

Abstract
Atrioventricular valve development commences with an EMT event whereby endocardial cells transform into mesenchyme. The molecular events that induce this phenotypic change are well understood and include many growth factors, signaling components, and transcription factors. Besides their clear importance in valve development, the role of these transformed mesenchyme and the function they serve in the developing prevalve leaflets is less understood. Indeed, we know that these cells migrate, but how and why do they migrate? We also know that they undergo a transition to a mature, committed cell, largely defined as an interstitial fibroblast due to their ability to secrete various matrix components including collagen type I. However, we have yet to uncover mechanisms by which the matrix is synthesized, how it is secreted, and how it is organized. As valve disease is largely characterized by altered cell number, cell activation, and matrix disorganization, answering questions of how the valves are built will likely provide us with information of real clinical relevance. Although expression profiling and descriptive or correlative analyses are insightful, to advance the field, we must now move past the simplicity of these assays and ask fundamental, mechanistic based questions aimed at understanding how valves are ‘built”. Herein we review current understandings of atrioventricular valve development and present what is known and what isn’t known. In most cases, basic, biological questions and hypotheses that were presented decades ago on valve development still are yet to be answered but likely hold keys to uncovering new discoveries with relevance to both embryonic development and the developmental basis of adult heart valve diseases. Thus, the goal of this review is to remind us of these questions and provide new perspectives on an old theme of valve development.
Keywords: Valve Development, EMT, Post-EMT, Valve Disease, cushions, Fibroblasts
Introduction
Normal function of the mammalian heart is dependent on unidirectional blood flow during the cardiac cycle. Maintaining a proper flow pattern in the heart is primarily due to the function of the cardiac valves, of which mammals have four sets. These include the inlet valves (mitral and tricuspid) and the outlet valves (aortic and pulmonic). Blood from the systemic circulation enters the right atrium via the sinus coronaries and the superior and inferior venae cava where it is pushed through the tricuspid valve into the right ventricle. During systole the blood exits the right ventricle through the pulmonic valve to enter the pulmonary circulation where it becomes oxygenated and returned to the left atrium via the pulmonary veins. During diastole, blood is then pushed through the mitral valve and into the left ventricle. Following left ventricular systolic contraction, the blood is ejected through the aortic leaflets into the aorta. Thus, all cardiac valves function to control unidirectional blood flow from one chamber to another, or from the ventricles to outside the heart.
The structure and function of all cardiac valves is largely dependent upon how these tissues are built during development. A schematic of the structural changes that occur during AV valve development is presented in Figure 1. In most cases, gene mutations, present at conception lead to altered developmental processes that are not clinically evident at birth, but result in increased susceptibility to disease as the individual ages. Thus, we and others have collectively forged the idea that many valve diseases can be traced back to developmental errors (Hinton et al., 2008; Hinton et al., 2006; Hinton and Yutzey, 2011; Lincoln et al., 2004; Lincoln and Yutzey, 2011; Wirrig and Yutzey, 2011; Markwald et al., 2011). Based on this concept, it seems logical to propose that studying valve development will provide insight into pathogenetic mechanisms underlying not only postnatal (pediatric) but also adult valve diseases. This is of great clinical significance since valve abnormalities contribute either directly or indirectly to a wide range of cardiac diseases (e.g. arrhythmia, regurgitation, valve prolapse, stenosis, myocardial hypertrophy, heart failure). Based on these valve related diseases, it is not surprising that valvular heart disease is a major public health burden and results in at least 23,000 deaths annually. Surgical intervention resulting in valve repair or replacement is the main mode of treatment (Rajamannan et al., 2003; Roger et al., 2011). Thus, one purpose of this review is to show that by understanding normal valve development, new insights can be gained in the pathogenesis of valve diseases with the ultimate goal of translating discoveries into non-surgical advances in treatment. Additionally, we pose several fundamental questions related to valve development that still need to be mechanistically answered.
Figure 1. Schematic of AV Valve Development.
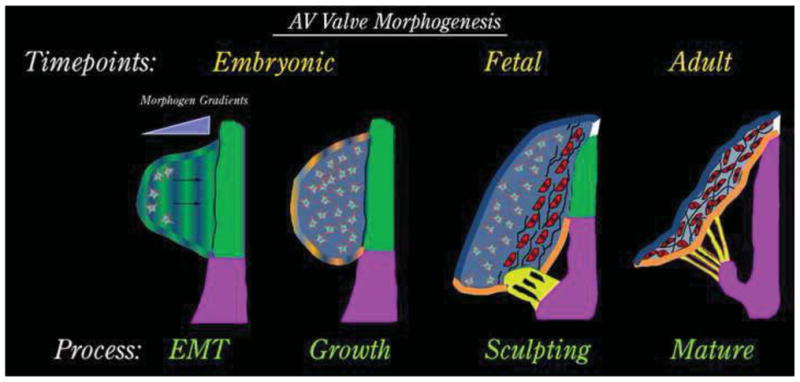
AV valve morphogenesis commences from an EMT event whereby morphogenic cues secreted by the AV junctional myocardium (green) diffuse through the cardiac jelly. Mesenchyme migrates towards these cues (arrows) in a directed fashion and undergoes a proliferative expansion to fill the cushion during the “growth phase”. As cells get closer to the myocardium (red cells), they change their secretory prolife and begin synthesizing fibrillar matrices such as collagens (black lines). During this fetal timepoint, cellular contraction and cell density facilitate valve sculpting. Development of the chordae tendineae (yellow) and the fibrous annulus (white) are markedly observed. The valve continues to attenuate and separate from the AV junctional myocardium (green), which is selectively removed yielding a tethered AV valve leaflet during adulthood.
Endothelial–Mesenchymal Transition
During early embryogenesis, the heart tube has an outer myocardial layer and an inner endocardial layer of cells separated by a largely acellular hydrated tissue filled with extra cellular matrix (ECM) called the cardiac jelly (Armstrong and Bischoff, 2004; de la Cruz and Markwald, 1998b; Krug et al., 1985; Markwald et al., 1977; Markwald et al., 1975; Markwald and Smith, 1972; Runyan and Markwald, 1983). Cardiac jelly is composed primarily of proteoglycan glycosaminoglycans of which hyaluronan and chondroitin sulfates are the major components (Person et al., 2005). After the heart tube begins rightward looping, the myocardium of specific regions of the primary heart tube, the atrioventricular (AV) junction and the ventricular outflow tract (OFT) upregulates secretion of ECM which causes the cardiac jelly to swell into the lumen of the heart tube, forming primordial structures termed “endocardial cushions” (Markwald et al., 1979; Bolender and Markwald, 1979; Combs and Yutzey, 2009; Eisenberg and Markwald, 1995; Person et al., 2005). Stimulated by local signals, a subset of AV and OFT endocardial cells loose cell-cell contact, resulting in their delamination into the cardiac jelly and their adoption of a migratory, mesenchymal-like phenotype. This phenotype is retained during the population of cardiac jelly of the AV and OFT cushions. The process whereby subsets of AV and OFT endocardial cells transform into a mesenchymal phenotype has been called an endocardial-to-mesenchymal transition (EMT), which determines the anatomical placement where the valves will form within the primary heart tube. This seminal finding was observed not only in vivo, but also recapitulated in vitro using collagen gel assays wherein subsets of isolated AV canal and OFT endocardial cells placed on top of a collagen gel surface transformed into cushion mesenchyme and invaded the collagen lattice. These assays confirmed that factors secreted by the AV junction or OFT myocardium were sufficient and necessary to induce a subset of endocardial cells to undergo EMT (Runyan and Markwald, 1983) (Lencinas et al., 2011). Over the past 3 decades, the application of this system has lead to the discovery of many key morphogenetic pathways and regulatory components required in promoting an EMT event, which has been reviewed in depth by Person et al. (Person et al., 2005).
As described in “The Ridge Hypothesis,” mesenchymal cells of the cushions following their delamination from the endocardium receive cues that promote their proliferation. These cues appear to be derived from the persisting cushion endocardium, which undergoes hypertrophy into a thickened, “ridge-like” cushion endocardium (de la Cruz and Markwald, 1998a; de la Cruz and Markwald, 1998b; de la Cruz et al., 1977) analogous to that of the apical ectodermal ridge of developing limb buds. The significance and precise number of proliferating cells is not well examined, but it appears that most of the proliferating cells are either of endocardial or sub-endocardial mesenchyme origin and that proliferation ceases as the cells move distally away from the endocardium. The molecular signals emanating from the cushion endocardium that promote proliferation include specific isoforms to FGF growth factors (Sugi et al., 2003). Additionally, expression studies and genetic manipulation of the transcription factors Twist, Msx-1, and Tbx20 within the “subridge” mesenchyme appear important for promoting proliferative pathways (Chen et al., 2007; Shelton and Yutzey, 2007; 2008). However, the mechanisms by which these transcription factors regulate cell cycle progression, mitosis and/or cytokinesis are unknown. Regardless, based on the expression of these HLH and homeobox genes and incorporation of BrdU, it appears that as the EMT derived cells migrate away from the endocardial ridge, the proliferative signals diminish creating two histological zones within the developing cushion tissue, a proximal (close to the myocardium), Msx1-positive zone of proliferation and a distal (close to the endocardium), Msx2-positive zone of differentiation adjacent to the AV or OFT myocardium (de la Cruz and Markwald, 1998a; de la Cruz and Markwald, 1998b; de la Cruz et al., 2001; de la Cruz et al., 1977). How the distal zone of post-EMT cushion mesenchyme migrate and differentiate are discussed in the following 2 sections.
