

Abstract
Purpose
The objectives of this study were to determine whether high-glucose-induced upregulation of heparanase (HPSE) expression and differential heparanase expression in human retinal vascular endothelial cells (HRECs) can alter HREC migration and proliferation. We also aimed to determine whether HREC migration and proliferation correlate with the levels of protein kinase B (Akt) and extracellular-signal-regulated kinase (ERK) phosphorylation and activation.
Methods
HRECs were treated with either 5 mM glucose (Glu5) or high (30 mM) glucose (Glu30) for 48 h. Untransfected HRECs were grown in human endothelial serum-free medium (HE-SFM) in the presence of 5 mM glucose and supplemented with 30 mM mannitol for 48 h as an osmotic control (mannitol). HRECs were also infected with a heparanase small interfering RNA recombinant lentiviral vector (HPSE-LV) or a control vector (Con-LV) at a multiplicity of infection (MOI) of 60 for three days. Then the con-LV and HPSE-LV-infected cells were treated with 30 mM glucose for 48 h (Con-LV-Glu30 and HSPE-LV-Glu30, respectively). The expression levels of heparanase mRNA and protein and HREC proliferation and migration were examined using quantitative real-time polymerase chain reaction (qRT–PCR), western blot analysis, 3-(4,5-dimethylthiahiazol-2-y1)-2,5-diphenyltetrazolium bromide assay, bromodeoxyuridine histochemical staining, and the Boyden chamber assay. The expression level of paxillin was examined using immunofluorescent staining. Akt and ERK phosphorylation was evaluated using western blot analysis.
Results
We successfully transfected the HPSE RNAi lentiviral vector into HRECs and demonstrated that it can suppress the expression of the heparanase gene in these cells. Western blot and qRT–PCR analyses showed that HRECs treated with a high concentration of glucose exhibited increased heparanase protein and mRNA levels, while the levels were decreased in HRECs that had been infected with HPSE-LV before treatment with high glucose (HPSE-LV-Glu30; p<0.05). The observed increase or decrease in the levels of heparanase correlated with increased or decreased HREC migration and proliferation, respectively (p<0.05). HREC proliferation and migration were found to correlate with Akt and ERK phosphorylation levels (p<0.5).
Conclusions
Our results indicate that heparanase plays a significant role in mediating retinal vascular endothelial cell proliferation and migration after the HRECs are exposed to high levels of glucose. Signaling inducing heparanase-stimulated HREC proliferation and migration appears to be related to the activation of Akt and ERK via their phosphorylation.
Introduction
Diabetes is the primary chronic systemic disease responsible for visual loss [1]. Diabetic retinopathy (DR) is the leading cause of preventable blindness in adults worldwide [2]. Developing countries with rapidly evolving economies face the challenge of a DR epidemic [3,4].
Angiopathy, a complication of diabetes mellitus, is characterized by microvascular pathology in the retina and renal glomerulus. Abnormal angiogenesis-induced vascular leakage, endothelial cell damage [5,6] and severely impaired retinal function are the consequences of retinal hypoxia and ischemia. The endothelial cells (ECs) that line the blood vessels appear to be the initial targets of the vascular damage due to hyperglycemia. Furthermore, changes due to hyperglycemia can cause vascular remodeling [7]. These abnormalities result in vasoconstriction, hypertension, tissue ischemia, and eventually infarction and an increase in vascular permeability [8].
Heparan sulfate (HS) is a glycosaminoglycan associated with the cell membrane, basement membrane, and extracellular matrix (ECM) [9]. The depletion of HS and/or alteration in its metabolism is considered a major mechanism of EC injury [10-12]. Heparanase is a mammalian endoglucuronidase responsible for HS degradation, and yields HS fragments with an appreciable size (5–10 kDa) and biologic potency [10,13]. HS is a major constituent of the ECM, and HS-degrading activity is thought to play a decisive role in the fundamental biologic processes associated with remodeling of the ECM, such as angiogenesis and cancer metastasis. Heparanase activity has generally been shown to correlate with cell invasion processes associated with cancer metastasis, which is a consequence of a structural modification that loosens the ECM barrier [14-16].
Studies have suggested that heparanase may be induced by hyperglycemia [17] and may contribute to EC dysfunction by degrading HS. Adding heparanase to the media of cultured endothelial cells results in injury to these cells [17]. Researchers have reported that heparanase induction correlates with increased tumor metastasis, vascular density, and a shorter post-operative survival rate, thus providing strong clinical support for the prometastatic and proangiogenic functions of this enzyme [18-21]. In addition to the well studied catalytic characteristics of heparanase, researchers have reported it exerts biologic functions that are apparently independent of its enzymatic activity. Recently, researchers reported that adding latent heparanase stimulates Akt-dependent endothelial cell invasion and migration activities that are independent of heparanase’s enzymatic activity [22]. Nonenzymatic functions of heparanase include enhanced adhesion of gliomas [23], lymphomas [23,24], and T cells [25]; these functions are mediated by β1-integrin and correlate with protein kinase B (Akt), proline-rich tyrosine kinase 2 (Pyk2), and extracellular-signal-regulated kinase (ERK) activation [23,25]. The ability of heparanase to function in a nonenzymatic manner and to elicit signal transduction cascades led us to hypothesize that its potent angiogenic functions observed under clinical and experimental settings are due to the release of HS–bound growth factors and the regulation of human retinal vascular endothelial cell (HREC) proliferation and migration. Thus, our objectives were to determine whether heparanase is upregulated in HRECs exposed to high glucose and whether heparanase can be downregulated using a specific RNA interference (RNAi) vector in HRECs grown under high-glucose conditions. Our results indicate heparanase expression levels correlate with HREC proliferation and migration; furthermore, our data suggest the putative signal transduction pathway by which this action occurs.
