

Abstract
Transforming growth factor beta 2 (TGF-β2) is elevated in the aqueous humor of patients with glaucoma. This growth factor is known to increase extracellular matrix (ECM) deposition in the trabecular meshwork (TM) as well as increase intraocular pressure (IOP) in perfused human cultured anterior eye segments. In addition overexpression of TGF-β2 in the mouse TM leads to elevated IOP. Exogenous TGF-β2 also increases tissue transglutaminase (TGM2) protein levels and enzyme activity in TM cells. TGM2 is a calcium-dependent enzyme that mediates cross-linking of ECM proteins, thus making ECM proteins resistant to enzymatic degradation and physical breakdown. We have investigated the signaling pathway by which TGF-β2 induces TGM2 in human TM cells. Human TM cells (N=6) were treated for 48hours with TGF-β2 (0-10ng/ml) in serum-free medium. TGM2 enzyme activity differences between non-treated and TGF-β2 treated TM cells were studied using a biotin cadaverine assay. Endogenous TGF-β2 protein levels were examined in normal trabecular meshwork (NTM) and glaucomatous trabecular meshwork (GTM) cell strains. Immunohistochemistry was used to evaluate the expression and co-localization of TGF-β2 and TGM2 in NTM and GTM tissues. Activation of Smad3 signaling pathway was evaluated by western immunoblot analysis using phospho-specific antibodies following exogenous TGF-β2 treatment. Pharmacological specific inhibitor of Smad3 (SIS3) and short interfering (si)RNAs were used to suppress Smad3 activity and CTGF gene expression respectively. Endogenous TGF-β2 levels were significantly elevated in cultured GTM cells (p<0.05) when compared to NTM cells. Immunohistochemistry studies also demonstrated elevated expression and co-localization of both TGF-β2 and TGM2 in glaucoma human TM tissues. Exogenous TGF-β2 increased both TGM2 protein levels and enzyme activity in TM cells. Phosphorylation of Smad3 was stimulated in TM cell strains by exogenous TGF-β2. TGF-β2 induction of TGM2 was not inhibited with selective siRNA knockdown of CTGF. In contrast, a specific inhibitor of Smad3 (SIS3) and siRNAs knockdown of Smad3 (p<0.05) suppressed TGF-β2 induction of TGM2. This study demonstrated that TGF-β2 induction of TGM2 can be mediated via the canonical Smad signaling pathway but does not appear to involve CTGF as a downstream mediator. Regulation of the Smad signaling pathway in the TM may be useful in the therapy for glaucoma associated with aberrant TGF-β2 signaling.
Keywords: Glaucoma, trabecular meshwork, tissue transglutaminase, transforming growth factor –β2
1. Introduction
Primary open-angle glaucoma (POAG) is a leading cause of irreversible visual impairment and blindness in the world (Quigley. 1996, Quigley and Broman. 2006). A significant risk factor in the development and progression of POAG is elevated intraocular pressure (IOP) (Kass et al. 2002, AGIS. 2000). Increased resistance to aqueous humor (AH) outflow has been correlated with elevated IOP (Rohen. 1983). Several morphological and biochemical changes in the trabecular meshwork (TM) have been associated with elevated IOP and resistance to aqueous humor outflow (Rohen et al. 1993)(Lutjen-Drecoll E. 1996).
Extracellular matrix (ECM) proteins play a major role in the architecture of the TM (Yue. 1996). Within the TM, there is a delicate balance of deposition and degradation of the ECM proteins. In POAG, there appears to be excessive amounts of cross-linked ECM proteins in the TM and this accumulation has been associated with increased AH outflow resistance and elevated IOP (Lutjen-Drecoll. 1999, Rohen and Witmer. 1972).
Tissue transglutaminase (TGM2) may play a role in modulating ECM protein turnover in glaucoma (Fuchshofer et al. 2005, Welge-Lussen et al. 2000, Tovar-Vidales et al. 2008). ) TGM2 (EC 2.3.2.13) is a member of the transglutaminase enzyme family and is known to catalyze the posttranslational modification of proteins by inserting highly stable bonds between ε-(γ-glutamyl) lysine or (γ-glutamyl) polyamine (Folk and Finlayson. 1977). This enzymatic reaction renders ECM proteins resistant to both physical and enzymatic degradation (Folk and Finlayson. 1977).
TGM2 expression and enzyme activity are present in human TM cells and tissues (Tovar-Vidales et al. 2008, Welge-Lussen et al. 2000). In addition, we demonstrated that glaucomatous TM cells and tissues expressed higher levels of TGM2 compared to age-matched controls, suggesting that TGM2 may play an important role in glaucoma by making ECM proteins more resistant to degradation (Tovar-Vidales et al. 2008). Other reports have indicated that exogenous TGF-β1 and TGF-β2 increased mRNA expression, protein levels, and enzymatic activity of TGM2 in cultured human TM cells (Welge-Lussen et al. 2000). In addition, Fuchshofer and co-workers reported TGF-β2 induction of TGM2 expression in optic nerve head (ONH) astrocytes. They further demonstrated that this TGF-β2 induction of TGM2 in ONH astrocytes was mediated through connective tissue growth factor (CTGF) (Fuchshofer et al. 2005). TGF-β2 can regulate ECM deposition by inducing CTGF (Leask and Abraham. 2004). Interestingly, Fuchshofer and co-workers also demonstrated that TGF-β2 induced CTGF mRNA expression and protein levels in HTM cells (Fuchshofer et al. 2007).
TGF-β2 is known to signal via both the canonical Smad pathway and non-canonical pathways (Javelaud and Mauviel. 2004a, Javelaud and Mauviel. 2004b, Javelaud and Mauviel. 2005, Massague and Chen. 2000). In the Smad canonical signaling pathway, binding of the ligand to the TGF-β receptor complex results in phosphorylation of receptor Smads (e.g. Smad2/Smad3). Phosphorylated Smads then bind Co-Smad4, and the complex is translocated to the nucleus (Javelaud and Mauviel. 2004a, Javelaud and Mauviel. 2004b, Javelaud and Mauviel. 2005, Massague and Chen. 2000). TGF-β2 has also been reported to signal via non-canonical pathways including p38, ERK and JNK (Javelaud and Mauviel. 2004a, Javelaud and Mauviel. 2004b, Javelaud and Mauviel. 2005, Massague and Chen. 2000). The signaling pathway(s) utilized by TGF-β2 to stimulate TGM2 expression in the TM has not been defined. The purpose of this study was to determine: (a) whether TGF-β2 regulates TGM2 expression in human TM cells via the Smad canonical signaling pathway and (b) if this regulation is mediated by CTGF.
2. Methods
2.1 Trabecular meshwork cell culture
Previously characterized non-transformed primary human TM cells were obtained from Alcon Research Ltd. (Fort Worth, Texas) (Wordinger et al. 1998). Human TM cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (low glucose) supplemented with 10% fetal bovine serum (FBS) (HyClone Labs, Logan, UT), L-glutamine (0.292 mg/ml), penicillin (100 units/ml), streptomycin (0.1 mg/ml), and amphotericin B (4 mg/ml). Antibiotics were purchased from Gibco BRL, Grand Island, NY. Fresh medium was exchanged every 2-3 days and maintained at 37°C in 5% CO2-95% air. Experimental studies performed in human TM cells were between passages 8-11 and there was no evidence of cellular senescence in these passages.