Migration of Prevalvular Mesenchyme Into the Cardiac Jelly
Cell migration is a broad term referring to the process that involves the translocation of cells from one position to another and has been the subject of many reviews. The cytoskeleton seems central to most cell translocation mechanisms including endocardially-derived mesenchyme (Funderburg and Markwald, 1986; Markwald and Funderburg, 1983; Bolender and Markwald, 1979). Based on histological observations, cushion mesenchyme migrates as single entities, and not as interconnected cohorts. However, based on expression of connexin 45, it appears that the cells have potential to communicate with each other (Nishii et al., 2003; Nishii et al., 2001; Kumai et al., 2000). Thus, based on tissue morphology, cushion cell migratory behavior appears to be a non-random, directed event in which the cells are programmed to move in a unifying manner. This concept has been further supported by mathematical modeling simulations, indicating that cell migration within a developing tissue is not random (Sepich et al., 2005; Hacker, 2011). Although computer models for cushion cell migration have not been tested experimentally, it seems logical to propose that loss of directed cell migration would compromise cushion post-EMT morphogenesis and its integration with other segments of the developing heart. Accordingly, how migration of prevalvular cushion mesenchyme is directed or “polarized” is one of the major unsolved riddles of valvulogenesis. Cell polarization refers to the tendency of migrating cells to have distinct leading vs. trailing cell surfaces or borders (i.e. a stable front and rear). The ability for cushion mesenchyme to adopt a polarized configuration was originally proposed by Markwald et al in 1977 (Markwald et al., 1977). Based on serial sections of chick and rat embryos, Markwald described transformed mesenchyme as adopting a “centrifugal migratory direction”. Studies by Krug et al (Krug et al., 1985), followed up on these observations and noted that lateral cell mobility and hypertrophy and polarization of intracellular structures occurred during mesenchyme transformation. These findings were the first to recognize cell polarity as integral to valvulogenesis.
Since the inception of this discovery, cell polarity has been described as arising and/or being reinforced due to local cues that stem from either: (i) chemoattractants (or morphogens), (ii) haptotactic stimuli caused by varying concentrations of matrix components, (iii) biomechanical signals emitted by physical forces (e.g. shear stress) and/or (iv) changes in the interaction between extracellular matrix and membrane bound receptors. Thus, the concept of directed cell migration is based on the notion that moving cells have sensors that respond to an evolving morphogenetic and biomechanical environment. While morphogens (MacGrogan et al.; Luna-Zurita et al.; de la Pompa, 2009; Timmerman et al., 2004; Jiao et al., 2006; Song et al., 2007; Townsend et al.; Townsend et al.; Frieden et al.; Townsend et al., 2008; Desgrosellier et al., 2005; Barnett and Desgrosellier, 2003; Lai et al., 2000; Brown et al., 1999; 1996; Azhar et al., 2012; Azhar et al., 2009; Azhar et al., 2003; Bartram et al., 2001; Boyer et al., 1999; Boyer and Runyan, 2001; Sugi et al., 2003; Sugi et al., 2004; Millan et al., 1991; Ma et al., 2005; Alfieri et al., 2010; Lincoln et al., 2006; Kruithof et al., 2006; Van Den Akker et al., 2005; Van den Akker et al., 2008), matrix (Camenisch and McDonald, 2000; Camenisch et al., 2002; Camenisch et al., 2000; Schroeder et al., 2003; Norris et al., 2008; Norris et al., 2009b; Tan et al., 2011; Krug et al., 1985; Markwald et al., 1977; Peacock et al., 2008; Kruzynska-Frejtag et al., 2001; Kruithof et al., 2007; Kitten et al., 1996; Klewer et al., 1998; Hurle et al., 1994; Gittenberger-de Groot et al., 2003; Akiyama et al., 2004; Bouchey et al., 1996) and biomechanics (Butcher and Markwald, 2007; Butcher et al., 2007a; Yalcin et al., 2011) are well known as playing roles in cushion development, specific molecular changes induced by each of these signals are poorly defined. Recent studies have begun to understand how morphogens, matrix and/or biomechanics can mediate downstream changes in gene expression (Hierck et al., 2008b; Poelmann et al., 2008b; Hierck et al., 2008a; Poelmann et al., 2008a; Groenendijk et al., 2007; Groenendijk et al., 2004). For example, Vangl2, a component of non-canonical Wnt “planar cell polarity pathway” is expressed in developing cushion tissue, which speaks to the importance of cell polarity during development (Phillips et al., 2005; Henderson et al., 2006; Phillips et al., 2007; Phillips et al., 2008). Cells deficient for Vangl2 fail to extend lamellipodia or filopodia into the cardiac jelly of cushion tissue and failed to generate actin stress-fibers required for mobility. As a result Vangl2 KO mice have profound developmental cardiac defects indicating a requirement for the planar cell polarity pathway mediated through members of the Wnt/β-catenin family and their potential to regulate actin cytoskeletal machinery. It is noteworthy that Wnts and components of the actomyosin cytoskeleton (Acta2 and Filamin-A) have each been associated with either cardiac development and/or pathogenic diseases that cause cardiac defects in humans (Kyndt et al., 1998; Kyndt et al., 2007; Lardeux et al., 2011; Guo et al., 2007; Milewicz et al., 2008; Guo et al., 2009). The function of these proteins in regulating polarized cell-fate decisions has yet to be experimentally defined.
Regardless of the mechanism, it is well-recognized that following EMT, the molecular and cellular constituents of the developing cushions define this structure as a primitive pre-valvular entity, which functions to prevent retrograde blood flow almost as soon as the heart begins to beat (Armstrong and Bischoff, 2004; Eisenberg and Markwald, 1995).
Post-EMT Valvulogenesis
A poorly understood process during valvulogenesis concerns the mechanisms regulating how mesenchymal cells derived by EMT differentiate into valve interstitial fibroblasts and remodel their local environment into the mature leaflets and their supporting apparatus (e.g. tendinous cords and the fibrous annulus). Maturation of cushion tissue into attenuated fibrous valve leaflets is one of the first remodeling events in the developing mammal (Chakraborty et al., 2010; de Lange et al., 2004; Norris et al., 2009a). As noted above, de la Cruz and Markwald (1998) proposed two signaling fields within the cushions: a proliferation field, located adjacent to the distended endocardium, and a differentiation field, adjacent to the myocardium. This concept of zonal boundaries within the cushion is reminiscent with findings in limb bud development whereby specific signals are secreted from different locations (e.g. progress zone vs. zone of polarizing activity) that inform and define the phenotype and function of these cells. If this zonal interface were to exist during valve development, one would expect to find preferential expression of specific genes and proteins in one zone versus the other. This, indeed, has been demonstrated for collagen proteins and may identify those cells that have matured from an undifferentiated mesenchyme into a fibroblastic-type cell (Norris et al., 2009b; Tan et al., 2011). We further show in Figure 2 that the enzyme transglutaminase-2 may serve as a quintessential marker of this zonal boundary whereby cells closest to the myocardium are molecularly distinct from those located subendocardially. This boundary demarcation is but only one similarity between cushion and limb bud development. Additionally, the expression of many of the same genes required for cartilage and tendon formation during limb development are also found expressed in the developing cardiac cushions (Hinton et al., 2006; Combs and Yutzey, 2009; Alfieri et al., 2010; Hinton and Yutzey, 2011; Lincoln and Yutzey, 2011) indicating these two seemingly divergent tissues may rely on similar molecular cues for their formation.
Figure 2. Expression of Transglutaminase-2 (TG2) Defines a Cushion Boundary Interface.
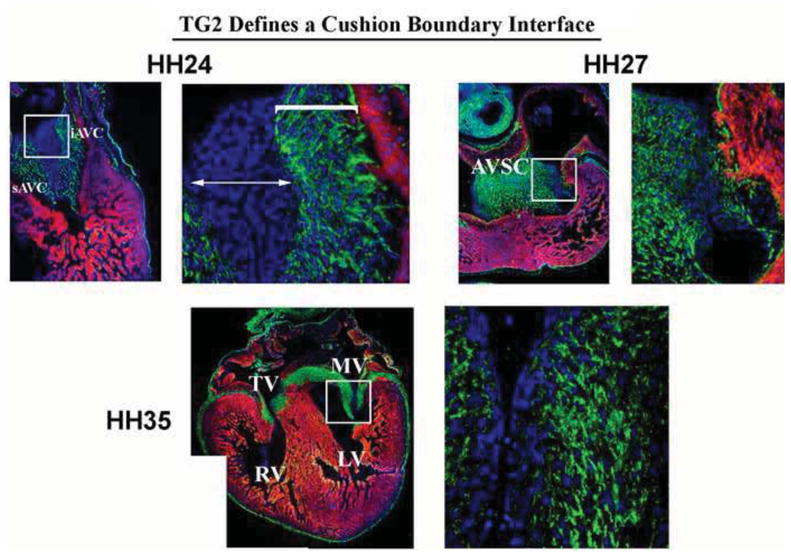
Immunohistochemical analyses for TG2 were performed on chick embryos through embryonic and fetal life. Expression of this enzyme at HH24 is restricted to a region close to the myocardium (bracket), whereas the distal half of the cushion is devoid of expression. Expression following cushion fusion (HH27) and during maturation (HH35) demonstrates nearly all interstitial cells stain positive. These data demonstrate a boundary interface is present in the developing cushions at early embryonic timepoints and may define a border of differentiation.