Methods
Human retinal vascular endothelial cell culture and infection
Our experiments followed the principles of the Declaration of Helsinki. Cultured HRECs from the eyes of eight donors were obtained from the Zhongshan Ophthalmic Center Eye Bank. The donors, whose mean age was 22.3 years, were otherwise healthy victims of accidents. To avoid significant pigment epithelium contamination, we harvested the retinas carefully, placed them in Hank’s balanced salt solution (137.93 mM NaCl, 5.33 mM KCl, 4.17 mM NaHCO3, 0.441 mM KH2PO4, 0.338 mM Na2HPO4, 5.56 mM D-Glucose), and washed them. After we gently minced them, the retinal pieces were digested in 2% trypsin for 20 min at room temperature, collected by centrifugation at 217.3392× g, and resuspended in 0.1% collagenase for 20 min at 37 °C. We centrifuged the homogenate (150.93× g, 10 min), and resuspended the pellet in HE-SFM supplemented with 10% fetal bovine serum (FBS), 5 ng/ml recombinant human β-endothelial cell growth factor (β-ECGF; R&D Systems, Minneapolis, MN), and 1% insulin-transferrin-selenium. We cultured the cells in fibronectin-coated flasks and incubated them at 37 °C in a humidified atmosphere containing 5% CO2. We characterized the cultured cells for endothelial homogeneity by testing for immunoreactivity to factor VIII antigen using light microscopy. To avoid age-dependent cellular modification, we used only cells at passages 2 and 3.
We purchased the Homo sapiens (human) heparanase RNAi lentiviral expression vector (RNAi TargetSeq 5′-CCA GGA TAT TTG CAA ATA T-3′) and the corresponding control expression vector pGCSIL from Shanghai Genechem Co. Ltd. (Shanghai, China). We seeded the lentivirus-infected HRECs in six-well plates in HE-SFM media plus FBS, β-endothelial cell growth factor (β-ECGF), and 1% insulin-transferrin-selenium. We cultured the cells in fibronectin-coated flasks and incubated at 37 °C in a humidified atmosphere containing 5% CO2 until the cells reached 40% confluence. Subsequently, we added 3×106 TU per well of the recombinant lentivirus to the cells and cultured them in an incubator at 37 °C in 5% CO2 for three days at a multiplicity of infection (MOI) of 60.
We used seven parallel wells for each group of HRECs: untransfected HRECs were grown in HE-SFM in the presence of 5 mM glucose medium supplemented with 30 mM mannitol for 48 h as an osmotic control (mannitol), untransfected HRECs were cultured in HE-SFM in the presence of either 5 mM (Glu5) or 30 mM glucose (Glu30) for 48 h, and control pGCSIL-transfected HRECs (Con-LV) and lentivirus RNAi heparanase-transfected HRECs (HPSE-LV) were cultured in the presence of 30 mM glucose for 48 h (Con-LV-Glu30 and HPSE-LV-Glu30, respectively). We evaluated the phosphorylation levels of Akt and ERK using western blotting and the level of heparanase expression using real-time reverse transcription PCR and western blotting. We evaluated the effects on cell proliferation using 3-(4,5-dimethylthiahiazol-2-y1)-2,5-diphenyltetrazolium bromide (MTT) assays and bromodeoxyuridine (BrdU) staining, and evaluated the effects on cell migration using the Boyden chamber assay and paxillin immunofluorescent staining. Quantification with quantitative real-time polymerase chain reaction
After transfection, we extracted the total RNA from the cells harvested from the different groups. We isolated the total RNA using TRIzol Reagent (Invitrogen, San Diego, CA). We determined the concentration and purity of the RNA samples using a spectrophotometer. We purified the RNA from the cells using Amplification Grade DNase I (AMPD1; Sigma-Aldrich, St. Louis, MO), and used RNA reverse transcriptase (PrimeScript RT Reagent Kit; DRR037A; TaKaRa, Tokyo, Japan) to synthesize cDNA. We performed quantitative real-time polymerase chain reaction (RT–PCR) assays using the SYBR Green Real-Time PCR Master Mix (SYBR Premix Ex Taq Kit, TaKaRa), and measured gene expression levels using SYBR Green II (Qiagen, Tpkyo, Japan) and the ABI PRISM 7000 (Applied Biosystems Company, Foster City, CA) real-time PCR system.
The PCR primers we used to detect heparanase and 18S were as follows: heparanase sense strand (5′-CTC GAA GAA AGA CGG CTA AGA-3′); heparanase reverse strand (5′-TGG TAG CAG TCC GTC CAT T-3′); 18S sense strand (5′-TTC CGA TAA CGA ACG AGA CTC T-3′); and 18S reverse strand (5′-TGG CTG ACA CGC CAC TTG TC-3′).