2.2 Treatment of human TM cells
Human TM cells were grown in DMEM plus 10% FBS until they were 80% to 90% confluent. The cells were washed with serum-free DMEM and cultured in serum-free DMEM for 24 hours and then treated with recombinant TGF-β2 protein (R&D#302-B2, R&D Systems, Minneapolis, MN) at selected concentrations. To examine the inhibiting effects of the specific inhibitor of Smad3 (SIS3, Sigma#S0447, Sigma-Aldrich, St. Louis, MO), cells were preincubated for 1hr prior to TGF-β2 treatment. Human TM cells were treated at selective SIS3 concentrations (0.5, 1.0, 1.5, 2.0, 3.0, and 5.0 um) with TGF-β2 or SIS3 alone (5.0um).
2.3 Cell enzyme activity
TGM2 enzyme activity was measured using a microscopic assay as previously described (Zhang et al. 1998)(Kim et al. 2001, Tovar-Vidales et al. 2008, Zhang et al. 1998). Briefly, cells were put in DMEM serum-less media for 24hours followed by treatment with TGF-β2 at 5ng/ml for 48 hours. TGM2 activity was assessed by exposing cells to 1mM biotin cadaverine, a pseudo-substrate of TGM2, for 1 hour at the end of the 48 hour treatment. Cells were fixed for 30 minutes in 3.7% volume per volume basis (v/v) formaldehyde in PBS and permeabilized with 0.2% (v/v) Triton X-100 in PBS for 30 minutes at room temperature. Cells were further incubated with an Alexa Fluor 488 streptavidin-conjugate (1:1000, v/v) in PBS for 1 hour at room temperature, washed, and mounted. The cells were observed with an Olympus Model BX51 trinocular research microscope (Leeds Instruments, Irving TX). TGM 2 activity was seen as increased fluorescence labeling. Analysis of staining intensity was performed using NIH Image J program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/).
2.4 Protein extraction and western blot analysis
Total cellular protein was isolated from cultured TM cells using (a) M-PER extraction buffer (#78501, Pierce Biotech, Rockford, IL.) and Protease Inhibitor Cocktail (#78415, Pierce Biotech) or (b) Laemmli Sample buffer with 5% beta-mercaptoethanol. Protein concentration was determined by using the Bio-Rad Dc Protein Assay System according to manufacturer’s instructions (Bio-Rad Laboratories, Richmond, CA) or the EZQ Protein Quantitation Kit according to manufacturer’s instructions (#R33200, Molecular Probes).
A total of 20 ug of protein was loaded per well and separated on a SDS-PAGE denaturing polyacrylamide gel and then transferred by electrophoresis to PVDF membranes. The PVDF membranes were incubated in 5% milk in Tris Buffered Saline Tween (TBST - 20mM Tris, 0.5M NaCl, and 0.05% Tween 20, pH 7.4) for approximately 60 minutes in order to block non-specific binding. Blots were processed using appropriate antibodies and appropriate secondary antibodies (Table 1). The Super Signal West Femto Maximus Sensitivity Substrate (#34095, Pierce Biotech) was used for detection and blots were exposed in a Fluorchem 8900 imager (Alpha Innotech, San Leandro, CA).
Table 1.
List of Antibodies for Western Immunoblots
| Antibody ( cat #) | Dilutions | Source |
|---|---|---|
| TGM2 ( MS-300-P1) | 1:1000 | NeoMarkers, Fremont, Ca. |
| CTGF (sc-14939) | 1:1000 | Santa Cruz Biotechnology, Inc. Santa Cruz, CA. |
| TGF-β2 (sc-90) | 1:500 | Santa Cruz Biotechnology, Inc. Santa Cruz, CA. |
| pSmad3 (#9520) | 1:500 | Cell Signaling Technology. Inc Danvers, MA |
| Smad3 (#9523s) | 1:500 | Cell Signaling Technology. Inc Danvers, MA |
| Fibronectin (AB1945) | 1:500 | Millipore Corporate, Billerica, MA |
| β-actin (1501) | 1:1000 | Millipore Corporate, Billerica, MA |
| Goat Anti Mouse (sc#2005) | 1:10000 | Santa Cruz Biotechnology, Inc. Santa Cruz, CA. |
| Anti Rabbit (#7074) | 1: 15000 | Cell Signaling Technology. Inc Danvers, MA |
2.5 Immunohistochemistry of TM Tissues
Details of the immunohistochemistry of TGM2 in TM tissues have been published previously (Tovar-Vidales et al. 2008). In order to demonstrate the presence of TGM2 and TGF-β2 in TM tissues, 3 pairs of normal and 3 pairs of glaucoma age-matched human eyes were used (e.g. normal donors of 72, 88 and 94 yrs; glaucomatous donors of 76, 87, and 92 yrs.). Paraffin sections were processed with primary antibodies for TGM2 (#MS-300-P1, Neomarkers) and TGF-β2 (#sc-90, Santa Cruz Biotechnology) (Table 2). The primary antibodies were detected with appropriate secondary antibodies (Alexa Fluor 488, and 633; Molecular Probes) for 45 minutes. The visualization of cell nuclei was performed by staining tissue sections with DAPI (300 μm) for 10 minutes. Controls sections consisted of omission of primary antibodies, IgG, and/or mouse ascites fluid. Images were captured using a Zeiss 410 confocal imaging system (Carl Zeiss, Thornwood, N.Y.). Immunofluorescence imaging precautions consisted of taking images in a mass manner and having the same settings for all slides including negative controls. Analysis of staining intensity was performed using NIH Image J program.
Table 2.
List of Antibodies for Staining
| Antibody ( cat #) | Dilutions | Source |
|---|---|---|
| TGM2 mab ( MS-300-P1) | 1:2000 | NeoMarkers, Fremont, Ca. |
| TGF-β2 ( sc-90 ) | 1:200 | Santa Cruz Biotechnology, Inc. Santa Cruz, CA. |
| Smad3 (#9513) | 1:200 | Cell Signaling Technology. Inc Danvers, MA |
2.6 Immunocytochemistry of TM Cells
Primary human TM cells were grown on glass coverslips in 24 well plates. At 80% confluent, cells were fixed with 3.5% formaldehyde (Fisher Scientific, Pittsburgh, PA) in 1 X PBS for 20 minutes. Cells were treated with 0.2% Triton X-100 in 1 X PBS for 20 minutes. Incubation took take place for 1 hour with 5% normal blocking serum and 0.3% in 1X PBS. Cells were then incubated with primary antibodies overnight at 4° C and secondary antibodies at 1:200 and 1%BSA in 1 X PBS for 1 hour at room temperature. Negative controls consisted of omission of primary antibody, IgG, and/or mouse ascites fluid. To visualize nuclei, cells were treated with 300 nM DAPI nuclear stain and mounted using Aqua-Mount (Lerner Laboratories, Pittsburgh, PA). Slides were stored in the dark at 4° C until visualized on a Zeiss 410 confocal imaging system (Carl Zeiss, Thornwood, N.Y.).