The molecular cues that cause zonal boundaries and cellular phenotypic changes during valvulogenesis are unknown but like the limb bud, likely invoke gradients in morphogens and/or differences in cell receptor profiles (Figure 1). In this example, at least three possibilities exist. First, the myocardium secretes a morphogen, or combination of morphogens, that directly stimulates the cells to differentiate (e.g. BMP 2,4). Second, the endocardium secretes a suppressive signal that is inhibitory to differentiation for cells closest to the endocardium. As the cells migrate away from the endocardial ridge, the dose of inhibitory morphogen is spatially diluted and the block to differentiation is removed. A third potential mechanism is that migrating mesenchymal cells secrete an autocrine signal, which, over time, promotes cell autonomous differentiation (as observed with the matricellular protein, periostin (Borg and Markwald, 2007; Norris et al., 2008; Snider et al., 2008; Tkatchenko et al., 2009). These hypotheses need to be experimentally defined, but data presented here (Figure 2) as well as previous data supports the notion that endocardial cushions have definitive boundaries of proliferation vs. differentiation (de la Cruz and Markwald, 1998b). Additionally, the persistence of subendocardial mesenchyme in the tips of adult leaflets (called Noduli Arantes) is broadly consistent with these hypotheses, especially the existence of a pro-proliferative, endocardial suppressive signal for differentiation. Loss of this signal in remodeling leaflets would also be consistent with the low rate of mitosis observed in mature valve leaflets (Mulholland and Gotlieb, 1996; Fayet et al., 2007; Liu et al., 2007; Chakraborty et al., 2010; Chakraborty et al., 2011; Shelton and Yutzey, 2008; 2007). This apparent reciprocal relationship between proliferation and differentiation correlates with the onset of active remodeling which we define as an adaptive reorganization of the microenvironment driven by molecular, cellular and mechanical cues that are required for maturation of cushion tissues into valve structures.
The septation of the OFT and AV junction into separate right and left ventricular outlets and inlets is also mediated by the fusion of cushion tissue. Thus the original mesenchymal septa of the early embryonic heart (membranous IVS, primary atrial septum) share a common endocardial origin with the valves. Specifically in the AV junction two major cushions, referred to as the superior and inferior endocardial cushions (Wessels and Sedmera, 2003) fuse centrally to form the AV septum (or “septum intermedium”) (Wessels et al., 1996; Wessels and Sedmera, 2003; Snarr et al., 2007; Snarr et al., 2008; Eisenberg and Markwald, 1995; Person et al., 2005). The AV septum (AVS) aligns and eventually merges with both the developing ventricular septum and the interatrial septum, the latter merging via a mesenchymal cap and the dorsal mesenchymal protrusion (DMP) to form a valvuloseptal complex that variably contributes to midline leaflets or supporting apparatus of the right (tricuspid) and left (mitral) valves (de la Cruz and Markwald, 1998a; de la Cruz and Markwald, 1998b; de la Cruz et al., 2001; Person et al., 2005; Wirrig and Yutzey, 2011; Snarr et al., 2007; Snarr et al., 2008). A second set of endocardial cushions also forms in the left and right AV junctions, the left and right lateral AV cushions (llAVC and rlAVC), which become the future mural (posterior) leaflet of the mitral valve and the anterosuperior leaflet of the tricuspid, respectively (de Lange et al., 2004; Lincoln and Yutzey, 2011; Snarr et al., 2008). The mesenchymal cells within the lateral and septal AV cushions, like those of the outflow cushions, progressively (and irreversibly) transition into interstitial cells characterized by increased synthesis of fibrillar matrices (i.e. collagens) and decreased presence of the mesenchymal cell markers twist and snail (Tan et al., 2011; Peacock et al., 2008; Lincoln et al., 2004; Tao et al., 2011 ; Chakraborty et al., 2011). Unlike the OFT cushions, the AV cushions develop into leaflets upon a myocardial template (de Lange et al., 2004). However the process by which these leaflets mature into fully functional and free-moving leaflets is poorly understood. As the AV primordial leaflets (especially the lateral or mural cushions) distend into the lumen, they are accompanied by a portion of the myocardium (Oosthoek et al., 1997; Oosthoek et al., 1998a; Oosthoek et al., 1998b; de Lange et al., 2004). This myocardial template is progressively removed and/or remodeled during fetal life, leaving a mobile leaflet. Mechanisms contributing to myocardial removal are poorly understood, but are an important and underappreciated facet of valve development. In the AV leaflets, it has been proposed that the attached myocardium undergoes programmed cell death leaving behind the mobile leaflet and remnant tissue that may contribute to the development of the chordae tendineae and papillary muscles (de Lange et al., 2004). However, it is not clear whether the modest amount of apoptosis in this region would be sufficient to remove all myocardium and brings into question whether additional mechanisms are in play. Regardless, the interesting questions, assuming that myocardial apoptosis is the main mechanism for removal of this subvalvular tissue, are: Why does this remodeling process occur primarily in this region of the heart and does this suggest a specified regulatory network in subvalvular myocardial removal? Although these questions have yet to be answered, recent work has demonstrated that the fasciclin related matricellular protein, βIG-H3 (TGFβ-induced gene-H3) may play a key role. βIG-H3 is expressed not only in the valves but also in the subvalvular myocardium during this “apoptotic phase” (Norris et al., 2005) and has been shown as an important mediator of anoikis (integrin-mediated apoptosis) in other cell types(Kim et al., 2003). Since anoikis is likely involved in pathological remodeling of cardiovascular tissues, including cardiac myocyte detachment in heart failure(Michel, 2003), it would not be surprising if this unique mode of apoptosis functions during normal development through a βIG-H3 pathway. Although the function of βIG-H3 in promoting anoikis in the subvavluar myocardium is logical to propose, a characterization of the βIG-H3 knockout mouse cardiac phenotype, and its contribution to myocardial remodeling has yet to be described. Regardless, this is a clinically relevant process to characterize and fully understand as retention of the myocardial template on the septal leaflet of the tricuspid valve can result in congenital heart defects e.g. Ebstein’s Anomaly or accessory conduction pathways as in WPW (Wolff-Parkinson-White) syndrome (Anderson et al., 1979; Zuberbuhler et al., 1979; Stein, 1946).
An important consideration for this timepoint in development is that post-EMT prevalvular cells secrete large amounts of extracellular matrix including collagens, proteoglycans and hyaluronan (Hinton et al., 2006; Lincoln et al., 2004; Lincoln and Yutzey, 2011; Levay et al., 2008; Peacock et al., 2008; Tan et al., 2011). However, aside from their ability to secrete matrix proteins, the function of these interstitial cells and molecular mechanisms driving matrix synthesis and remodeling are not well understood. It is clear that this remodeling phase is a critical window of time during which prevalvular cushions undergo a transition from a largely stochastic mesenchymal cell/matrix milieu into a tightly compacted, highly ordered, lamellar array of interstitial cells and ECM. Indeed, a fundamental question of valve development is not just what regulates synthesis of extracellular matrix but also how the secreted matrix is organized to assume its final trilaminar structure. Recent data have suggested that fibroblasts under tensile strain develop unique cell structures call “fibripositors”, which promote the secretion and alignment of collagen fibrils (Kadler et al., 1996; Canty and Kadler, 2002; Canty et al., 2004; Kadler, 2004; Canty and Kadler, 2005; Canty et al., 2006; Kapacee et al., 2008). Tension appears to be a fundamental driving force in the formation of fibripositors. It is therefore interesting to note that during valvulogenesis, unique, tension points are created within the annulus fibrosis at the base of the leaflet and in the chordae tendineae where they attach to the free edges of leaflet. In our studies, we have identified these structures as being present in the developing valve, specifically at times of fetal life when matrix is being synthesized and aligned (Figure 3). Thus, through the generation of tension points and their resultant effect upon formation of fibripositor provide one potential mechanism by which the prevalvular mesenchymal cells or fibroblasts contribute to matrix secretion and alignment. Further work is needed to identify the true contribution/importance of fibripositors to the remodeling phase of valvulogenesis when valve tissue needs to reorganize its matrix and cellular constituents to attain compacted, attenuated and highly organized histological structure.
Figure 3. Fibripositor number during mitral valve maturation is temporally regulated.
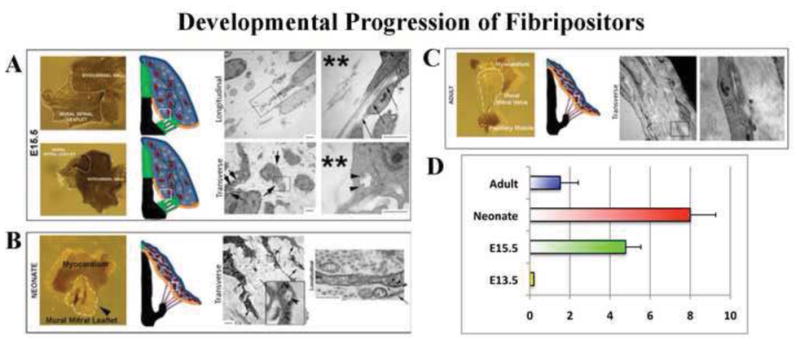
(A–C) Transmission Electron Micrographs of mitral leaflets through valve maturation. Left panels depict gross whole-mount views of the leaflets processed for TEM with the timepoints denoted at the left and leaflet tissue depicted with white lines. Middle panels represent a schematic of various stages of AV valve development (red cells depict fibroblasts, green cells represent mesenchyme, black lines are collagen). Small white boxes denote where TEMs were taken. Right panel denotes TEM images. (A) Note very little collagen present at this stage (**), and randomized cellular distribution. Close up images of boxed regions show collagen fibrils present within cellular organelles (arrow heads). Longitudinal views show collagen fibrils initiating inside cell processes/fibripositors (arrows). (B) By neonatal development, parallel collagen fibrils are seen coincident with cellular alignment and an increase in fibripositor formation (arrows). (C) By adulthood, fibripositors and intracellular collagen containing organelles are not observed. (D) Fibripositors are first observed at E15.5 with a substantial increase at postnatal day 2 (neonate). By adulthood, fibripositors are scarcely observed within mature mitral valve fibroblasts. N>30 cells for each timepoint.