Western blotting
We prepared cell lysates from the cells grown in six-well plates (including seven sample groups: Glu5, mannitol, Glu30, Con-LV, HPSE-LV, Con-LV-Glu30, and HPSE-LV-Glu30) by adding to each well 100 µl of lysis buffer containing 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.5% Triton X-100, and a protease inhibitor cocktail (Sigma-Aldrich, Inc.). We centrifuged the harvested samples at 11,000× g at 4 °C for 30 min. We collected the supernatants, and evaluated the protein supernatants using bicinchoninic acid protein and assay kit (Bio-Rad, Hercules, CA). We mixed lysates containing 100 mg of protein with loading buffer containing 5% β-mercaptoethanol and heated them for 8 min at 100 °C. We separated the samples with sodium dodecyl sulfate–PAGE and subsequently transferred them to polyvinylidene difluoride membranes (Bio-Rad). We incubated the membranes in blocking buffer (Tris-buffered saline [TBS], 0.1% Tween-20, and 5% nonfat dry milk) for 1 h at room temperature, followed by hybridization with a rabbit antiheparanase-1 polyclonal antibody (1:1,000 dilution; Abcam, Cambridge, UK), a rabbit antiphospho-Akt monoclonal antibody (1:1,000 dilution; Cell Signaling Technology, Danvers, MA), a rabbit anti-Akt (pan) monoclonal antibody (1:1,000 dilution; Cell Signaling Technology), a rabbit antiphospho-ERK monoclonal antibody (1:1,000 dilution; Cell Signaling Technology), a rabbit anti-ERK (pan) monoclonal antibody (1:1,000 dilution; Cell Signaling Technology), or a mouse antiactin monoclonal antibody (1:1,000 dilution; Lab Vision, Fremont, CA) at 4 °C overnight. After washing the membranes three times in TBS/0.1% Tween-20, we hybridized the membranes with a horseradish peroxidase-conjugated antirabbit or antimouse immunoglobulin G secondary antibody (1:1000 dilution; Cell Signaling Technology) for 1 h at room temperature. We washed the membranes three times in TBS/0.1% Tween-20, and detected signals using chemiluminescence with luminol (Santa Cruz Biotechnology, Santa Cruz, CA). We performed semiquantitative analysis by measuring the optical densities of the bands.
Cell migration assay
We performed a cell migration assay using a specialized Boyden migration chamber that included a 24-well tissue culture plate with 12 cell culture inserts (Chemicon, Temecula, CA). The inserts contained an 8-µm pore size polycarbonate membrane with a thin precoated layer of basement membrane matrix (EC Matrix). Briefly, we starved HRECs, Con-LV, and HPSE-LV cells in 2% calf serum overnight and then seeded them at a concentration of 5×103 cells/well in Transwell plates. We added culture medium containing 10% FBS supplemented with either 5 mM glucose (Glu5 group) or 30 mM glucose (Glu30, Con-LV-Glu30, and HPSE-LV-Glu30 groups) to each upper chamber and lower compartment. After 48 h of incubation at 37 °C, we removed the HRECs from the upper surface of the membrane using a moist cotton-tipped swab. We stained the migrating cells on the lower surface of the membrane, which had migrated through the polycarbonate membrane, with crystal violet staining solution for 30 min and rinsed them three times with distilled water. We quantified migration by selecting 10 different views repeated 400 times and calculating the number of migrating cells.
Cell proliferation assay
We used an MTT assay to quantify cell proliferation. We initially grew HRECs to confluence before seeding them into a 96-well plate at a density of 4×103 cells/well for 48 h. We determined cell viability by adding 200 µl of 5 mg/ml MTT assay to each well followed by incubation for an additional 4 h. We solubilized precipitates by adding 150 µl of dimethyl sulfoxide (DMSO) to each well. After 10 min of shaking, we assessed the absorbance value (A value) at 570 nm. The observed optical density directly correlated to the number of cells. We repeated these experiments three times.
Immunofluorescent staining
For immunofluorescent staining, we fixed the cells with 4% paraformaldehyde for 20 min, washed them with phosphate buffer solution (PBS, KH2PO4 0.27 g,Na2HPO4 1.42 g, NaCl 8 g, KCl 0.2 g, and add the whole liquid is 1000 ml, pH is 7.4), and permeabilized them with 0.5% Triton X-100 in PBS for 15 min. We then incubated the cells in PBS containing 10% normal goat serum for 1 h at room temperature, followed by 1 h of incubation with the indicated primary antibody (rabbit antipaxillin monoclonal antibody; 1:250 dilution; Abcam). We thoroughly washed the cells with PBS and incubated them with the appropriate Cy3-conjugated secondary antibody (1:250 dilution; Jackson ImmunoResearch, West Grove, PA) for 1 h. Then we washed the cells with PBS, stained them with Hoechst 33342 (Sigma-Aldrich) for 5 min, washed them with PBS again, and mounted them. We observed staining under a fluorescence microscope.
To assess cell growth using bromodeoxyuridine (BrdU) staining, we counted the cells every day for four days after trypsinization using a Coulter counter, and confirmed the cell numbers by counting with a hemocytometer. Additionally, we analyzed cell proliferation by measuring BrdU (10 μmol/l; Sigma-Aldrich) incorporation using a cell proliferation labeling reagent. Briefly, we incubated subconfluent cells grown on glass coverslips in complete growth medium supplemented with 5 or 30 mM glucose for 48 h in the presence of BrdU, fixed them with 4% paraformaldehyde for 20 min, permeabilized them with 1.0% Triton X-100 in PBS for 15 min, washed them with PBS, and incubated them with PBS containing 10% normal goat serum for 1 h at room temperature. We then incubated the cells with a mouse anti-BrdU antibody (1:20 dilution; Abcam) for 1 h at room temperature, thoroughly washed them with PBS, and incubated them with the appropriate Cy3-conjugated secondary antibody (1:250 dilution; Jackson ImmunoResearch) for 1 h at room temperature. We subsequently washed and stained the cells with Hoechst 33342 (Sigma-Aldrich) for 5 min, washed with them PBS, and mounted them. We observed staining under a fluorescence microscope. We determined the mitotic index by calculating the number of BrdU-positive nuclei as a percentage of the total number of cells.
Statistical analysis
Data are presented as the mean±SD. We analyzed statistical significance with one-way ANOVA using the GraphPad Prism 4.0 software system (GraphPad, San Diego, CA) and the statistical software program SPSS 16.0 for Windows (Chicago, IL). Values of p<0.05 were considered significant in all cases.