2.7 Transfection with siRNA
Slight modifications of previously published methods for siRNA transfection (Kobayashi et al. 2005, Zode et al. 2009) were performed. The siRNA mediated gene suppression experiments were conducted in primary human TM cells using siGenome On-Target plus SMART pool duplex siRNAs (pools of 4 siRNA duplexes) from Dharmacon. (Lafayette, CO). . Controls included a RISC-free siRNA which impairs loading uptake of siRNA into RISC free complex and a non-targeted siRNA which will not recognize any human gene sequence in the human genome. ). siRNA for Smad3, CTGF, and TGM2 siRNA were purchased from Dharmacon (SMARTpool). Briefly, confluent primary human TM cells were trypsinized, counted, and plated at a density of 8000 cells/well in a 12 well plate in DMEM media containing 10% serum. Primary human TM cells were allowed to incubate overnight at 37°C in 5% CO2-95% air. The following day selected siRNAs were transfected to cells using DharmaFECT 1 (T-2001-01, Dharmacon) at the desired concentrations (e.g. 0.1, 1.0, and 10nM siRNAs) in OPTI-MEM media without FBS and antibiotics (Gibco, Grand Island, NY). Two separate tubes were used, one containing 4ul of DharmaFECT 1 was mixed gently with 200ul of Opti-MEM for 5 minutes at room temperature. The second tube contained the desired siRNA mixed gently with 200ul of Opti-MEM medium for 5 minutes at room temperature. After, the two tubes were combined, gently mixed, and incubated for 20 minutes at room temperature. At the end of the incubation period, Opti-MEM was added to a final volume of 2ml for each siRNA and placed into the appropriate well for 24 hours. At the end of 24 hours, media was changed and cells were cultured with either TGF-β2 in serum free media or serum free media alone for an additional 48 hours.
3. Results
3.1 TGM2 protein levels in response to TGF-β2
Six individual primary human TM cell strains were exposed to various concentrations of TGF-β2 (e.g. 0.5, 1.25, 2.5, 5.0 or 10.0 ng/ml) for 48 hours. Cell lysates were subsequently examined by Western blot analysis for TGM2 protein level. A representative Western blot is shown as Figure 1. The presence of TGM2 protein in the serum-free media (SFM) lane indicates that human TM cells normally express TGM2 at low levels. A substantial increase in TGM2 protein levels was observed beginning with 1.25 ng/ml of TGF-β2. All Western blots were re-probed for β-actin that served as a loading control (Fig. 2).
Fig. 1. Dose Response of TGM2 Following Treatment with TGF-β2.
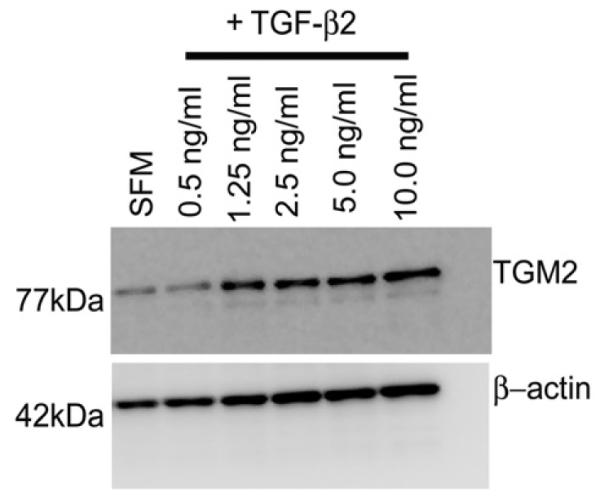
A representative western blot from one of six independent TM cell strains is shown for expression of TGM2 (77kDa). Human TM cells were treated with TGF-β2 at 0.5ng/ml, 1.25ng/ml, 2.5ng/ml, 5.0ng/ml, or 10.0ng/ml for 48 hours and then cell lysates obtained and probed for TGM2. Blots were stripped, washed thoroughly and re-probed for β-actin.
Fig. 2. TGM2 Enzyme Activity measured by a Biotinylated Cadaverine Assay Following Treatment with TGF-β2.

A representative biotinlated cadaverine assay of TM cells. Four different human TM cell strains were incubated with vehicle (DMSO) control (A), biotin-labeled cadaverine (1mM) (B), and biotin-labeled cadaverine with TGF-β2 (5ng/ml) (C) for 48 hours. Transamidated and cross-linked cadaverine (green) was detected by Alexa Fluor 488 streptavidin-conjugate. TGM2 induction was observed with treatment of TGF-β2. Immunohistochemical staining analysis revealed statistical elevated levels for the biotin-labeled cadaverine with TGF-β2 compared to the biotin-labeled cadaverine alone. Image analysis was performed on a PC computer using the public domain NIH Image J program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/). Image J Analysis was performed to quantitate the intensity levels, * statistical difference at the p<0.05 level (+/− SEM) (D).
3.2 TGM2 enzyme activity in response to TGF-β2
Having shown that TGM2 protein levels are induced by TGF-β2, we next wanted to determined whether TGM2 enzyme activity is also elevated in TM cell strains following TGF-β2 treatment. TGM2 activity was analyzed utilizing a biotin labeled cadaverine-streptavidin immunohistochemical assay. Briefly, cells were labeled with biotin cadaverine for 1 hour after the 48 hours treatment with TGF-β2 before fixation and staining, and TGM2 enzyme activity was detected by the AlexaFluor 488 streptavidin conjugate. This enzyme activity works by biotin cadaverine being incorporated into TM cells and cross-linked to proteins, then streptavidin will bind to the biotin and label the cross-linked proteins. Therefore, in this assay increased labeling shows increased TGM2 activity. Figure 2A-C is a representative example of one human TM cell strain. Figure 2B indicates that there is a low amount of TGM2 enzyme activity in human TM cells in the absence of exogenous TGF-β2. This correlates to Western blot results from Figure 1. Treatment with exogenous TGF-β2 (5ng/nl) for 48 hours dramatically increased TGM2 enzyme activity (Figure 2C). Vehicle DSMO control is shown as Figure 2A. The enzyme activity of TGM2 following TGF-β2 treatment of 4 TM cell strains was significantly elevated (p<0.05) compared to untreated control with cadaverine alone (Fig. 2D).
3.3 TGF-β2 protein levels are elevated in glaucoma cell strains
The protein expression of TGF-β2 in the lysates of three NTM and two GTM was examined by Western blot analysis (Figure 3A). GTM cells appeared to have increased TGF-β2 protein levels compared with the NTM cells. TGF-β2 protein was expressed at approximately 60 kDa on the Western blot and β-actin served as a loading control (Fig. 3A). To determine if the 60 kDa was TGF-β2 specific, the antibody was preincubated with its peptide and a Western blot performed eliminating the 60 kDa protein band (data not shown). Statistical analysis was performed on densitometry results from 3 NTM and 2 GTM. These data indicate that protein levels of TGF-β2 were significantly elevated (p<0.05) in the GTM cells (Fig. 3B).
Fig. 3. Western blot of TGF- β2 in NTM and GTM cells.

(A) Up-regulation of TGF- β2 in glaucomatous cells compared to normal TM cells. Five different trabecular meshwork cell strains, 3 normal (NTM) and 2 glaucomatous (GTM), were grown in DMEM containing serum. Cells were rinsed with PBS and collected with cell lysis buffer. Cell lysate were subjected to 10% SDS-PAGE and western blot analysis using anti-TGF-β2 antibody. The molecular weight for the TGF-β2 monomer is 12.5 kDa and the dimer is 25kDa,however, our Western blot analysis shows a molecular weight at approximately 60 kDa. β -actin was used as a loading control. (B) Densitometric reading of TGF-β2 normalized to β-actin for 3NTM and 2 GTM cell strains. * Statistical difference at the p<0.05 level (+/− SEM) were calculated with GraphPad software Prism 5.
