am Chem Soc. 1996;118:2297–2298. doi: 10.1021/ja953574q. [DOI] [Google Scholar]
- 21.Jencks W P. Proc Natl Acad Sci U S A. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hubble J. Mol Immunol. 1999;36:13–18. doi: 10.1016/S0161-5890(98)00116-3. [DOI] [PubMed] [Google Scholar]
- 23.Gargano J M, Ngo T, Kim J Y, Acheson D W K, Lees W J. J Am Chem Soc. 2001;123:12909–12910. doi: 10.1021/ja016305a. [DOI] [PubMed] [Google Scholar]
- 24.Ercolani G. J Am Chem Soc. 2003;125:16097–16103. doi: 10.1021/ja038396c. [DOI] [PubMed] [Google Scholar]
- 25.Kitov P I, Bundle D R. J Am Chem Soc. 2003;125:16271–16284. doi: 10.1021/ja038223n. [DOI] [PubMed] [Google Scholar]
- 26.Krishnamurthy V M, Semetey V, Bracher P J, Shen N, Whitesides G M. J Am Chem Soc. 2007;129:1312–1320. doi: 10.1021/ja066780e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diestler D J, Knapp E W. J Phys Chem C. 2010;114:5287–5304. doi: 10.1021/jp904258c. [DOI] [Google Scholar]
- 28.Zanini D, Roy R. Bioconjugate Chem. 1997;8:187–192. doi: 10.1021/bc970005u. [DOI] [PubMed] [Google Scholar]
- 29.Furuike T, Aiba S. Chem Lett. 1999;28:69–70. doi: 10.1246/cl.1999.69. [DOI] [Google Scholar]
- 30.García-López J J, Hernández-Mateo F, Isac-García J, Kim J M, Roy R, Santoyo-González F, Vargas-Berenguel A. J Org Chem. 1999;64:522–531. doi: 10.1021/jo981576y. [DOI] [Google Scholar]
- 31.Furuike T, Aiba S, Nishimura S-I. Tetrahedron. 2000;56:9909–9915. doi: 10.1016/S0040-4020(00)00962-5. [DOI] [Google Scholar]
- 32.Consoli G M L, Cunsolo F, Geraci C, Sgarlata V. Org Lett. 2004;6:4163–4166. doi: 10.1021/ol0485767. [DOI] [PubMed] [Google Scholar]
- 33.Yamada A, Hatano K, Koyama T, Matsuoka K, Takahashi N, Hidari K I P J, Suzuki T, Suzuki Y, Terunuma D. Bioorg Med Chem. 2007;15:1606–1614. doi: 10.1016/j.bmc.2006.12.030. [DOI] [PubMed] [Google Scholar]
- 34.Maierhofer C, Rohmer K, Wittmann V. Bioorg Med Chem. 2007;15:7661–7676. doi: 10.1016/j.bmc.2007.08.063. [DOI] [PubMed] [Google Scholar]
- 35.Misawa Y, Masaka R, Maeda K, Yano M, Murata T, Kawagishi H, Usui T. Carbohydr Res. 2008;343:434–442. doi: 10.1016/j.carres.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 36.Schwefel D, Maierhofer C, Beck J G, Seeberger S, Diederichs K, Möller H M, Welte W, Wittmann V. J Am Chem Soc. 2010;132:8704–8719. doi: 10.1021/ja101646k. [DOI] [PubMed] [Google Scholar]
- 37.Wittmann V, Seeberger S. Angew Chem, Int Ed. 2004;43:900–903. doi: 10.1002/anie.200352055. [DOI] [PubMed] [Google Scholar]
- 38.Wittmann V, Seeberger S. Angew Chem, Int Ed. 2000;39:4348–4352. doi: 10.1002/1521-3773(20001201)39:23<4348::AID-ANIE4348>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 39.Wright C S. J Mol Biol. 1980;141:267–291. doi: 10.1016/0022-2836(80)90181-3. [DOI] [PubMed] [Google Scholar]
- 40.Mirelman D, Galun E, Sharon N, Lotan R. Nature. 1975;256:414–416. doi: 10.1038/256414a0. [DOI] [PubMed] [Google Scholar]