Results
Heparanase expression increased in human retinal vascular endothelial cells under high-glucose conditions
HREC cultures were established successfully, and the percentage of factor VIII-positive cells in the cultured cells was 96.7%. We conducted in vitro cell culture studies to determine whether heparanase expression was upregulated in HRECs under high-glucose conditions. As shown in Figure 1, heparanase expression was significantly higher (2.6 fold) in high-glucose-treated HRECs than in HRECs grown in normal medium (p<0.01). HRECs grown in normal medium supplemented with 30 mM mannitol (as an osmotic control) showed no change in heparanase protein levels compared with cells grown in normal medium.
Figure 1.
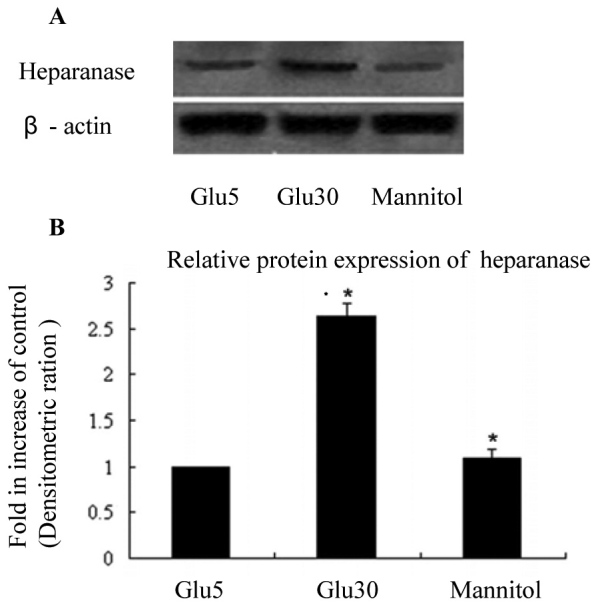
Heparanase expression in human retinal vascular endothelial cells under high-glucose conditions for 48 h is increased than in the Glu5 and mannitol control groups. A, B: We used western blotting to determine that the expression of heparanase in human retinal vascular endothelial cells (HRECs) grown in conditioned medium. The band that represents heparanase expression was more intense in the Glu30 group than in the Glu5 and mannitol control groups. Each group experiment was repeated 3 times (n=3). * compared with the Glu30 group (p<0.01); ** compared with the Glu5 and mannitol control groups (p<0.01).
Expression of heparanase in human retinal vascular endothelial cells is suppressed by RNA interference
To determine the effects of the transfection of heparanase RNAi on the expression of HREC heparanase, we observed green fluorescent protein expression under fluorescence microscopy in HRECs 72 h after infection with HPSE-RNAi-LV. We performed real-time PCR and western blotting to determine the mRNA and protein levels of heparanase in the HPSE-LV and Con-LV groups and HRECs. These analyses demonstrated that HPSE-LV significantly inhibited the expression of heparanase mRNA (p=0.006, p=0.007) and protein compared with uninfected HRECs and the Con-LV group (Figure 2).
Figure 2.

Expression of heparanase in human retinal vascular endothelial cells is suppressed by RNAi. A: Human retinal vascular endothelial cells (HRECs) were infected with Con-LV or heparanase small interfering RNA recombinant lentiviral vector (HPSE-LV) at a multiplicity of infection (MOI) of 60. Green fluorescent protein (GFP) expression and phase contrast images were captured after 72 h (original magnification × 200). B: The heparanase mRNA levels were detected using real-time PCR after the HRECs were treated with HPSE-LV and Con-LV. HPSE-LV significantly inhibited the expression of HREC heparanase mRNA (●p<0.01, compared with HRECs; ▲p<0.01, compared with the Con-LV group). Each group experiment was repeated 3 times (n=3). C: western blotting analysis showed that the heparanase protein was markedly inhibited by HPSE-LV in HRECs.
Heparanase expression in human retinal vascular endothelial cells
To determine the effects of heparanase RNAi treatment on the expression of heparanase mRNA and protein in high-glucose-treated HRECs, we performed real-time PCR and western blotting analysis on samples from the Glu5, Glu30, Con-LV-Glu30, and HPSE-LV-Glu30 groups. Heparanase RNAi significantly reduced the expression of heparanase mRNA (p=0.004, Figure 3C) and protein (p<0.001, Figure 3A,B) in high-glucose-treated HRECs compared with the Glu30 and Con-LV-Glu30 groups. Heparanase mRNA (p<0.001, Figure 3C) and protein (p<0.001, Figure 3A,B) expression in Glu30 cells was significantly higher than in the Glu5 and HPSE-LV-Glu30 groups. Therefore, the heparanase-specific RNAi treatments used in our study are efficient at downregulating the increased expression of heparanase in high-glucose-treated HRECs. We next examined whether heparanase levels correlate with the proliferation and migration of HRECs.
Figure 3.

Expression of heparanase protein and mRNA in high-glucose-treated human retinal vascular endothelial cells is suppressed by RNAi. A, B: HPSE-LV significantly inhibited human retinal vascular endothelial cells (HRECs) heparanase protein expression in the heparanase small interfering RNA recombinant lentiviral vector (HPSE-LV)-Glu30 group compared with HRECs in the Glu30 and Con-LV-Glu30 groups (●p<0.01 versus Glu5; ▲p<0.01 versus HPSE-LV-Glu30 group). Each group experiment was repeated 3 times (n=3). C: Real-time PCR analysis showed that heparanase mRNA was markedly inhibited in the HPSE-LV-Glu30 group compared with HRECs in the Glu30 and Con-LV-Glu30 groups (●p<0.01 versus Glu5; ▲p<0.01 versus HPSE-LV-Glu30). Each group experiment was repeated 3 times (n=3).