3.4 Immunohistochemical localization of TGF-β2 and TGM2 in human TM tissues
Having shown that human TM cells have endogenous levels of both TGF-β2 and TGM2 proteins in vitro, and that exogenous TGF-β2 increases both TGM2 protein levels and enzyme activity, we next demonstrated co-localization of both proteins in human TM tissues. Trabecular meshwork tissues from 3 normal and 3 glaucomatous human donors were utilized.
Immunostaining of TM tissues demonstrated that TGF-β2 (Figure A and D) and TGM2 (Figure B and E) were expressed in human TM tissue samples. The expression of TGF-β2 was uniformly localized throughout all TM tissues examined (Figure 4A and 4D). In two of the three GTM human tissues, TGF-β2 appeared to be expressed at a higher level than in NTM tissues (Compare Figure 4A and Figure 4D). TGM2 was present in all human TM samples (Figures 4B and 4E). All 3 GTM human TM samples had elevated TGM2 expression compared to age-matched controls (Compare Figure 4B versus 4E). Co-localization of TGF-β2 and TGM2 are shown in Figures 4C and 4F. Statistical analysis was performed on staining intensities for TGF-β2 and TGM2 in 3 NTM and 3 GTM tissues (Figure 4G). The relative intensity of TGF-β2 and TGM2 measured by Image J software (NIH) indicated that TGM2 protein was statistically increased in age matched glaucomatous TM tissues (p<0.05) (Fig. 4E) when compared to age-matched controls.
Fig. 4. Immunohistochemical localization of TGF-β2 and TGM2 in NTM and GTM tissues.


Age-matched tissues NTM (88 yrs.) and GTM (87 yrs.), were fixed, sectioned and stained with antibodies for TGF-β2 and TGM2. Negative controls consisted of PBS-BSA without primary antibody, IgG, and mouse ascites (data not shown). GTM tissues showed an increased expression of co-localization of TGM2 and TGF- β2. (A-C) NTM (40X), (D-F) GTM (40X). (G) Image J Analysis was performed to quantitate the intensity levels, * statistical difference at the p<0.05 level (+/− SEM). Arrowhead points to the endothelium of Schlemm’s canal. abbreviations: AC- anterior chamber, TM- trabecular meshwork
3.5 Phosphorylation of Smad3
The canonical downstream signaling pathway for TGF- β2 primarily utilizes intracellular Smad protein3 (Smad3). We next wanted to verify that Smad3 protein is present in human TM cells and whether exogenous TGF-β2 results in Smad3 phosphorylation (p-Smad3).
Six separate human TM cell strains were treated with TGF-β2 (5ng/ml) for various time points (e.g. 5, 15, 30 or 60 minutes or 2 hours). Western blot analysis detected phosphorylated Smad3 protein in cell lysates beginning at 15 minutes. A substantial elevation in p-Smad3 protein levels was observed by 30 and 60 minutes (Figure 5A). Densitometric analysis of p-Smad3 protein levels from 6 human TM cell strains is presented as Figure 5B. These results demonstrated that phosphorylation of Smad3 was increased at 15 minutes, with a maximum activation at 30 and 60 minutes. This was followed by a decrease at 2 hours for all TM cells (Figure 5B). The level of p-Smad3 was statistically significant at 60 minutes compared to five minutes (p<0.05) (Fig 5B). It was also apparent that the canonical signaling pathway is enhanced in two of the three GTM cell strains (data not included).
Fig. 5. Western Blot analysis of phosphorylated Smad3 in TGF-β2 treated TM cells.

(A) Western blot analysis for phosphorylated Smad3 (p-Smad3) in TM cells treated with exogenous TGF-β2 (5ng/mL) for various times at 0 minutes, 5 minutes, 15 minutes, 30 minutes, 60 minutes, 2 hours. Control shown was at 2 hours (2hrCo). Blots were stripped, washed thoroughly and re-probed with total Smad3. (B) Phosphorylated Smad3 were measured by western blot and analyzed by densitometry for TM cell strains. * Statistical difference at the p<0.05 level (+/− SEM) at 60 minutes compared to 5 minutes were calculated with GraphPad software Prism 5.
3.6 Inhibition of Smad3
Specific inhibitor of Smad3 (SIS3) blocks TGF-β signaling via specific inhibition of Smad3 phosphorylation. We treated 3 primary human TM cell strains with exogenous TGF-β2 and various concentrations of SIS3 (e.g. 0.5, 1.0, 1.5, 2.0, 3.0 or 5.0 μM) for 48 hours. Subsequently, Western blotting was performed to determine if blocking Smad3 would reduce TGM2 protein levels following exogenous TGF-β2 treatment. Figure 6 is a representative Western blot that demonstrates an apparent reduction of TGM2 protein levels following SIS3 treatment. SIS3 concentrations between 2.0-5.0 μM had the greatest effect. All Western blots were re-probed for β-actin that served as a loading control (Fig. 6).
Fig. 6. Western Blot analysis of TGM2 Following SIS3 Treatment.
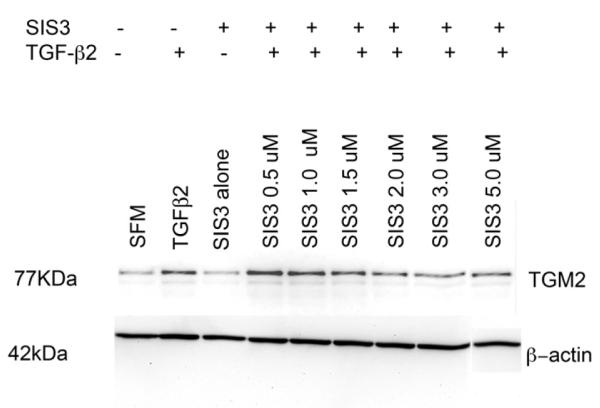
Western blot analysis in TM cells for TGM2 in three different cell strains. TM cells were pre-treated with SIS3 (an inhibitor of Smad3) at various concentrations (0.5 μM – 5 μM) for 1 hour and then treated with TGF-β2 for 48 hours. Untreated TM cells served as controls. β-actin was used as a loading control.
3.7 Immunostaining of TGM2 in Primary Human TM Cells following siSmad3 knockdown
Smad3 siRNA mediated gene suppression experiments were conducted in 3 primary human TM cell strains. Treatments included, control cells in serum-less medium (SFM) (Fig. 7A), TGF-β2 treatment alone (Fig. 7B), a siRNA control that impairs RISC uptake (Co#1) (Fig. 7C) in combination with TGF-β2, and a non-targeted gene (Co#2) (Fig. 7D) in combination with TGF-β2. Knockdown of Smad3 was performed with siRNA treatment of 1.0 nM, 10.0 nM, and 20.0 nM (Fig. 7 E-G) in combination with TGF-β2.
Fig. 7. Immunohistochemical Staining of NTM and GTM Cells after Smad3 siRNA.