To understand the mechanisms driving this remodeling process, previous studies from our laboratory and other groups have demonstrated that during post-EMT, valve fibroblasts acquire a contractile state due to various stimuli (Butcher and Nerem, 2004; Butcher et al., 2007a; Butcher et al., 2007b). As a result, the cells are able to “pull” on the surrounding matrix environment promoting condensation/compaction and alignment of the cells and matrix. This event occurs during a time of increased cell density (Kruithof et al., 2007), which may additionally facilitate the remodeling process and thereby influence the establishment of the adult AV valve configuration. These structural and cellular changes in the tissue are essential to adaptively confer increased biomechanical stability to the valve, which is required for protecting against the increasing hemodynamic load of a growing heart (Kruithof et al., 2007; Rabkin-Aikawa et al., 2004; Hinton and Yutzey, 2011; Hinton et al., 2008; Yalcin et al., 2011). The contractile force responsible for matrix condensation and tissue compaction is well recognized as a cytoskeletal dependent event, requiring the organization and function of an actin-network and its interacting proteins (Sandbo and Dulin, 2011; Butcher et al., 2007b; Canty et al., 2006). Additionally, cell surface membrane receptors (e.g. integrins) serve as crucial elements in tethering the actin cytoskeleton to the extracellular matrix. Thus, any candidate regulatory mechanisms for promoting fibroblast contractility would likely invoke components of the actin cytoskeletal machinery and/or integrin receptors. Identification of such regulatory mechanisms is being pursued through in vitro approaches whose relevance to in vivo, post-EMT processes largely remains to be established. Based on previous data (Kapacee et al., 2008), we developed and applied a novel methodology, which takes into consideration the anatomy of the AV valve leaflets in vivo as well as permits evaluation of de novo collagen secretion and cellular contractility. This method, which we term “Fibrin Post Assay” is a tool for understanding molecular regulation of post-EMT AV valve development in vitro which holds real potential for understanding in situ mechanisms by which collagen secretion occurs, cell-cell communication is achieved and how cell-cell and cell-matrix organization and alignment proceeds (Figure 4).
Figure 4. Fibrin gel assay.
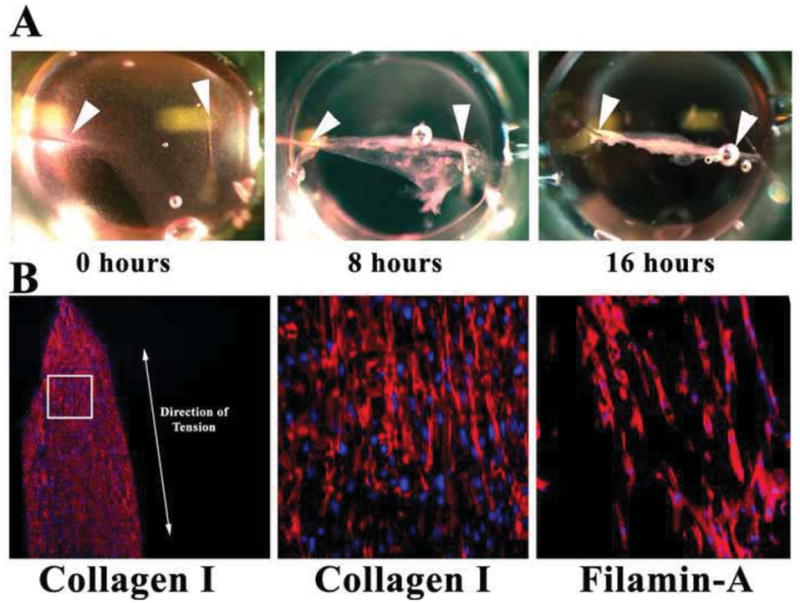
(A) Pictures of fibrin gels compacting over time in 96-well dishes. Arrowheads demarcate where needles (“pillars”) were placed. Gel compacts to a linear construct in <1 day. (B) IHC of sectioned constructs showing de novocollagen I (red) and FLNA (red) align along the axis of tension (double-headed arrow). Nuclei-blue.
There is considerable evidence that congenital valve malformations in humans have a genetic basis and therefore represent abnormalities in development. In most cases, gene mutations, present at conception lead to altered developmental processes that are not clinically evident at birth, but result in increased susceptibility to disease as the individual ages. As most defects associated with the EMT phase of valve development result in embryonic lethality, we hypothesize that the majority of congenital cardiac anomalies are associated with post-EMT events. If true, this underlies the importance for studying post-EMT processes and their relevance to pediatric and adult diseases.
Lineage as a key to understanding AV Valve morphogenesis
The integration of multiple mesenchymal building blocks is required for the formation of the AV septum (or septal complex) and the AV valves. Thus an important question to answer is what are the cell lineages that contribute to the building of these structures. It is still not widely recognized that the lineages of the different AV mesenchymal primordial (including the AV valves) are not all the same. In addition to the dorsal mesenchymal protrusion (DMP) of secondary heart field lineage, the mesenchymal cells of the two large midline AV cushions (sAVC, iAVC), the atrial cap, and the post-fusion AV valve apparatus (leaflet, annulus, chordae tendineae) appear to be derived from at least four distinct lineages. One of these lineages, or subsets of mesenchyme, is derived from the endocardium, which has been confirmed by the transformation of a Tie2+cre endocardial endothelium to mesenchyme (Lincoln et al., 2004; Rivera-Feliciano et al., 2006). These Tie2+ cre cells not only give rise to components of the major midline cushions and AV valve leaflets, but also appear to contribute to the chordae tendineae (de Lange et al., 2004; Lincoln et al., 2004). A second cellular subset that contributes to the developing AV septal complex is the epicardium and epicardial-derived cells (EPDCs) (Perez-Pomares et al., 2002; Wessels and Perez-Pomares, 2004; Gittenberger-de Groot et al., 1998). Although early reports had indicated that the proepicardium/epicardium and/or EPDC’s do not appreciably contribute to the forming valves (de Lange et al., 2004), recent work using more robust mouse genetics has demonstrated an epicardial-contribution not only to the annulus fibrosis and AV sulcus/groove, but also to the mural mitral and tricuspid leaflets (Zhou et al., 2010; Wessels et al., 2012; Gittenberger-de Groot et al., 1998; Kolditz et al., 2007). A third cellular subset that contributes to the development of the AV septal complex and the AV valves is the neural crest cells. Whereas early reports stated the lack of Wnt-1 Cre/R26R positive cells in the AV valves (de Lange et al., 2004), more recent studies using 2 Cre models (Wnt-1Cre/R26R and POCre/R26R) have demonstrated neural crest cells do, in fact, migrate into the septal tricuspid leaflet and the aortic leaflet of the mitral valve while sparing the mural leaflets (Nakamura et al., 2006). These neural crest cells appear to penetrate through the sinus venosus mesocardium and proceed to the AV canal at E12.5. By E17.5, many of these Wnt1-Cre/R26R positive cells are found in the AV node and cardiac conduction tissue as well as in the septal leaflet of the tricuspid valve and aortic leaflet of the mitral valve where they also stain positive for the melanocyte marker, TRP1, further substantiating the presence of neural crest cells within the AV valves (Nakamura et al., 2006; Mjaatvedt et al., 2005). Recently, a fourth contributing cell population has been identified. Transplantation of an EGFP positive bone marrow cell into a lethally irradiated host resulted in EGFP positive cells within the adult cardiac valves that were also positive for collagen I expression. This demonstrates that cells of a bone marrow hematopoietic stem cell origin are able to contribute to valvular interstitial cells (or VICs) (Visconti et al., 2006; Hajdu et al., 2011). This population of cells was also found to be positive for the hematopoietc stem cell marker, CD45, demonstrating that these cells arise (and arrive) from the blood and may play a key role in valve homeostasis. The precise timing of CD45+ cell entry into the valves has not been reported to date. Defining the temporal and spatial contribution of CD45+ cells to the valves may lead to new and exciting discoveries of how the valve leaflets develop as well as provide novel homeostatic mechanisms by which postnatal maintenance of the valves during normal life proceeds with age.
Except for the timing of endocardial, neural crest, and epicardial-derived cell contribution to the developing cushions/leaflets, the engraftment mechanisms by which the epicardial and CD45-derived cells enter the AV (and OFT) valves are not understood. Additionally, the ratios of these cells of different origins, their precise localization within the valves and their developmental or postnatal functions represent largely unanswered questions. With the development of new and applicable genetic mouse models such as the CD45-Cre and WT-1-Cre, answers to these questions may finally be realized, especially when combined with genetic models of AV valvuloseptal defects and diseases. For example, does each of the progenitor populations truly bring something unique to the development of the valve tissue? This is intriguing to ponder, as cells placed in a foreign environment will, in many instances, obtain the phenotype and function of resident cells simply based on their local cues. As we demonstrate in Figure 5 cushion mesenchyme dissected from a quail heart and placed in the limb bud of a developing chick will indeed assume the phenotypic characteristics of cells in the forming limb. These experiments demonstrate that undifferentiated cushion mesenchyme is multipotential and can be influenced by its microenvironment. This is consistent with recent findings demonstrating the matricellular protein, periostin and an upstream negative regulator, SRF, promote cardiogenic mesoderm into prevalvular mesenchyme, which ultimately differentiate into fibroblasts (Niu et al., 2008; Norris et al., 2008; Norris et al., 2009b). Global deletion of periostin permits aberrant differentiation of cushion mesenchyme into alternative cell lineages (e.g. bone, myocytes, etc.). Whether the valve progenitor cells of different origins respond similarly to periostin or other differentiation stimuli are unknown but an important question to address as cardiovascular diseases may reflect the function of one progenitor cell type more significantly than another. Alternatively, one valve progenitor cell type may be more susceptible to genetic, pharmacological and/or environmentally induced disease. Thus, by understanding the lineage question, there is potential for revealing new pathogenetic mechanisms for both congenital and adult heart diseases whose root causes (e.g. mutations) extend back into embryonic life.