Effects of heparanase expression levels on human retinal vascular endothelial cell proliferation
To determine whether heparanase expression levels correlate with the proliferation of HRECs, we examined the effects of the high-glucose and RNAi treatments on HREC growth using MTT and BrdU assays. We found that the growth of cells infected with HPSE-LV was markedly inhibited compared with the Con-LV-Glu30 and Glu30 groups. However, the growth of cells in the Glu30 group was significantly higher than in the HPSE-LV-Glu30 and Glu5 groups (p<0.001, Figure 4A-C). These results show that heparanase overexpression due to high-glucose conditions correlates with HREC proliferation. Furthermore, our results demonstrate that the RNAi method used in our study to knock down high-glucose-induced heparanase expression is also efficient at inhibiting increased HREC proliferation.
Figure 4.

We used 3-(4,5-dimethylthiahiazol-2-y1)-2,5-diphenyltetrazolium bromide and bromodeoxyuridine staining assays to determine that effects of heparanase expression on human retinal vascular endothelial cell proliferation. A: 3-(4,5-dimethylthiahiazol-2-y1)-2,5-diphenyltetrazolium bromide (MTT) assays revealed that cell proliferation in the heparanase small interfering RNA recombinant lentiviral vector (HPSE-LV)-Glu30 group was significantly suppressed by HPSE-LV compared with the Con-LV-Glu30 and Glu30 groups. Human retinal vascular endothelial cell (HREC) proliferation in the Glu30 group was significantly higher than in the Glu5 and HPSE-LV-Glu30 groups (●p<0.01 versus Glu5; ▲p<0.01 versus HPSE-LV-Glu30). Each group experiment was repeated 3 times (n=3). B, C: Bromodeoxyuridine (BrdU) staining assays revealed that HPSE-LV-Glu30 cell growth was significantly lower than in the Con-LV-Glu30 and Glu30 groups. The HREC growth of the Glu30 group was significantly higher than in the Glu5 and HPSE-LV-Glu30 groups. (●p<0.01 versus Glu5; ▲p<0.01 versus HPSE-LV-Glu30). Data are presented as the mean±SD. Each group experiment was repeated 3 times (n=3).
Effects of heparanase expression levels on human retinal vascular endothelial cells’ migration ability
To evaluate whether heparanase expression levels correlate with the migration of HRECs, we performed an in vitro cell migration assay using paxillin, and determined the number of cells that migrate across Boyden chambers. Heparanase RNAi markedly reduced the number of cells that migrated through the chamber compared with the Con-LV-Glu30 (p<0.001) and the Glu30 groups, while the Glu30 group’s migration ability was significantly higher than the HPSE-LV-Glu30 and Glu5 groups (p<0.001, Figure 5A,C).
Figure 5.

We used Boyden chambers and paxillin immunofluorescence staining assays to determine that effects of heparanase expression levels on human retinal vascular endothelial cell migration. A: The human retinal vascular endothelial cells (HRECs) that are stained with crystal violet are those that migrated across the extracellular matrix (ECM) in the specialized migration chamber by migrating through the polycarbonate membrane to the lower surface of the membrane (original magnification 200×). B: Paxillin immunofluorescence staining indicated positive paxillin protein expression in every group. HPSE-LV significantly inhibited HREC paxillin protein expression compared with the Con-LV-Glu30 and Glu30 groups. The paxillin protein expression in the Glu30 group was significantly higher than in the HPSE-LV-Glu30 and Glu5 groups (the yellow arrow indicates paxillin). C: Heparanase small interfering RNA recombinant lentiviral vector (HPSE-LV) significantly decreased the HRECs’ migration ability. Bars indicate the mean±SD (p<0.05). The HPSE-LV-Glu30 group showed a markedly decreased migration ability compared to the Con-LV-Glu30 (p<0.01) and Glu30 groups, which migrated significantly more than the HPSE-LV-Glu30 and Glu5 groups (●p<0.01 versus Glu5; ▲p<0.01 versus HPSE-LV-Glu30). Each group experiment was repeated 3 times (n=3).
Paxillin acts as an adaptor protein that localizes to focal adhesions during cell migration. Immunofluorescent staining showed that in the Glu30 and Con-LV-Glu30 groups, paxillin expression in the HRECs was significantly higher than in the HPSE-LV-Glu30 and Glu5 groups (Figure 5B). However, in the HPSE-LV-Glu30 group, paxillin expression was markedly reduced compared with the Con-LV-Glu30 (Figure 5B) and Glu30 groups.
Enhanced cell proliferation by heparanase is mediated by Akt and extracellular-signal-regulated kinase
To pinpoint the signal transduction pathway by which the effects of heparanase-induced endothelial cell proliferation and migration may occur, we analyzed the levels of ERK and Akt phosphorylation using western blotting. We noted significant activation of Akt and ERK phosphorylation in the Glu30 group cells stimulated by heparanase overexpression, while Akt and ERK activation was abolished in the HPSE-LV-Glu30 group (p<0.001, Figure 6 A,B). Additionally, cell proliferation stimulated by heparanase overexpression in the Glu30 group was significantly increased, but in the HPSE-LV-Glu30 group, cell proliferation was inhibited. Taken together, these results indicate that Akt and ERK phosphorylation and activation due to heparanase overexpression correlate with increased cell proliferation and migration.
Figure 6.

Effects of heparanase expression levels on human retinal vascular endothelial cell AKT and extracellular signal regulated kinase (ERK) protein expression western blotting analyses showed that the levels of phosphorylated ERK and Akt were enhanced by heparanase overexpression. A, B: HPSE-LV-Glu30 human retinal vascular endothelial cells (HRECs) showed significantly reduced ERK and Akt phosphorylation levels compared with the Con-LV-Glu30 and Glu30 groups. The ERK and Akt phosphorylation levels of the Glu30 HRECs were significantly higher than the HPSE-LV-Glu30 and Glu5 groups. (●p<0.01 versus Glu5 p-Akt; ▲p<0.01 versus HPSE-LV-Glu30 p-Akt; ★p<0.01 versus Glu5 p-ERK; ■p<0.01 versus HPSE-LV-Glu30 p-ERK). Bars indicate the mean±SD, p<0.05. Each group experiment was repeated 3 times (n=3).