A representative immunohistochemical evaluation of TGM2 and Smad3 expression in human TM cells. As previously described, TM cells were transfected with Smad3 siRNA (1.0nM, 10.0nM, and 20.0nM) and then treated with TGF-β2 for 48 hours. Human TM cells were fixed and stained with antibodies for Smad3 (red), TGM2 (green), and co-localization (merged) expression. (A) Smad3 endogenous expression was located primarily in the cytoplasm and upon TGF-β2 treatment (B) there is more Smad3 in the nucleus. (C-D) siRNA controls (20nm) had no effects on Smad3 expression. (E-G) siRNA transfections at 1.0, 10.0, and 20.0nM suppressed Smad3 expression regardless of the presence of TGF-β2. (A) TGM2 expression was located primarily in the cytoplasm and was elevated upon TGF-β2 treatment (B). (C-D) siRNA controls (20nm) had no effects on TGM2 expression. (E-G) siRNA transfections at 1.0, 10.0, and 20.0nm decreased TGM2 proteins levels regardless of the presence of TGF-β2.
Human primary TM cells grown in SFM are shown in Figure 7A. Endogenous Smad3 is localized to the cytoplasm. Endogenous TGM2 expression is also evident. Upon treatment with exogenous TGF-β2, there is more Smad3 in the nucleus and TGM2 protein levels are increased (Figure 7B). Both the RISC uptake and non-targeted siRNA controls (Figure 7C and Figure 7D) were similar to TGF-β2 treated cells displaying increased nuclear accumulation of Smad3 and increased cytoplasmic TGM2 protein levels. siRNA knockdown of Smad3 is shown in Figures 7E-G. Smad3 siRNA down-regulated Smad3 expression and reduced TGM2 expression compared to TGF-β2 treatment and Smad3 siRNA controls plus TGF-β2.
3.8 Involvement of CTGF in Regulating TGM2
We next determined whether connective tissue growth factor (CTGF), which can act as a downstream mediator of TGF-β2activity , is involved in increased TGM2 protein levels seen following TGF-β2 treatment. CTGF mediated gene suppression experiments were conducted in 3 primary human TM cell strains. Control experiments consisted of primary human TM cells cultured in serum-less media (SFM), exogenous TGF-β2 treatment (5ng/ml) for 48 hours, a siRNA control that impairs RISC uptake (Co#1) in combination with TGF-β2, and a non-targeted gene (Co#2) in combination with TGF-β2. Human TM cells were treated with CTGF siRNA at 0.1, 1.0 nM, and 10.0 nM with TGF-β2. Western blot analysis for CTGF, TGM2 and fibronectin (FN) was performed (Figure 8A).
Fig. 8. Effects of CTGF siRNA Treatment in TM Cells.

(A) Western blot analysis of CTGF and TGM2 with treatment of CTGF siRNA followed by TGF-β2 (5ng/ml) for 48 hours. Representative blots are shown from one TM cell strain. Blots were stripped, washed thoroughly and re-probed for TGM2, FN and β-actin. β-actin served as a loading control. (B) Densitometric analysis was done for CTGF and TGM2 normalized to β-actin. * Statistical difference at the p<0.05 level (one tailed t-test, compared to Co#1) were calculated with GraphPad software Prism 5.
As shown in Figure 8A, there was a reduction in CTGF and FN protein levels in all cell strains examined following treatment with siRNA concentrations of 1.0nM, 10.0nM, and 20.0nM. All western blots were re-probed to determine CTGF siRNA effects on TGM2. There was no apparent effect on TGM2 with CTGF suppression (Fig 8A). In contrast, fibronectin (FN) induction with TGF-β2 was reduced with CTGF siRNA (Fig. 8A). β-actin was used as a loading control (Fig. 8A). Both CTGF and TGM2 protein band density normalized to actin is shown in Figure 8B. CTGF protein levels were significantly reduced (p<0.05, one tailed t-test) as compared to siRNA controls (Fig. 8B). Protein levels of TGM2 were not significantly changed following siRNA knockdown of CTGF.
4. Discussion
A major risk factor for the development of POAG is elevated IOP (Kass et al. 2002, AGIS. 2000). Elevated IOP is primarily due to increased AH outflow resistance within the juxtacanalicular region (JCT) of the TM. This resistance is associated with local morphologic and biochemical changes in the JCT (Rohen. 1983). In the normal JCT there are adequate homeostatic control mechanisms with respect to ECM protein deposition, turnover and modification. However in POAG, distinct structural changes occur in the JCT including the accumulation of fibrillar proteins (Lutjen-Drecoll. 2005, Rohen. 1983). These changes are thought to lead to decreased outflow facility and increased IOP. The accumulation of ECM proteins may be the result of abnormal regulation of ECM protein turnover by MMPs and PAI-1 (Bradley et al. 1998, Fleenor et al. 2006, Fuchshofer et al. 2003, Pang et al. 2003). It is also possible that elevated expression of cross-linking enzymes within the TM may lead to ECM proteins that are resistant to enzymatic digestion by MMPs. Growth factors present in the TM and AH are known to regulate both MMPs and cross-linking enzymes in TM cells. Thus growth factors are considered important regulators of ECM homeostasis and altered growth factor expression may result in deleterious consequences to the TM and/or regulation of AH outflow pathways.
TGF-β2 influences many aspects of cellular behavior including proliferation, differentiation, migration, and ECM synthesis and breakdown (Zhu and Burgess. 2001)(Verrecchia and Mauviel. 2002). With respect to glaucoma, a number of studies have reported elevated levels of TGF-β2 in the AH of glaucoma patients compared to age-matched normal eyes (Inatani et al. 2001, Ochiai and Ochiai. 2002, Ozcan et al. 2004, Picht et al. 2001, Tripathi et al. 1994). Experimentally, perfusion of TGF-β2 in ex-vivo anterior segment organ culture models causes an accumulation of fibrillar material in the TM (Gottanka et al. 2004). Fleenor et al. (2006), also reported that perfusion of TGF-β2 in ex-vivo anterior segment organ culture models elevated IOP in a time dependent manner and also increased fibronectin and PAI-I levels in the elutes while decreasing outflow facility (Fleenor et al. 2006). Intraocular injection of bioactive TGF-β2 has been reported to induce ocular hypertension in the rat and mouse and to reduce AH outflow facility in the mouse (Shepard et al., 2010). Thus, it is reasonable to suspect TGF-β2 may have a direct role in deposition and accumulation of ECM proteins in the TM of glaucoma patients.
TGM2 is a cross-linking enzyme that may contribute to the increase resistance of aqueous humor outflow. We speculate that cross-linking of proteins by TGM2 would stiffen the trabecular meshwork. In fact, recent evidence by atomic force microscopy have shown that the GTM tissue is significantly stiffer compared to NTM tissues (Last et al., 2011). Welge-Lüssen and co-workers (2000) first reported the presence and induction of TGM2 by TGF-β1 and β2 in cultured HTM. Our laboratory previously reported that TGM2 protein levels and enzyme activities were elevated in GTM cells and tissues (Tovar-Vidales et al., 2008). Despite the fact that TGF-β1 and β2 induces TGM2 and TGM2 is elevated in glaucomatous TM cells, the exact role or molecular mechanism for TGM2 in the pathogenesis of glaucoma is not clear. Therefore, the purpose of this study was three-fold: (a) to examine the induction of TGM2 by TGF-β2, (b) to identify if the canonical TGF-β2 signaling pathway is utilized to regulate TGM2 in cultured TM cells and (c), to determine if CTGF acts as a downstream mediator for TGF-β2 - regulation of TGM2 in human TM cells.