Figure 5. Multipotentiality of Cushion Mesenchyme.
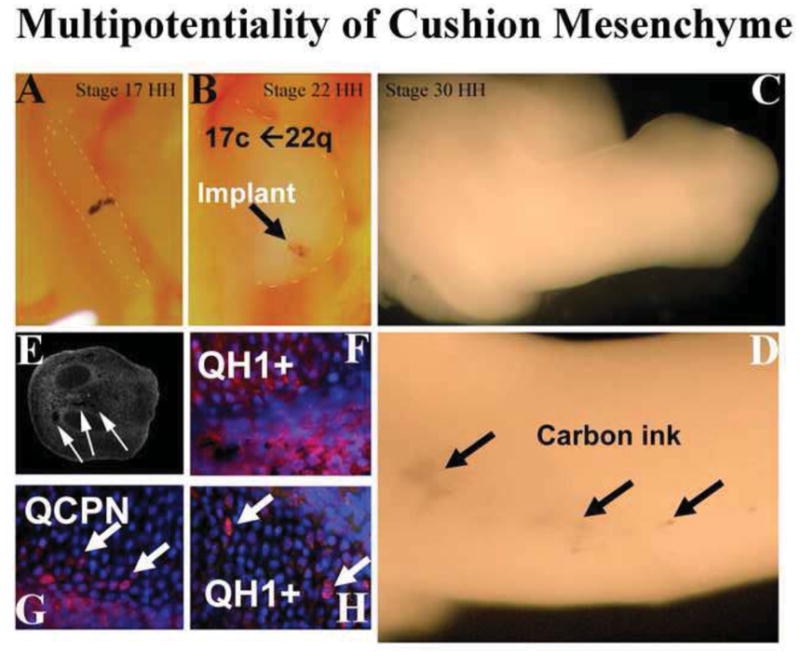
Stage 22 quail AV cushion was explanted, tattooed with carbon ink and placed in the developing wing-bud of a stage 15 chick (A). As the wind bud grew out, the carbon ink permitted visualization of the quail tissues/cells (arrows in B–D). Chimeric chick limbs were sectioned and carbon ink was visualized (arrows in E). Staining for quail-specific markers QH1 (red) and QCPN1 (red) identified cells of quail origin (F–H). When cushion tissue from quail was implanted into chick, these cells integrated with existing tissue and assumed a bone phenotype demonstrating that cushion mesenchyme is multipotential and its phenotype can be influenced by the microenvironment.
Structure of the Valves: Matrix Components and Organization
A detailed description of the structural components and integration of AV cushions with the developing heart has been detailed by our colleagues (Kruithof et al., 2012; Briggs et al., 2012). Thus, we focus in this section primarily on the structure of the fully mature adult AV and SL valves in mouse and human systems. The mouse has become an important model for valve development and comparative studies of pathogenesis in the human heart as they share many similarities. In both, the heart has four chambers: two atria and two ventricles. The mitral and tricuspid AV valves maintain unidirectional blood flow from the atria into the ventricles, while the aortic and pulmonary semilunar (SL) valves regulate blood flow from the ventricles into the outflow arteries. The AV valves are composed of asymmetrical leaflets attached to a ring-shaped annulus and tethered by a sub-valvular apparatus consisting of the chordae tendineae and papillary muscles (Hinton and Yutzey, 2011). The mitral valve separates the left atrium and left ventricle and is composed of an anterior and posterior leaflet. Each leaflet is secured to the annulus fibrosis, a fibrous and tough elastic tissue that encircles the AV canal and OFT orifices and provides structural support to the valves while also dividing the ventricular and atrial myocardium (Snarr et al., 2008; Kruithof et al., 2007). The free edges of the valves are tethered to either an anterior or posterior papillary muscle at the base of the ventricle via the chordae tendineae (CT). CT provides tension and prevent prolapse of the valves back into the atria during AV valve closure, or ventricular systole. The tricuspid valve is situated between the right atrium and ventricle and has three leaflets: the anterior, posterior, and septal leaflets. The leaflets are attached to the annulus fibrosa and tethered via CT to several papillary muscles continuous with the right ventricular myocardium.
In comparison, the SL valves are self-supporting and attach to their crown-shaped arterial roots. The aortic valve segregates the left ventricle from the aortic outlet artery and consists of three cusps. The cusps, left coronary, right coronary, and noncoronary, are named in relation to their associated coronary arteries. Each cusp inserts into the aortic root via the fibrous annulus. The pulmonary valve separates the right ventricle from the pulmonary artery and also has three leaflets termed the left, right, and anterior cusps. These cusps insert into the pulmonary root not through a fibrous annulus, but primarily through a freestanding, muscular sleeve termed the right ventricular infindibulum.
Human valve leaflets are of course much larger and thicker than their mouse counterparts, human valves being typically in the millimeter range whereas mouse leaflets are measured in microns with the AV valves being normally thicker than SL in both species. The leaflets in both species are composed of an ECM that is produced and organized based on the specific developmental programs and cellular events described earlier. The inception of the formation of this structure truly begins during post-EMT valvulogenesis when the matrix is being actively synthesized and organized. In addition, their molecular make-up and organization may be largely dictated by the biomechanical stresses that they encounter(Butcher and Markwald, 2007).
Although the types of ECM produced and the organization of this ECM have been largely viewed as the same across species, we believe the mature valve is organized quite differently between mouse and humans. Apart from their obvious differences in thickness, there are significant physiological and biomechanical differences between these two species (Wessels and Sedmera, 2003). Nonetheless, investigators often portray the matrix and its organization to be similar. Herein we will present some significant differences in the molecular organization of the mature valves and demonstrate data to substantiate these findings.
Human valves have typically been described as containing a highly organized ECM distributed into three layers, the fibrosa, spongiosa, and either ventricularis or atrialis depending upon whether it is an inlet or outlet valve leaflet (Aikawa et al., 2006; Hinton and Yutzey, 2011). The fibrosa consists mostly of fibrillar collagens that provide tensile stiffness; the spongiosa is enriched in proteoglycans that allows for flexibility and compression; and, the atrialis (for inlet valves)/ventricularis (for outlet valves) is composed of filamentous elastic fibers that facilitate retraction of the leaflet back to coaptation (Padala et al., 2009; Sacks and Yoganathan, 2007; Sacks et al., 2006; Ritchie et al., 2006; Sacks et al., 2002). This classical viewpoint for human valves is based on histological stainings and does not take into account potential molecular differences between the boundaries. Based on our further inspection, human heart valves actually consist of at least six molecularly distinct zones (Figure 6). Starting from the ventricularis going towards the atrialis, here is a description of each of the zones: 1) Along the ventricular portion of the AV valves (or arterial portion of SL) there is a subendocardial zone rich in periostin, hyaluronan, and collagen I, 2) a collagen rich, periostin and hyaluronan free zone, 3) collagen and hyaluronan-rich, 4) collagen rich mid-region (based on Movats stain) 5) primarily proteoglycan containing (based on Movats stain), and 6) atrial aspect of the AV valves (or ventricular aspect of SL) which consists mostly of elastin (based on Movats staining) and appreciable amounts of periostin (data not shown). These differences are important to consider since each molecular zone likely has it own, unique biomechanical properties that are important to the normal function of the valve. It is also important to consider the relevance this may have to understanding valve diseases. Valve disease, especially myxomatous mitral valve disease, is largely viewed as having an expansion of the spongiosa layer. Thus, if we are to understand and study clinically evident valve disease, it will be critical to appreciate how the spongiosa layer is defined and what type of matrix and cells reside there.
Figure 6. Histological and Molecular Profile of Human Mitral Valve.
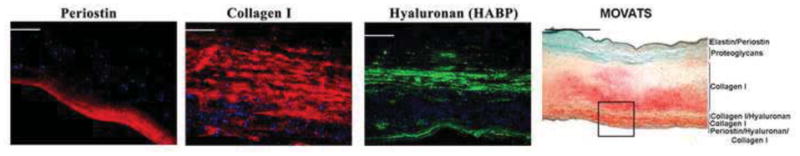
An adult human mitral valve was histologically stained for Movats Pentachrome stain (far right panel) and revealed multiple layers of collagen (yellow), proteoglycans (blue/green), and elastin (black). IHC analyses of boxed region in far right panel was evaluated for expression of hyaluronan (via hyaluronan binding protein-HABP), periostin, and collagen I. The molecular partitioning of this valve is indicated in the Movats stained panel. The IHC data indicates that the fibrosa layer of the mitral valve can be dissected into 3 layers, and the leaflet in its entirety can be comprised of at least 6 molecular distinct zones.
In comparison, the mouse heart valves (inlet and outlet) do not have these 5 molecularly distinct regions. This is important to note, since mouse valves have been historically viewed as being structured like the human valves. Due to the size difference between species and the requirement of the valve structure in maintaining such massive differences in biomechanical stresses, it would be illogical to assume that the mouse valve is structured in the same manner as the human. Indeed the mouse valve is only 2–5 cell-layers thick, precluding its ability to contain 5 molecularly distinct matrix layers. Upon performing histological stains and immunohistochemical analyses, we are able to define clearly only two zones, not 3 layers (Figure 7). Unlike the human and conventional descriptions in mouse, these zones are not organized in layers, but rather provide boundary interfaces along a proximal-distal axis of the leaflet. Indeed, in the mitral leaflets we observe the base and mid-region of the leaflet as a proteoglycan-free, collagen-rich zone whereas the tip of the leaflet is proteoglycan rich and collagen free. Although, RT-PCR analyses have defined elastin as being expressed in murine cardiac valves, its precise protein localization and/or organization within a defined boundary is difficult to ascertain (Hinton et al., 2011 ). One study has demonstrated weak expression of elastin in a portion of the atrial aspect of the mitral leaflet, appearing confined to the endocardial cells (Peacock et al., 2008). Although this may represent a third layer, this report shows that elastin staining does not transverse the entire atrial aspect of the leaflet, further supporting our concept of zonation along a proximal-distal axis in the mouse valve. Of course, the mouse model, due to the ability to manipulate its genetics, is the best model for studying human related valvular diseases. But, the structural discrepancies between mouse and human valve leaflets needs to be carefully considered when comparing mouse defects to human counterparts, especially in the context of gene deletions, mutation analyses, and/or valve remodeling diseases.