Discussion
In addition to the traditional enzymatic function of ECM degradation, heparanase facilitates the adhesion, spreading, and migration of several cell types in a manner that appears to be independent of its enzymatic activity [22,23,25]. Researchers have previously reported that the nonenzymatic functions of heparanase include enhanced adhesion of gliomas [23], lymphomas [23,24], and T cells [25]; these functions are mediated by β1-integrin and correlate with Akt, Pyk2, and ERK activation [23,25]. Increased expression of heparanase mRNA and protein in tumors is evident in tissue specimens derived from adenocarcinomas of the ovary, metastatic melanomas, oral squamous cell carcinomas, hepatocellular carcinomas, and carcinomas of the prostate, bladder, and pancreas [26-35]. Studies have reported that inhibiting the expression of heparanase can impede tumor invasion, metastasis, and angiogenesis; therefore, heparanase is a critical cancer target [30,36].
Heparanase is also an important vasculopathy target. In diabetes, the ECs lining all vessels appear to be the initial target of vascular damage due to hyperglycemia. Previous studies have shown that heparanase mRNA and activity were detected in ECs treated with high glucose and H2O2, which cause EC injury. An increase in osmolarity is not likely the cause of heparanase upregulation because heparanase mRNA and activity was not found in mannitol-treated cells [37]. ECs in the macrovessels and microvessels have different properties, but both are characterized by the same pathological features in patients with diabetes mellitus, such as exaggerated proliferation of ECs and thickening of the basement membrane. This proliferation and thickening result in the narrowing of the vessel lumen, which contributes to premature thrombosis and ischemia, leading to vascular remodeling [38,39].
To determine the effects of heparanase on HREC proliferation and migration, we used a heparanase RNAi lentiviral vector to deplete heparanase expression (Figure 2). Our results show that while the heparanase gene was highly expressed in high-glucose-treated HRECs (the Glu30 group, Figure 3), RNAi effectively inhibited heparanase gene expression in high-glucose-treated HRECs (the HPSE-LV-Glu30 group; Figure 3). Heparanase gene expression did not change in the 30 mM mannitol-treated HRECs (the mannitol group, Figure 2). Moreover, the proliferation and migration of HPSE-LV-infected cells were lower than those of the Con-LV-Glu30 and Glu30 groups (Figure 4 and Figure 5). Our results show that in the Glu30 and Con-LV-Glu30 groups, HREC paxillin expression is significantly higher than in the HPSE-LV-Glu30 and Glu5 groups (Figure 5B). Paxillin acts as an adaptor protein that localizes to focal adhesions and is regulated during cell migration via phosphorylation of its tyrosine, serine, and threonine residues. Most of these phosphorylation sites have been implicated in regulating the different steps of cell migration.
The results of the present study show that decreased heparanase expression inhibits HREC proliferation and migration. Importantly, HREC proliferation and migration may contribute to increased angiogenesis in established diabetic retinopathy. Therefore, there is a strong relationship between heparanase levels and HREC migration and proliferation. The present work also revealed a relationship between HREC heparanase expression levels and Akt and ERK phosphorylation levels; the observed upregulation or downregulation of heparanase expression enhanced or inhibited ERK and Akt phosphorylation, respectively, thereby mediating HREC proliferation and migration (Figure 6). Importantly, this result further supports the hypothesis that in correlation with ERK- and Akt-dependent HREC proliferation and migration, heparanase overexpression accelerates the angiogenic processes. Activation of the ERK and Akt pathways plays a major role in regulating cell growth, proliferation, differentiation, and angiogenesis and provides a protective effect against apoptosis and vascular remodeling [40,41].
Heparanase upregulation might also be common in injured ECs from large and small vessels. Heparanase activates endothelial cells and elicits angiogenic responses. Adding the 65-kDa latent heparanase protein to endothelial cells enhances Akt and ERK signaling. Heparanase-mediated Akt and ERK phosphorylation is independent of heparanase enzymatic activity and of the presence of cell membrane HS proteoglycans. Heparanase also stimulates phosphatidylinositol 3-kinase-dependent endothelial cell migration and invasion [22].
In conclusion, numerous ocular diseases result in the growth of new blood vessels within the eye. Diabetic vascular complications are primarily caused by injury to ECs. ECs play a pivotal role in regulating vascular tone, and injury of these cells is the initial step leading to irreversible structural abnormalities. Furthermore, HREC proliferation and migration may contribute to increased angiogenesis in established diabetic retinopathy. Our results reveal that high glucose levels induced the upregulation of heparanase expression in HRECs. This study further demonstrated that upregulation or downregulation of heparanase expression enhanced or inhibited ERK and Akt phosphorylation, respectively; these changes in ERK and Akt phosphorylation correlate with changes in HREC proliferation and migration. Therefore, these findings provide the basis for further studies focusing on understanding and treating diabetic vascular complications.
Acknowledgments
This work was supported by grants from the Key Technology Introduction Projection of Guangdong Province/The International Cooperation Project of Guangdong Province (Grant No. 2008B050100038); the Fundamental Research Funds of State Key Lab of Ophthalmology, Sun Yat-Sen University (Grant No. 2010C04). Dr. Shibo Tang (Tangsb@sysu.edu.cn) and Dr. Jie Hu contributed equally to the research project and can be considered co-corresponding authors.