Within the glaucomatous eye, TGF-β2 has been reported to be as high as 2.7ng/ml compared to 1.48ng/ml in normal eyes (Tripathi et al. 1994). For our purposes we performed a dose response experiment using TGF-β2 at concentrations of 0.5ng/ml, 1.25ng/ml, 2.5ng/ml, 5.0ng/ml, and 10.0ng/ml. Interestingly, our data did not show a dose dependent increase of TGM2 with TGF-β2. We observed an induction of TGM2 by TGF-β2 from 1.25 to 10.0 ng/ml concentrations when compared to untreated controls. Thus even a very low concentration of TGF-β2 (1.25ng/ml) is capable of stimulating TGM2 expression in TM cells.
However the presence of TGM2 protein does not always correlate with enzyme activity. Therefore we examined the induction of TGM2 enzyme activity by TGF-β2 in cultured TM cells. Enzyme activity was measured by the incorporation of biotin-cadaverine, a pseudo-substrate for TGM2 (Tovar-Vidales et al. 2008). Our results demonstrated significantly increased enzymatic activity of TGM2 in cultured TM cells following exogenous treatment by TGF-β2. These results demonstrate that TGM2 is induced by TGF-β2 in primary TM cells and is biologically active.
We had previously reported that TGM2 protein levels and enzyme activities were elevated in GTM cells and tissues (Tovar-Vidales et al., 2008). Previous reports had indicated that TGF-β2 levels were also elevated in the aqueous humor of glaucomatous patients (Inatani et al. 2001, Picht et al. 2001, Tripathi et al. 1994). However previous reports had not examined endogenous TGF-β2 protein levels in glaucomatous TM cells nor did they co-localize TGF-β2 and TGM2 in glaucomatous TM tissues. Our results demonstrated significantly increased protein levels of TGF-β2. , Although, the molecular weight of the TGF-β2 protein band was detected at 60 kDa, we presume the antibody is detecting the latent form of TGF-β2, thus there is greater expression of the TGF-β2 latent form in glaucomatous TM cells when compared to normal TM cells. Interestingly, Tritschler et al., (2009) demonstrated a similar molecular weight of the latent form of TGF-β1 and TGF-β2 in the conditioned media of human malignant glioma cells. (Tritschler et al. 2009). Secondly, to verify that co-localization of TGF-β2 with TGM2 was increased in glaucoma, normal TM and glaucomatous TM tissues were evaluated with immunohistochemistry. Our immunohistochemical results indicated increased expression and increased co-localization for both TGF-β2 and TGM2 in glaucoma TM tissues in comparison to normal TM tissues. This is of interest since the endothelium of Schlemm’s canal is considered to be the site of aqueous humor outflow resistance (Tamm and Fuchshofer. 2007). The increase co-localization for both TGF-β2 and TGM2 are interesting since there is evidence that TGM2 can increase the conversion of latent TGF-β to its biologically active form, and then TGF-β can further increase TGM2 providing a positive feedback mechanism in glaucomatous TM (Kojima et al., 1993, Kojima S et al., 1995, Nunes et al., 1997). These results suggest a direct role of TGM2 induction by TGF-β2 in the TM of human glaucomatous eyes.
The canonical signaling pathway utilized by TGF-β2 involves ligand binding to a receptor complex and subsequent downstream signaling via receptor Smad proteins (Massague. 1998, Massague and Chen. 2000, Massague. 2000). Specific Smad proteins for TGF-β2 include Smad2 and Smad3 (Massague. 1998, Massague and Chen. 2000, Massague. 2000). However several reports indicate that Smad3 is mainly utilized for fibrotic responses to TGF-β2 (Flanders. 2004, Roberts et al. 2003). As a first step to evaluate TGF-β2 regulation of TGM2, we wanted to determine if isolated TM cells express Smad3 and respond to exogenous TGF-β2 via Smad3 phosphorylation in a time dependent manner. Our data demonstrated that TM cells express endogenous phosphorylated Smad3 (p-Smad3) and respond to exogenous TGF-β2 with increased protein levels of p-Smad3. The endogenous presence of p-Smad-3 indicates an autocrine loop is present in TM cells. We also showed that SIS3, a inhibitor of the phosphorylation of Smad3, down-regulated TGM2 protein levels in TM cells. In addition, silencing of Smad3 gene expression via siRNA also decreased TGM2 protein expression. Our immunocytochemistry results indicated that upon TGF-β2 treatment, there is more Smad3 in the nucleus and there is none to little staining in the Smad3 siRNA. Treatment with Smad3 siRNA also reduced TGM2 protein levels in TM cells. These results indicate that human TM cells can respond to exogenous TGF-β2 via phosphorylation of Smad3 and that blocking Smad 3 protein phosphorylation or reducing the Smad3 protein levels, prevents TGF-β2 upregulation of TGM2.
Connective tissue growth factor (CTGF) is highly expressed in the TM (Tomarev et al. 2003) and has been shown to be an important downstream mediator of fibrotic changes induced by TGF-β2. (Leask and Abraham. 2004). Specifically, Junglas et al., (2009) demonstrated that CTGF is a critical mediator of the effects of TGF-β2 on the synthesis of ECM proteins in TM cells (Junglas et al. 2009). Fuchshofer et al., (2007), previously demonstrated that TGF-β2 induces CTGF in TM cell cultures (Fuchshofer et al. 2007). Our current results verify and are in agreement with their study. We also determined if the induction of TGM2 by TGF-β2 was mediated by CTGF in TM cell strains. We first demonstrated reduction of endogenous CTGF protein levels in TM cells following treatment with CTGF siRNA. While this reduction in CTGF blocked TGF-β2 upregulation of FN, there was no reduction of TGM2 protein levels in TM cells. Interestingly, using optic nerve head astrocytes (ONHA) , Fuchshofer and coworkers (2005), demonstrated that TGF-β2 did induce TGM2 mRNA expression and protein levels and that this action was mediated via CTGF (Fuchshofer et al. 2005). Priglinger has reported that TGM2 is present in PVR membranes and the level is related to the state of differentiation of retinal pigmented epithelial (RPE) cells and TGF-β2 stimulation. They further showed that the TGF-β2-mediated increase of TGM2 was also independent of CTGF. Thus, ONA regulation of TGM2 is CTGF dependent and in RPE and TM cells it is not dependent on CTGF. Therefore, it appears that the regulation of endpoints may be cell context dependent.
In conclusion, our results demonstrated that cultured glaucomatous TM cells and tissues from human donors have elevated levels of TGF-β2. We have shown that TM cells express TGM2 and expression and enzyme activity are stimulated by TGF-β2. In addition, we have demonstrated that the canonical pathway is stimulated by TGF-β2 in TM cells. We also showed Smad3 siRNA and SIS3 down-regulated TGM2 protein levels. Lastly we demonstrated that TGF-β2 regulation of TGM2 in human TM cells is independent of CTGF action.
 Exogenous TGF-β2 increased both TGM2 protein levels and enzyme activity in TM cells.
Exogenous TGF-β2 increased both TGM2 protein levels and enzyme activity in TM cells. Phosphorylation of Smad3 was stimulated in TM cell strains by exogenous TGF-β2.