Figure 7. Histological and Molecular Analyses of Mouse Mitral Valve.
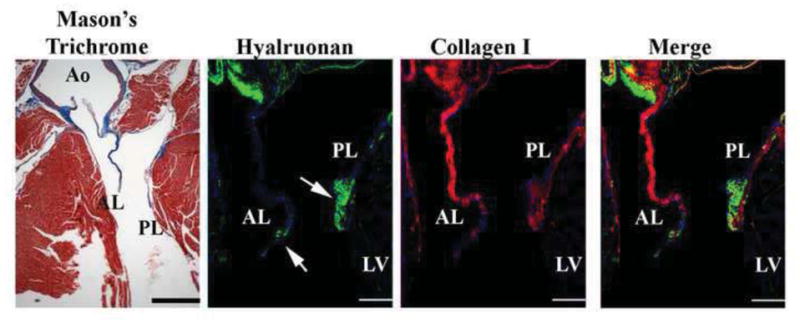
A 2 month old adult heart was stained for Masson’s trichrome which demonstrates the anterior (AL) and posterior (PL) leaflets are primarily collagen with only a small region at the distal tip showing lack of expression. This was confirmed by IHC stainings for collagen. The distal tip of the leaflets (arrows) appears to be only comprised of proteoglycans. Thus the mouse leaflet is divided into 2 zones along a proximal-distal axis of the valve. Scale bars=200 μm.
Biomechanics
In an average lifespan the valves will cycle approximately three billion times to maintain unidirectional blood flow through the heart (Sacks and Yoganathan, 2007). Valve opening and closing is passively directed by blood pressures within the heart chambers and the heart itself (Sacks and Yoganathan, 2007; Butcher et al., 2007a; Yalcin et al., 2011). Before atrial contraction occurs, passive flow allows for the main mass of blood to enter the ventricles. Once the pressure in the ventricles rises above that in the atria, the AV valves close for systole and the SL valves open, allowing blood to flow from the high-pressured and contracting ventricles into their corresponding great artery. Also, because of the load capacity differences between the left and right side of the heart, the left side normally maintains higher pressures; thus, both hemodynamics and valve location are important for understanding valve biomechanics and associated force loads.
The forces exerted upon the valve tissues during opening and closing are those characterized as flexure, shear, and tension (Sacks and Yoganathan, 2007). Flexure occurs as the valves open and close, shear stress results from the passing blood-flow, and tension occurs during coaptation or full loading. These loading forces vary between the valves depending on local pressures and supporting apparatuses (Sacks and Yoganathan, 2007). Therefore, the cellular content and matrix composition are important for considering valve dynamics and loading force endurance. Using smooth muscle α-actin (SMA) and heat shock protein 47 (Hsp47) to quantify cellular stiffness and collagen biosynthesis, Sacks and Yoganathan (2007) found the left-sided AV valve interstitial cells (VICs) to have higher quantities of both SMA and Hsp47, indicating that these VICs, are exposed to greater forces and respond or adapt to local tissue stress by increasing collagen and cellular (cytoskeletal) stiffness in order to conserve homeostasis. This is also consistent with findings from Aikawa et al (2006) who demonstrate that during the postnatal period, activated interstitial valve fibroblasts gradually become quiescent, whereas collagen matures (Aikawa et al., 2006). This maturation of collagen is likely due to increased collagen accretion and cross-linking and is a required step for biomechanically mediated adaptation. These studies demonstrate that valves have plasticity, viz. they are adaptive and reactive to their local environment. The ability of the valves to adapt has also recently been demonstrated in both sheep and humans primarily in cases of left ventricular infarction, dilation, and/or hypertrophy. In these cases, mitral leaflet tethering by displaced papillary muscles frequently induces mitral regurgitation and can significantly increase morbidity (Granier et al., 2012; Hagege et al., 2012; Judge et al., 2012; Messas et al., 2012). Dal-Bianco et al. has demonstrated in a sheep model of papillary tethering, that mechanical stresses imposed by papillary muscle tethering causes the mitral valve to adaptively respond. Significant increases in area and thickness of the mitral leaflets are observed in this model, concomitant with reactivation of developmental pathways (Dal-Bianco et al., 2009). Additionally, retrospective analyses in humans having undergone the Ross procedure have further demonstrated the ability for valve leaflets/cusps to adaptively respond to changes in their biomechanical environment. The Ross procedure uses a patients’ pulmonary valve to replace a diseased aortic valve/root with a homograft inserted into the right ventricular outflow tract (Ross, 1962). The Ross procedure is the only option with potential for annulus growth and is thus arguably the best technique of choice for children needing an aortic valve replacement. The fact that this procedure caries a relatively low early and mid-term mortality in patients who require aortic valve replacement (McBrien et al., 2012) speaks to the ability of the pulmonary valve to adapt to the significant increase in hemodynamic pressure of the left ventricular outflow tract. Thus, it is clear that biomechanics play a central role in not only the formation of the cardiac valves but also their response to changes in the microenvironment.
Developmental Basis of Disease and Concluding Remarks
The association of valve disease with a vast number of genetic disorders, coupled with the large number of family disease studies in the literature, defines adult-onset valve disease as having a genetic basis in much the same way as hypertrophic cardiomyopathy, diabetes or Alzheimer’s disease. In all cases, there are gene mutations, present at conception in affected individuals, which segregate in families and only manifest as clinically relevant diseases later in life. These mutations are thought to lead to altered developmental processes not evident at birth but result in increased susceptibility to disease or organ dysfunction as an individual ages. Defining the genes and their function in familial disease is crucial to understanding the pathways that lead to all forms of a disease. Genome-wide association studies in large populations of individuals with ‘sporadic’ disease have revealed, in all cases to date, specific genetic changes that are associated with adult-onset diseases. Mitral and aortic valve dystrophies, like most other genetic diseases, show variation in age at onset in families, despite the fact that the affected individuals all carry the same mutation. In fact, children with clinically relevant mitral valve prolapse (MVP) have been diagnosed as young as six years of age. In these families, mutations in the Filamin-A gene can cause juvenile or adult onset disease, and the onset of MVP in Marfan Syndrome patients is variable (Dietz, 1993). Taken together, this suggests that genetic defects present at the time of valve morphogenesis, in combination with the individual’s genetic background, may lead to subtle or more drastic dysfunction that progresses to clinically relevant disease, perhaps in only a few years or perhaps over many decades. Therefore, we propose that genetic factors play a substantial role in aging-related valve diseases, and only through the identification and characterization of disease causing genes and the subsequent elucidation of the relevant developmental pathways can we begin to understand, and someday prevent, progression of valve dystrophies.
To summarize
The development of the heart valves is a complex, progressive process that occurs as the heart is growing. This remarkable feat is even more incredible when considering this all happens as hemodynamic pressures increase. However, it is becoming clear that biomechanical signals are but one of multiple stimuli that are critical to forming the valve. Rather, it is the integration of biomechanical stimuli with molecular and cellular changes that promote how the valve transforms from a rudimentary cushion into a highly organized leaflet with supporting structures. Although much is known about valve development, our knowledge is still lacking in key areas of mesenchymal cell migration, matrix synthesis, matrix deposition, matrix organization, as well as the role of distinct cell populations (lineages) that integrate within the developing and adult valve. Thus, understanding how the valve is built may provide answers as to why and how valve disease progresses. Time may prove that degenerative (myxomatous) valve diseases are, in fact, the outcome of the valve being built improperly. Because mutations occur at conception, perhaps myxomatous valve diseases should be considered the result of generating a degenerative phenotype. Nonetheless, it will be important to study in greater detail the mechanistic underpinnings of how molecular pathways integrate with cellular changes and how mutations affect their integration or interaction. With the advent of human genetic studies, the identification of gene mutations in patients with valve diseases should open doors to understanding new, key regulatory pathways essential for cardiac valve development and disease pathogenesis.