References
- 1.Rowe S, MacLean C, Shekelle P. Preventing visual loss from chronic eye disease in primary care: Scientific review. JAMA. 2004;291:1487–95. doi: 10.1001/jama.291.12.1487. [DOI] [PubMed] [Google Scholar]
- 2.Resnikoff S, Pascolini D, Etya'ale D, Kocur I, Pararajasegaram R, Pokharel GP, Mariotti SP. Global data on visual impairment in the year 2002. Bull World Health Organ. 2004;82:844–51. [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Adsani AM. Risk factors for diabetic retinopathy in Kuwaiti type 2 diabetic patients. Saudi Med J. 2007;28:579–83. [PubMed] [Google Scholar]
- 4.Agarwal S, Raman R, Paul PG, Rani PK, Uthra S, Gayathree R, McCarty C, Kumaramanickavel G, Sharma T. Sankara Nethralaya-Diabetic Retinopathy Epidemiology and Molecular Genetic Study (SN-DREAMS 1): study design and research methodology. Ophthalmic Epidemiol. 2005;12:143–53. doi: 10.1080/09286580590932734. [DOI] [PubMed] [Google Scholar]
- 5.Chibber R, Ben-Mahmud BM, Chibber S, Kohner EM. Leukocytes in diabetic retinopathy. Curr Diabetes Rev. 2007;3:3–14. doi: 10.2174/157339907779802139. [DOI] [PubMed] [Google Scholar]
- 6.Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001;158:147–52. doi: 10.1016/S0002-9440(10)63952-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kefalides NA. Basement membrane research in diabetes mellitus. Coll Relat Res. 1981;1:295–9. doi: 10.1016/s0174-173x(81)80006-4. [DOI] [PubMed] [Google Scholar]
- 8.Colwell JA, Lopes-Virella MF. A review of the development of largevessel disease in diabetes mellitus. Am J Med. 1988;85:113–8. doi: 10.1016/0002-9343(88)90403-2. [DOI] [PubMed] [Google Scholar]
- 9.Kjellén L, Lindahl U. Proteoglycans. structures and interactions. Annu Rev Biochem. 1991;60:443–75. doi: 10.1146/annurev.bi.60.070191.002303. [DOI] [PubMed] [Google Scholar]
- 10.Parish CR, Freeman C, Hulett MD. Heparanase: a key enzyme involved in cell invasion. Biochim Biophys Acta. 2001;1471:M99–108. doi: 10.1016/s0304-419x(01)00017-8. [DOI] [PubMed] [Google Scholar]
- 11.Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest. 2001;108:341–7. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dempsey LA, Brunn GJ, Platt JL. Heparanase, a potential regulator of cell-matrix interactions. Trends Biochem Sci. 2000;25:349–51. doi: 10.1016/s0968-0004(00)01619-4. [DOI] [PubMed] [Google Scholar]
- 13.Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest. 2001;108:341–7. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogishima T, Shiina H, Breault JE, Tabatabai L, Bassett WW, Enokida H, Li LC, Kawakami T, Urakami S, Ribeiro-Filho LA, Terashima M, Fujime M, Igawa M, Dahiya R. Increased heparanase expression is caused by promoter hypomethylation and up-regulation of transcriptional factor early growth response-1 in human prostate cancer. Clin Cancer Res. 2005;11:1028–36. [PubMed] [Google Scholar]
- 15.Beckhove P, Helmke BM, Ziouta Y, Bucur M, Dorner W, Mogler C, Dyckhoff G, Herold-Mende C. Heparanase expression at the invasion front of human head and neck cancers and correlation with poor prognosis. Clin Cancer Res. 2005;11:2899–906. doi: 10.1158/1078-0432.CCR-04-0664. [DOI] [PubMed] [Google Scholar]
- 16.Mikami S, Ohashi K, Usui Y, Nemoto T, Katsube K, Yanagishita M, Nakajima M, Nakamura K, Koike M. Loss of syndecan-1 and increased expression of heparanase in invasive esophageal carcinomas. Jpn J Cancer Res. 2001;92:1062–73. doi: 10.1111/j.1349-7006.2001.tb01061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maxhimer JB, Somenek M, Rao G, Pesce CE, Baldwin D, Jr, Gattuso P, Schwartz MM, Lewis EJ, Prinz RA, Xu X. Heparanase-1 gene expression and regulation by high glucose in renal epithelial cells: a potential role in the pathogenesis of proteinuria in diabetic patients. Diabetes. 2005;54:2172–8. doi: 10.2337/diabetes.54.7.2172. [DOI] [PubMed] [Google Scholar]
- 18.Ilan N, Elkin M, Vlodavsky I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int J Biochem Cell Biol. 2006;38:2018–39. doi: 10.1016/j.biocel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Miao HQ, Liu H, Navarro E, Kussie P, Zhu Z. Development of heparanase inhibitors for anti-cancer therapy. Curr Med Chem. 2006;13:2101–11. doi: 10.2174/092986706777935230. [DOI] [PubMed] [Google Scholar]
- 20.Sanderson RD, Yang Y, Suva LJ, Kelly T. Heparan sulfate proteoglycans and heparanase–partners in osteolytic tumor growth and metastasis. Matrix Biol. 2004;23:341–52. doi: 10.1016/j.matbio.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 21.Vlodavsky I, Abboud-Jarrous G, Elkin M, Naggi A, Casu B, Sasisekharan R. The impact of heparanese and heparin on cancer metastasis and angiogenesis. Pathophysiol Haemost Thromb. 2006;35:116–27. doi: 10.1159/000093553. [DOI] [PubMed] [Google Scholar]
- 22.Gingis-Velitski S, Zetser A, Flugelman MY, Vlodavsky I, Ilan N. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J Biol Chem. 2004;279:23536–41. doi: 10.1074/jbc.M400554200. [DOI] [PubMed] [Google Scholar]