Phosphorylation of Smad3 was stimulated in TM cell strains by exogenous TGF-β2. TGF-β2 induction of TGM2 was not inhibited with selective siRNA knockdown of CTGF.
TGF-β2 induction of TGM2 was not inhibited with selective siRNA knockdown of CTGF. A specific inhibitor of Smad3 (SIS3) and siRNAs knockdown of Smad3 suppressed TGF- β2 induction of TGM2.
A specific inhibitor of Smad3 (SIS3) and siRNAs knockdown of Smad3 suppressed TGF- β2 induction of TGM2.
Acknowledgments
The authors would like to thank Ann-Marie Brun and I-fen Chang, Microscopy Core Center, Department of Cell Biology and Anatomy, University of North Texas Health Science Center at Fort Worth for technical assistance. The authors would also like to thank Alcon Resarch, Ltd. and the Central Florida Eye and Tissue Bank for acquisition of donor eyes.
Grant Support: NIH Grant EY-012783 (RJW). This study is taken in part from a dissertation submitted to the UNT Health Science Center in partial fulfillment of the requirements for the degree Doctor of Philosophy for Tara Tovar-Vidales.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- AGIS Investigators The advanced glaucoma intervention study (AGIS): 7. The relationship between control of intraocular pressure and visual field deterioration. Am J Ophthalmol. 2000;130:429–440. doi: 10.1016/s0002-9394(00)00538-9. [DOI] [PubMed] [Google Scholar]
- Acott TS, Kelley MJ. Extracellular matrix in the trabecular meshwork. Exp. Eye Res. 2008;86:543–561. doi: 10.1016/j.exer.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley JM, Vranka J, Colvis CM, Conger DM, Alexander JP, Fisk AS, Samples JR, Acott TS. Effect of matrix metalloproteinases activity on outflow in perfused human organ culture. Invest. Ophthalmol. Vis. Sci. 1998;39:2649–2658. [PubMed] [Google Scholar]
- Flanders KC. Smad3 as a mediator of the fibrotic response. Int. J. Exp. Pathol. 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleenor DL, Shepard AR, Hellberg PE, Jacobson N, Pang IH, Clark AF. TGFbeta2-induced changes in human trabecular meshwork: implications for intraocular pressure. Invest. Ophthalmol. Vis. Sci. 2006;47:226–234. doi: 10.1167/iovs.05-1060. [DOI] [PubMed] [Google Scholar]
- Folk JE, Finlayson JS. The epsilon-(gamma-glutamyl)lysine crosslink and the catalytic role of transglutaminases. Adv. Protein Chem. 1977;31:1–133. doi: 10.1016/s0065-3233(08)60217-x. [DOI] [PubMed] [Google Scholar]
- Fuchshofer R, Birke M, Welge-Lussen U, Kook D, Lutjen-Drecoll E. Transforming growth factor-beta 2 modulated extracellular matrix component expression in cultured human optic nerve head astrocytes. Invest. Ophthalmol. Vis. Sci. 2005;46:568–578. doi: 10.1167/iovs.04-0649. [DOI] [PubMed] [Google Scholar]
- Fuchshofer R, Welge-Lussen U, Lutjen-Drecoll E. The effect of TGF-beta2 on human trabecular meshwork extracellular proteolytic system. Exp. Eye Res. 2003;77:757–765. doi: 10.1016/s0014-4835(03)00220-3. [DOI] [PubMed] [Google Scholar]
- Fuchshofer R, Yu AH, Welge-Lussen U, Tamm ER. Bone morphogenetic protein-7 is an antagonist of transforming growth factor-beta2 in human trabecular meshwork cells. Invest. Ophthalmol. Vis. Sci. 2007;48:715–726. doi: 10.1167/iovs.06-0226. [DOI] [PubMed] [Google Scholar]
- Gottanka J, Chan D, Eichhorn M, Lutjen-Drecoll E, Ethier CR. Effects of TGF-beta2 in perfused human eyes. Invest. Ophthalmol. Vis. Sci. 2004;45:153–158. doi: 10.1167/iovs.03-0796. [DOI] [PubMed] [Google Scholar]
- Inatani M, Tanihara H, Katsuta H, Honjo M, Kido N, Honda Y. Transforming growth factor-beta 2 levels in aqueous humor of glaucomatous eyes. Graefes Arch. Clin. Exp. Ophthalmol. 2001;239:109–113. doi: 10.1007/s004170000241. [DOI] [PubMed] [Google Scholar]
- Javelaud D, Mauviel A. Mammalian transforming growth factor-betas: Smad signaling and physio-pathological roles. Int. J. Biochem. Cell Biol. 2004a;36:1161–1165. doi: 10.1016/S1357-2725(03)00255-3. [DOI] [PubMed] [Google Scholar]
- Javelaud D, Mauviel A. Transforming growth factor-betas: smad signaling and roles in physiopathology. Pathol. Biol. (Paris) 2004b;52:50–54. doi: 10.1016/j.patbio.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Javelaud D, Mauviel A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene. 2005;24:5742–5750. doi: 10.1038/sj.onc.1208928. [DOI] [PubMed] [Google Scholar]
- Junglas B, Yu AH, Welge-Lussen U, Tamm ER, Fuchshofer R. Connective tissue growth factor induces extracellular matrix deposition in human trabecular meshwork cells. Exp. Eye Res. 2009;88:1065–1075. doi: 10.1016/j.exer.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Kass MA, Heuer DK, Higginbotham EJ, Johnson CA, Keltner JL, Parrish RK, 2nd, Wilson MR, Gordon MO. The ocular hypertension treatment study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002;120:701–713. doi: 10.1001/archopht.120.6.701. discussion 829–830. [DOI] [PubMed] [Google Scholar]
- Kim SW, Lee ZW, Lee C, Im KS, Ha KS. The role of tissue transglutaminase in the germinal vesicle breakdown of mouse oocytes. Biochem. Biophys. Res. Commun. 2001;286:229–234. doi: 10.1006/bbrc.2001.5381. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Liu X, Wen FQ, Fang Q, Abe S, Wang XQ, Hashimoto M, Shen L, Kawasaki S, Kim HJ, Kohyama T, Rennard SI. Smad3 mediates TGF-beta1 induction of VEGF production in lung fibroblasts. Biochem. Biophys. Res. Commun. 2005;327:393–398. doi: 10.1016/j.bbrc.2004.12.032. [DOI] [PubMed] [Google Scholar]
- Kojima S, Nara K, Rifkin DB. Requirement for transglutaminase in the activation of latent transforming growth factor-beta in bovine endothelial cells. J Cell Biol. 1993;121:439–448. doi: 10.1083/jcb.121.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S, Vernooy R, Moscatelli D, Amanuma H, Rifkin DB. Lipo-polysaccharide inhibits activation of latent transforming growth factor-beta in bovine endothelial cells. J Cell Physiol. 1995;163:210–219. doi: 10.1002/jcp.1041630124. [DOI] [PubMed] [Google Scholar]