Acknowledgments
This work was conducted in a facility constructed with support from the National Institutes of Health, Grant Number C06 RR018823 from the Extramural Research Facilities Program of the National Center for Research Resources. Other funding sources: NIH NHLBI: RO1-HL33756 (RRM), NIH-NCRR: COBRE P20RR016434-07 (RRM, RAN), P20RR016434-09S1 (RRM and RAN); American Heart Association: 11SDG5270006 (RAN), National Science Foundation: EPS-0902795 (RRM and RAN); The Foundation Leducq (Paris, France) Transatlantic Mitral Network of Excellence grant 07CVD04 (RAN, RRM, RAL, SAS, AH, AC, ZH, RV)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- Aikawa E, Whittaker P, Farber M, Mendelson K, Padera RF, Aikawa M, Schoen FJ. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation. 2006;113:1344–1352. doi: 10.1161/CIRCULATIONAHA.105.591768. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Chaboissier MC, Behringer RR, Rowitch DH, Schedl A, Epstein JA, de Crombrugghe B. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc Natl Acad Sci U S A. 2004;101:6502–6507. doi: 10.1073/pnas.0401711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfieri CM, Cheek J, Chakraborty S, Yutzey KE. Wnt signaling in heart valve development and osteogenic gene induction. Dev Biol. 2010;338:127–135. doi: 10.1016/j.ydbio.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KR, Zuberbuhler JR, Anderson RH, Becker AE, Lie JT. Morphologic spectrum of Ebstein’s anomaly of the heart: a review. Mayo Clin Proc. 1979;54:174–180. [PubMed] [Google Scholar]
- Armstrong EJ, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circ Res. 2004;95:459–470. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azhar M, Brown K, Gard C, Chen H, Rajan S, Elliott DA, Stevens MV, Camenisch TD, Conway SJ, Doetschman T. Transforming growth factor Beta2 is required for valve remodeling during heart development. Dev Dyn. 2012;240:2127–2141. doi: 10.1002/dvdy.22702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azhar M, Runyan RB, Gard C, Sanford LP, Miller ML, Andringa A, Pawlowski S, Rajan S, Doetschman T. Ligand-specific function of transforming growth factor beta in epithelial-mesenchymal transition in heart development. Dev Dyn. 2009;238:431–442. doi: 10.1002/dvdy.21854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azhar M, Schultz Jel J, Grupp I, Dorn GW, 2nd, Meneton P, Molin DG, Gittenberger-de Groot AC, Doetschman T. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 2003;14:391–407. doi: 10.1016/s1359-6101(03)00044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett JV, Desgrosellier JS. Early events in valvulogenesis: a signaling perspective. Birth Defects Res C Embryo Today. 2003;69:58–72. doi: 10.1002/bdrc.10006. [DOI] [PubMed] [Google Scholar]
- Bartram U, Molin DG, Wisse LJ, Mohamad A, Sanford LP, Doetschman T, Speer CP, Poelmann RE, Gittenberger-de Groot AC. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation. 2001;103:2745–2752. doi: 10.1161/01.cir.103.22.2745. [DOI] [PubMed] [Google Scholar]
- Bolender DL, Markwald RR. Epithelial-mesenchymal transformation in chick atrioventricular cushion morphogenesis. Scan Electron Microsc. 1979:313–321. [PubMed] [Google Scholar]
- Borg TK, Markwald R. Periostin: more than just an adhesion molecule. Circ Res. 2007;101:230–231. doi: 10.1161/CIRCRESAHA.107.159103. [DOI] [PubMed] [Google Scholar]
- Bouchey D, Argraves WS, Little CD. Fibulin-1, vitronectin, and fibronectin expression during avian cardiac valve and septa development. Anat Rec. 1996;244:540–551. doi: 10.1002/(SICI)1097-0185(199604)244:4<540::AID-AR12>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Boyer AS, Ayerinskas II, Vincent EB, McKinney LA, Weeks DL, Runyan RB. TGFbeta2 and TGFbeta3 have separate and sequential activities during epithelial-mesenchymal cell transformation in the embryonic heart. Dev Biol. 1999;208:530–545. doi: 10.1006/dbio.1999.9211. [DOI] [PubMed] [Google Scholar]
- Boyer AS, Runyan RB. TGFbeta Type III and TGFbeta Type II receptors have distinct activities during epithelial-mesenchymal cell transformation in the embryonic heart. Dev Dyn. 2001;221:454–459. doi: 10.1002/dvdy.1154. [DOI] [PubMed] [Google Scholar]
- Briggs LE, Kakarla J, Wessels A. The pathogenesis of atrial and atrioventricular septal defects with special emphasis on the dorsal mesenchymal protrusion. Differentiation. 2012 doi: 10.1016/j.diff.2012.05.006. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CB, Boyer AS, Runyan RB, Barnett JV. Antibodies to the Type II TGFbeta receptor block cell activation and migration during atrioventricular cushion transformation in the heart. Dev Biol. 1996;174:248–257. doi: 10.1006/dbio.1996.0070. [DOI] [PubMed] [Google Scholar]
- Brown CB, Boyer AS, Runyan RB, Barnett JV. Requirement of type III TGF-beta receptor for endocardial cell transformation in the heart. Science. 1999;283:2080–2082. doi: 10.1126/science.283.5410.2080. [DOI] [PubMed] [Google Scholar]
- Butcher JT, Markwald RR. Valvulogenesis: the moving target. Philos Trans R Soc Lond B Biol Sci. 2007;362:1489–1503. doi: 10.1098/rstb.2007.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher JT, McQuinn TC, Sedmera D, Turner D, Markwald RR. Transitions in early embryonic atrioventricular valvular function correspond with changes in cushion biomechanics that are predictable by tissue composition. Circ Res. 2007a;100:1503–1511. doi: 10.1161/CIRCRESAHA.107.148684. [DOI] [PubMed] [Google Scholar]
- Butcher JT, Nerem RM. Porcine aortic valve interstitial cells in three-dimensional culture: comparison of phenotype with aortic smooth muscle cells. Journal of Heart Valve Disease. 2004;13:478–485. discussion 485–476. [PubMed] [Google Scholar]
- Butcher JT, Norris RA, Hoffman S, Mjaatvedt CH, Markwald RR. Periostin promotes atrioventricular mesenchyme matrix invasion and remodeling mediated by integrin signaling through Rho/PI 3-kinase. Dev Biol. 2007b;302:256–266. doi: 10.1016/j.ydbio.2006.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camenisch TD, McDonald JA. Hyaluronan: is bigger better? Am J Respir Cell Mol Biol. 2000;23:431–433. doi: 10.1165/ajrcmb.23.4.f201. [DOI] [PubMed] [Google Scholar]
- Camenisch TD, Schroeder JA, Bradley J, Klewer SE, McDonald JA. Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2-ErbB3 receptors. Nat Med. 2002;8:850–855. doi: 10.1038/nm742. [DOI] [PubMed] [Google Scholar]
- Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A, Jr, Kubalak S, Klewer SE, McDonald JA. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest. 2000;106:349–360. doi: 10.1172/JCI10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canty EG, Kadler KE. Collagen fibril biosynthesis in tendon: a review and recent insights. Comp Biochem Physiol A Mol Integr Physiol. 2002;133:979–985. doi: 10.1016/s1095-6433(02)00212-x. [DOI] [PubMed] [Google Scholar]
- Canty EG, Kadler KE. Procollagen trafficking, processing and fibrillogenesis. J Cell Sci. 2005;118:1341–1353. doi: 10.1242/jcs.01731. [DOI] [PubMed] [Google Scholar]
- Canty EG, Lu Y, Meadows RS, Shaw MK, Holmes DF, Kadler KE. Coalignment of plasma membrane channels and protrusions (fibripositors) specifies the parallelism of tendon. J Cell Biol. 2004;165:553–563. doi: 10.1083/jcb.200312071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canty EG, Starborg T, Lu Y, Humphries SM, Holmes DF, Meadows RS, Huffman A, O’Toole ET, Kadler KE. Actin filaments are required for fibripositor-mediated collagen fibril alignment in tendon. J Biol Chem. 2006;281:38592–38598. doi: 10.1074/jbc.M607581200. [DOI] [PubMed] [Google Scholar]
- Chakraborty S, Combs MD, Yutzey KE. Transcriptional regulation of heart valve progenitor cells. Pediatr Cardiol. 2010;31:414–421. doi: 10.1007/s00246-009-9616-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Wirrig EE, Hinton RB, Merrill WH, Spicer DB, Yutzey KE. Twist1 promotes heart valve cell proliferation and extracellular matrix gene expression during development in vivo and is expressed in human diseased aortic valves. Dev Biol. 2011;347:167–179. doi: 10.1016/j.ydbio.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Ishii M, Sun J, Sucov HM, Maxson RE., Jr Msx1 and Msx2 regulate survival of secondary heart field precursors and post-migratory proliferation of cardiac neural crest in the outflow tract. Dev Biol. 2007;308:421–437. doi: 10.1016/j.ydbio.2007.05.037. [DOI] [PubMed] [Google Scholar]
- Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res. 2009;105:408–421. doi: 10.1161/CIRCRESAHA.109.201566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal-Bianco JP, Aikawa E, Bischoff J, Guerrero JL, Handschumacher MD, Sullivan S, Johnson B, Titus JS, Iwamoto Y, Wylie-Sears J, Levine RA, Carpentier A. Active adaptation of the tethered mitral valve: insights into a compensatory mechanism for functional mitral regurgitation. Circulation. 2009;120:334–342. doi: 10.1161/CIRCULATIONAHA.108.846782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz M, Markwald R. Embryological development of the ventricular inlets. Septation and atrioventricular valve apparatus 1998a [Google Scholar]
- de la Cruz MV, Markwald RR. Living Morphogenesis of the Heart. Birkhauser; Boston: 1998b. [Google Scholar]