- 41.Laroy K, Weeks G. J Cell Sci. 1983;59:203–212. doi: 10.1242/jcs.59.1.203. [DOI] [PubMed] [Google Scholar]
- 42.Asensio J L, Cañada F J, Siebert H-C, Laynez J, Poveda A, Nieto P M, Soedjanaamadja U M, Gabius H-J, Jiménez-Barbero J. Chem Biol. 2000;7:529–543. doi: 10.1016/S1074-5521(00)00136-8. [DOI] [PubMed] [Google Scholar]
- 43.Aub J C, Sanford B H, Cote M N. Proc Natl Acad Sci U S A. 1965;54:396–399. doi: 10.1073/pnas.54.2.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aub J C, Sanford B H, Wang L-H. Proc Natl Acad Sci U S A. 1965;54:400–402. doi: 10.1073/pnas.54.2.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Braun P, Nägele B, Wittmann V, Drescher M. Angew Chem, Int Ed. 2011;50:8428–8431. doi: 10.1002/anie.201101074. [DOI] [PubMed] [Google Scholar]
- 46.Wright C S, Kellogg G E. Protein Sci. 1996;5:1466–1476. doi: 10.1002/pro.5560050803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bains G, Lee R T, Lee Y C, Freire E. Biochemistry. 1992;31:12624–12628. doi: 10.1021/bi00165a012. [DOI] [PubMed] [Google Scholar]
- 48.Beckmann H S G, Wittmann V. Org Lett. 2007;9:1–4. doi: 10.1021/ol0621506. [DOI] [PubMed] [Google Scholar]
- 49.Rostovtsev V V, Green L G, Fokin V V, Sharpless K B. Angew Chem, Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 50.Tornøe C W, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 51.Huisgen R. Angew Chem, Int Ed Engl. 1963;2:565–598. doi: 10.1002/anie.196305651. [DOI] [Google Scholar]
- 52.Meldal M, Tornøe C W. Chem Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 53.Pieters R J, Rijkers D T S, Liskamp R M J. QSAR Comb Sci. 2007;26:1181–1190. doi: 10.1002/qsar.200740075. [DOI] [Google Scholar]
- 54.Dedola S, Nepogodiev S A, Field R A. Org Biomol Chem. 2007;5:1006–1017. doi: 10.1039/b618048p. [DOI] [PubMed] [Google Scholar]
- 55.Chan T R, Hilgraf R, Sharpless K B, Fokin V V. Org Lett. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 56.Merkel L, Beckmann H S G, Wittmann V, Budisa N. ChemBioChem. 2008;9:1220–1224. doi: 10.1002/cbic.200800050. [DOI] [PubMed] [Google Scholar]
- 57.Masaka R, Ogata M, Misawa Y, Yano M, Hashimoto C, Murata T, Kawagishi H, Usui T. Bioorg Med Chem. 2010;18:621–629. doi: 10.1016/j.bmc.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 58.André S, Specker D, Bovin N V, Lensch M, Kaltner H, Gabius H-J, Wittmann V. Bioconjugate Chem. 2009;20:1716–1728. doi: 10.1021/bc900152w. [DOI] [PubMed] [Google Scholar]
- 59.Corbell J B, Lundquist J J, Toone E J. Tetrahedron: Asymmetry. 2000;11:95–111. doi: 10.1016/S0957-4166(99)00589-3. [DOI] [Google Scholar]
- 60.Muraki M, Ishimura M, Harata K. Biochim Biophys Acta. 2002;1569:10–20. doi: 10.1016/S0304-4165(01)00231-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and analytical data for all new compounds.