- Last JA, Pan T, Ding Y, Reilly C, Keller K, Acott TS, Fautsch MP, Murphy CJ, Russell P. Elastic modulus determination of normal and glaucomatous human trabecular meshwork. Invest. Ophthalmol. Vis. Sci. 2011 doi: 10.1167/iovs.10-6342. (In-Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- Lutjen-Drecoll ERJ. Morphology of Aqueous Outflow Pathways in Normal and Glaucomatous Eyes. In: Ritch R, Shields MB, Krupin T, editors. The Glaucomas. Mosby; St. Louis: 1996. pp. 89–123. [Google Scholar]
- Lutjen-Drecoll E. Functional morphology of the trabecular meshwork in primate eyes. Prog. Retin. Eye Res. 1999;18:91–119. doi: 10.1016/s1350-9462(98)00011-1. [DOI] [PubMed] [Google Scholar]
- Lutjen-Drecoll E. Morphological changes in glaucomatous eyes and the role of TGFbeta2 for the pathogenesis of the disease. Exp. Eye Res. 2005;81:1–4. doi: 10.1016/j.exer.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Massague J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Massague J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- Massague J, Chen YG. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–644. [PubMed] [Google Scholar]
- Nunes I, Gleizes PE, Metz CN, Rifkin DB. Latent transforming growth factor-beta binding protein domains involved in activation and transglutaminase-dependent cross-linking of latent transforming growth factor-beta. J Cell Biol. 1997;136:1151–1163. doi: 10.1083/jcb.136.5.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai Y, Ochiai H. Higher concentration of transforming growth factor-beta in aqueous humor of glaucomatous eyes and diabetic eyes. Jpn. J. Ophthalmol. 2002;46:249–253. doi: 10.1016/s0021-5155(01)00523-8. [DOI] [PubMed] [Google Scholar]
- Ozcan AA, Ozdemir N, Canataroglu A. The aqueous levels of TGF-beta2 in patients with glaucoma. Int. Ophthalmol. 2004;25:19–22. doi: 10.1023/b:inte.0000018524.48581.79. [DOI] [PubMed] [Google Scholar]
- Pang IH, Fleenor DL, Hellberg PE, Stropki K, McCartney MD, Clark AF. Aqueous outflow-enhancing effect of tert-butylhydroquinone: involvement of AP-1 activation and MMP-3 expression. Invest. Ophthalmol. Vis. Sci. 2003;44:3502–3510. doi: 10.1167/iovs.02-0758. [DOI] [PubMed] [Google Scholar]
- Picht G, Welge-Luessen U, Grehn F, Lutjen-Drecoll E. Transforming growth factor beta 2 levels in the aqueous humor in different types of glaucoma and the relation to filtering bleb development. Graefes Arch. Clin. Exp. Ophthalmol. 2001;239:199–207. doi: 10.1007/s004170000252. [DOI] [PubMed] [Google Scholar]
- Quigley HA. Number of people with glaucoma worldwide. Br. J. Ophthalmol. 1996;80:389–393. doi: 10.1136/bjo.80.5.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006;90:262–267. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AB, Russo A, Felici A, Flanders KC. Smad3: a key player in pathogenetic mechanisms dependent on TGF-beta. Ann. N. Y. Acad. Sci. 2003;995:1–10. doi: 10.1111/j.1749-6632.2003.tb03205.x. [DOI] [PubMed] [Google Scholar]
- Rohen JW. Why is intraocular pressure elevated in chronic simple glaucoma? Anatomical considerations. Ophthalmology. 1983;90:758–765. doi: 10.1016/s0161-6420(83)34492-4. [DOI] [PubMed] [Google Scholar]
- Rohen JW, Lutjen-Drecoll E, Flugel C, Meyer M, Grierson I. Ultrastructure of the trabecular meshwork in untreated cases of primary open-angle glaucoma (POAG) Exp. Eye Res. 1993;56:683–692. doi: 10.1006/exer.1993.1085. [DOI] [PubMed] [Google Scholar]
- Rohen JW, Witmer R. Electrn microscopic studies on the trabecular meshwork in glaucoma simplex. Albrecht Von Graefes Arch. Klin. Exp. Ophthalmol. 1972;183:251–266. doi: 10.1007/BF00496153. [DOI] [PubMed] [Google Scholar]
- Tamm ER, Fuchshofer R. What increases outflow resistance in primary open-angle glaucoma? Surv Ophthalmol. 2007;52:S101–S104. doi: 10.1016/j.survophthal.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Tomarev SI, Wistow G, Raymond V, Dubois S, Malyukova I. Gene expression profile of the human trabecular meshwork: NEIBank sequence tag analysis. Invest. Ophthalmol. Vis. Sci. 2003;44:2588–2596. doi: 10.1167/iovs.02-1099. [DOI] [PubMed] [Google Scholar]
- Tovar-Vidales T, Roque R, Clark AF, Wordinger RJ. Tissue transglutaminase expression and activity in normal and glaucomatous human trabecular meshwork cells and tissues. Invest. Ophthalmol. Vis. Sci. 2008;49:622–628. doi: 10.1167/iovs.07-0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi RC, Li J, Chan WF, Tripathi BJ. Aqueous humor in glaucomatous eyes contains an increased level of TGF-beta 2. Exp. Eye Res. 1994;59:723–727. doi: 10.1006/exer.1994.1158. [DOI] [PubMed] [Google Scholar]
- Tritschler I, Gramatzki D, Capper D, Mittelbronn M, Meyermann R, Saharinen J, Wolfgang W, Keski-Oja J, Weller M. Modulation of TGF-β activity by latent TGF-β-binding protein 1 in human malignant glioma cells. Int. J. Cancer. 2009;125:530–540. doi: 10.1002/ijc.24443. [DOI] [PubMed] [Google Scholar]
- Verrecchia F, Mauviel A. Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J. Invest. Dermatol. 2002;118:211–215. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- Welge-Lussen U, May CA, Lutjen-Drecoll E. Induction of tissue transglutaminase in the trabecular meshwork by TGF-beta1 and TGF-beta2. Invest. Ophthalmol. Vis. Sci. 2000;41:2229–2238. [PubMed] [Google Scholar]
- Wordinger RJ, Clark AF, Agarwal R, Lambert W, McNatt L, Wilson SE, Qu Z, Fung BK. Cultured human trabecular meshwork cells express functional growth factor receptors. Invest. Ophthalmol. Vis. Sci. 1998;39:1575–1589. [PubMed] [Google Scholar]
- Yue BY. The extracellular matrix and its modulation in the trabecular meshwork. Surv. Ophthalmol. 1996;40:379–390. doi: 10.1016/s0039-6257(96)80066-x. [DOI] [PubMed] [Google Scholar]
- Zhang J, Lesort M, Guttmann RP, Johnson GV. Modulation of the in situ activity of tissue transglutaminase by calcium and GTP. J. Biol. Chem. 1998;273:2288–2295. doi: 10.1074/jbc.273.4.2288. [DOI] [PubMed] [Google Scholar]
- Zhu HJ, Burgess AW. Regulation of transforming growth factor-beta signaling. Mol. Cell Biol. Res. Commun. 2001;4:321–330. doi: 10.1006/mcbr.2001.0301. [DOI] [PubMed] [Google Scholar]
- Zode GS, Clark AF, Wordinger RJ. Bone morphogenetic protein 4 inhibits TGF-beta2 stimulation of extracellular matrix proteins in optic nerve head cells: role of gremlin in ECM modulation. Glia. 2009;57:755–766. doi: 10.1002/glia.20803. [DOI] [PubMed] [Google Scholar]