- 23.Zetser A, Bashenko Y, Miao H-Q, Vlodavsky I, Ilan N. Heparanase affects adhesive and tumorigenic potential of human glioma cells. Cancer Res. 2003;63:7733–41. [PubMed] [Google Scholar]
- 24.Goldshmidt O, Zcharia E, Cohen M. Heparanase mediates cell adhesion independent of its enzymatic activity. FASEB J. 2003;17:1015–25. doi: 10.1096/fj.02-0773com. [DOI] [PubMed] [Google Scholar]
- 25.Sotnikov I, Hershkoviz R, Grabovsky V. Enzymatically quiescent heparanase augments T cell interactions with VCAM-1 and extracellular matrix components under versatile dynamic contexts. J Immunol. 2004;172:5185–93. doi: 10.4049/jimmunol.172.9.5185. [DOI] [PubMed] [Google Scholar]
- 26.Xu X, Quiros RM, Maxhimer JB, Jiang P, Marcinek R, Ain KB, Platt JL, Shen J, Gattuso P, Prinz RA. Inverse correlation between heparan sulfate composition and heparanase-1 gene expression in thyroid papillary carcinomas: a potential role in tumor metastasis. Clin Cancer Res. 2003;9:5968–79. [PubMed] [Google Scholar]
- 27.Nadir Y, Brenner B, Gingis-Velitski S, Levy-Adam F, Ilan N, Zcharia E, Nadir E, Vlodavsky I. Heparanase induces tissue factor pathway inhibitor expression and extracellular accumulation in endothelial and tumor cells. Thromb Haemost. 2008;99:133–41. [PubMed] [Google Scholar]
- 28.McKenzie E, Tyson K, Stamps A, Smith P, Turner P, Barry R, Hircock M, Patel S, Barry E, Stubberfield C, Terrett J, Page M. Cloning and expression profiling of Hpa2, a novel mammalian heparanase family member. Biochem. Biophys. Res. Commun. 2000;276:1170–7. doi: 10.1006/bbrc.2000.3586. [DOI] [PubMed] [Google Scholar]
- 29.Friedmann Y, Vlodavsky I, Aingorn H, Aviv A, Peretz T, Pecker I, Pappo O. Expression of heparanase in normal, dysplastic, and neoplastic human colonic mucosa and stroma. Evidence for its role in colonic tumorigenesis. Am J Pathol. 2000;157:1167–75. doi: 10.1016/S0002-9440(10)64632-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zcharia E, Metzger S, Chajek-Shaul T, Friedmann Y, Pappo O, Aviv A, Elkin M, Pecker I, Peretz T, Vlodavsky I. Molecular properties and involvement of heparanase in cancer progression and mammary gland morphogenesis. J Mammary Gland Biol Neoplasia. 2001;6:311–22. doi: 10.1023/a:1011375624902. [DOI] [PubMed] [Google Scholar]
- 31.Koliopanos A, Friess H, Kleeff J, Shi X, Liao Q, Pecker I, Vlodavsky I, Zimmermann A, Buchler MW. Heparanase expression in primary and metastatic pancreatic cancer. Cancer Res. 2001;61:4655–9. [PubMed] [Google Scholar]
- 32.Cohen I, Pappo O, Elkin M, San T, Bar-Shavit R, Hazan R, Peretz T, Vlodavsky I, Abramovitch R. Heparanase promotes growth, angiogenesis and survival of primary breast tumors. Int J Cancer. 2006;118:1609–17. doi: 10.1002/ijc.21552. [DOI] [PubMed] [Google Scholar]
- 33.El-Assal ON, Yamanoi A, Ono T, Kohno H, Nagasue N. The clinicopathological significance of heparanase and basic fibroblast growth factor expressions in hepatocellular carcinoma. Clin Cancer Res. 2001;7:1299–305. [PubMed] [Google Scholar]
- 34.Bar-Sela G, Kaplan-Cohen V, Ilan N, Vlodavsky I, Ben-Izhak O. Heparanase expression in nasopharyngeal carcinoma inversely correlates with patient survival. Histopathology. 2006;49:188–93. doi: 10.1111/j.1365-2559.2006.02469.x. [DOI] [PubMed] [Google Scholar]
- 35.Ikeguchi M, Hirooka Y, Kaibara N. Heparanase gene expression and its correlation with spontaneous apoptosis in hepatocytes of cirrhotic liver and carcinoma. Eur J Cancer. 2003;39:86–90. doi: 10.1016/s0959-8049(02)00558-0. [DOI] [PubMed] [Google Scholar]
- 36.Sato T, Yamaguchi A, Goi T, Hirono Y, Takeuchi K, Katayama K, Matsukawa S. Heparanase expression in human colorectal cancer and its relationship to tumor angiogenesis, hematogenous metastasis, and prognosis. J Surg Oncol. 2004;87:174–81. doi: 10.1002/jso.20097. [DOI] [PubMed] [Google Scholar]
- 37.Han J, Woytowich AE, Mandal AK, Hiebert LM. Heparanase upregulation in high glucose-treated endothelial cells is prevented by insulin and heparin. Exp Biol Med (Maywood) 2007;232:927–34. [PubMed] [Google Scholar]
- 38.Nieuwdorp M, Haeften TW, Gouverneur MC, Mooij HL, van Lieshout MH, Levi M, Meijers JC, Holleman F, Hoekstra JB, Vink H, Kastelein JJ, Stroes ES. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes. 2006;55:480–6. doi: 10.2337/diabetes.55.02.06.db05-1103. [DOI] [PubMed] [Google Scholar]
- 39.Basta G, Schmidt AM, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res. 2004;63:582–92. doi: 10.1016/j.cardiores.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 40.Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res. 2002;90:1243–50. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 41.Gingis-Velitski S, Zetser A, Flugelman MY, Vlodavsky I, Ilan N. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J Biol Chem. 2004;279:23536–41. doi: 10.1074/jbc.M400554200. [DOI] [PubMed] [Google Scholar]