- de la Cruz MV, Markwald RR, Krug EL, Rumenoff L, Sanchez Gomez C, Sadowinski S, Galicia TD, Gomez F, Salazar Garcia M, Villavicencio Guzman L, Reyes Angeles L, Moreno-Rodriguez RA. Living morphogenesis of the ventricles and congenital pathology of their component parts. Cardiol Young. 2001;11:588–600. doi: 10.1017/s1047951101000932. [DOI] [PubMed] [Google Scholar]
- de la Cruz MV, Sanchez Gomez C, Arteaga MM, Arguello C. Experimental study of the development of the truncus and the conus in the chick embryo. J Anat. 1977;123:661–686. [PMC free article] [PubMed] [Google Scholar]
- de la Pompa JL. Notch signaling in cardiac development and disease. Pediatr Cardiol. 2009;30:643–650. doi: 10.1007/s00246-008-9368-z. [DOI] [PubMed] [Google Scholar]
- de Lange FJ, Moorman AF, Anderson RH, Manner J, Soufan AT, de Gier-de Vries C, Schneider MD, Webb S, van den Hoff MJ, Christoffels VM. Lineage and morphogenetic analysis of the cardiac valves. Circ Res. 2004;95:645–654. doi: 10.1161/01.RES.0000141429.13560.cb. [DOI] [PubMed] [Google Scholar]
- Desgrosellier JS, Mundell NA, McDonnell MA, Moses HL, Barnett JV. Activin receptor-like kinase 2 and Smad6 regulate epithelial-mesenchymal transformation during cardiac valve formation. Dev Biol. 2005;280:201–210. doi: 10.1016/j.ydbio.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Dietz HC. Marfan Syndrome. 1993. [DOI] [PubMed] [Google Scholar]
- Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res. 1995;77:1–6. doi: 10.1161/01.res.77.1.1. [DOI] [PubMed] [Google Scholar]
- Fayet C, Bendeck MP, Gotlieb AI. Cardiac valve interstitial cells secrete fibronectin and form fibrillar adhesions in response to injury. Cardiovascular Pathology. 2007;16:203–211. doi: 10.1016/j.carpath.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Frieden LA, Townsend TA, Vaught DB, Delaughter DM, Hwang Y, Barnett JV, Chen J. Regulation of heart valve morphogenesis by Eph receptor ligand, ephrin-A1. Dev Dyn. 239:3226–3234. doi: 10.1002/dvdy.22458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funderburg FM, Markwald RR. Conditioning of native substrates by chondroitin sulfate proteoglycans during cardiac mesenchymal cell migration. J Cell Biol. 1986;103:2475–2487. doi: 10.1083/jcb.103.6.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittenberger-de Groot AC, Bartram U, Oosthoek PW, Bartelings MM, Hogers B, Poelmann RE, Jongewaard IN, Klewer SE. Collagen type VI expression during cardiac development and in human fetuses with trisomy 21. Anat Rec A Discov Mol Cell Evol Biol. 2003;275:1109–1116. doi: 10.1002/ar.a.10126. [DOI] [PubMed] [Google Scholar]
- Gittenberger-de Groot AC, Vrancken Peeters MP, Mentink MM, Gourdie RG, Poelmann RE. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ Res. 1998;82:1043–1052. doi: 10.1161/01.res.82.10.1043. [DOI] [PubMed] [Google Scholar]
- Granier M, Jensen MO, Honge JL, Bel A, Menasche P, Nielsen SL, Carpentier A, Levine RA, Hagege AA. Consequences of mitral valve prolapse on chordal tension: ex vivo and in vivo studies in large animal models. J Thorac Cardiovasc Surg. 2012;142:1585–1587. doi: 10.1016/j.jtcvs.2011.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenendijk BC, Hierck BP, Gittenberger-De Groot AC, Poelmann RE. Development-related changes in the expression of shear stress responsive genes KLF-2, ET-1, and NOS-3 in the developing cardiovascular system of chicken embryos. Dev Dyn. 2004;230:57–68. doi: 10.1002/dvdy.20029. [DOI] [PubMed] [Google Scholar]
- Groenendijk BC, Van der Heiden K, Hierck BP, Poelmann RE. The role of shear stress on ET-1, KLF2, and NOS-3 expression in the developing cardiovascular system of chicken embryos in a venous ligation model. Physiology (Bethesda) 2007;22:380–389. doi: 10.1152/physiol.00023.2007. [DOI] [PubMed] [Google Scholar]
- Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, Kim DH, Pannu H, Willing MC, Sparks E, Pyeritz RE, Singh MN, Dalman RL, Grotta JC, Marian AJ, Boerwinkle EA, Frazier LQ, LeMaire SA, Coselli JS, Estrera AL, Safi HJ, Veeraraghavan S, Muzny DM, Wheeler DA, Willerson JT, Yu RK, Shete SS, Scherer SE, Raman CS, Buja LM, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84:617–627. doi: 10.1016/j.ajhg.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker A. A mathematical model for mesenchymal and chemosensitive cell dynamics. J Math Biol. 2011 doi: 10.1007/s00285-011-0415-7. [DOI] [PubMed] [Google Scholar]
- Hagege AA, Bruneval P, Levine RA, Desnos M, Neamatalla H, Judge DP. The mitral valve in hypertrophic cardiomyopathy: old versus new concepts. J Cardiovasc Transl Res. 2012;4:757–766. doi: 10.1007/s12265-011-9319-6. [DOI] [PubMed] [Google Scholar]
- Hajdu Z, Romeo SJ, Fleming PA, Markwald RR, Visconti RP, Drake CJ. Recruitment of bone marrow-derived valve interstitial cells is a normal homeostatic process. J Mol Cell Cardiol. 2011;51:955–965. doi: 10.1016/j.yjmcc.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson DJ, Phillips HM, Chaudhry B. Vang-like 2 and noncanonical Wnt signaling in outflow tract development. Trends Cardiovasc Med. 2006;16:38–45. doi: 10.1016/j.tcm.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Hierck BP, Van der Heiden K, Alkemade FE, Van de Pas S, Van Thienen JV, Groenendijk BC, Bax WH, Van der Laarse A, Deruiter MC, Horrevoets AJ, Poelmann RE. Primary cilia sensitize endothelial cells for fluid shear stress. Dev Dyn. 2008a;237:725–735. doi: 10.1002/dvdy.21472. [DOI] [PubMed] [Google Scholar]
- Hierck BP, Van der Heiden K, Poelma C, Westerweel J, Poelmann RE. Fluid shear stress and inner curvature remodeling of the embryonic heart. Choosing the right lane! ScientificWorldJournal. 2008b;8:212–222. doi: 10.1100/tsw.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton RB, Adelman-Brown J, Witt S, Krishnamurthy VK, Osinska H, Sakthivel B, James JF, Li DY, Narmoneva DA, Mecham RP, Benson DW. Elastin haploinsufficiency results in progressive aortic valve malformation and latent valve disease in a mouse model. Circ Res. 2011;107:549–557. doi: 10.1161/CIRCRESAHA.110.221358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton RB, Jr, Alfieri CM, Witt SA, Glascock BJ, Khoury PR, Benson DW, Yutzey KE. Mouse heart valve structure and function: echocardiographic and morphometric analyses from the fetus through the aged adult. Am J Physiol Heart Circ Physiol. 2008;294:H2480–2488. doi: 10.1152/ajpheart.91431.2007. [DOI] [PubMed] [Google Scholar]
- Hinton RB, Jr, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, Yutzey KE. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. 2006;98:1431–1438. doi: 10.1161/01.RES.0000224114.65109.4e. [DOI] [PubMed] [Google Scholar]
- Hinton RB, Yutzey KE. Heart valve structure and function in development and disease. Annu Rev Physiol. 2011;73:29–46. doi: 10.1146/annurev-physiol-012110-142145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurle JM, Kitten GT, Sakai LY, Volpin D, Solursh M. Elastic extracellular matrix of the embryonic chick heart: an immunohistological study using laser confocal microscopy. Dev Dyn. 1994;200:321–332. doi: 10.1002/aja.1002000407. [DOI] [PubMed] [Google Scholar]
- Jiao K, Langworthy M, Batts L, Brown CB, Moses HL, Baldwin HS. Tgfbeta signaling is required for atrioventricular cushion mesenchyme remodeling during in vivo cardiac development. Development. 2006;133:4585–4593. doi: 10.1242/dev.02597. [DOI] [PubMed] [Google Scholar]
- Judge DP, Markwald RR, Hagege AA, Levine RA. Translational research on the mitral valve: from developmental mechanisms to new therapies. J Cardiovasc Transl Res. 2012;4:699–701. doi: 10.1007/s12265-011-9320-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadler K. Matrix loading: assembly of extracellular matrix collagen fibrils during embryogenesis. Birth Defects Res C Embryo Today. 2004;72:1–11. doi: 10.1002/bdrc.20002. [DOI] [PubMed] [Google Scholar]
- Kadler KE, Holmes DF, Trotter JA, Chapman JA. Collagen fibril formation. Biochem J. 1996;316 ( Pt 1):1–11. doi: 10.1042/bj3160001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapacee Z, Richardson SH, Lu Y, Starborg T, Holmes DF, Baar K, Kadler KE. Tension is required for fibripositor formation. Matrix Biol. 2008;27:371–375. doi: 10.1016/j.matbio.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Kim JE, Kim SJ, Jeong HW, Lee BH, Choi JY, Park RW, Park JY, Kim IS. RGD peptides released from beta ig-h3, a TGF-beta-induced cell-adhesive molecule, mediate apoptosis. Oncogene. 2003;22:2045–2053. doi: 10.1038/sj.onc.1206269. [DOI] [PubMed] [Google Scholar]
- Kitten GT, Kolker SJ, Krob SL, Klewer SE. Type VI collagen in the cardiac valves and connective tissue septa during heart development. Braz J Med Biol Res. 1996;29:1189–1193. [PubMed] [Google Scholar]
- Klewer SE, Krob SL, Kolker SJ, Kitten GT. Expression of type VI collagen in the developing mouse heart. Dev Dyn. 1998;211:248–255. doi: 10.1002/(SICI)1097-0177(199803)211:3<248::AID-AJA6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Kolditz DP, Wijffels MC, Blom NA, van der Laarse A, Markwald RR, Schalij MJ, Gittenberger-de Groot AC. Persistence of functional atrioventricular accessory pathways in postseptated embryonic avian hearts: implications for morphogenesis and functional maturation of the cardiac conduction system. Circulation. 2007;115:17–26. doi: 10.1161/CIRCULATIONAHA.106.658807. [DOI] [PubMed] [Google Scholar]
- Krug EL, Runyan RB, Markwald RR. Protein extracts from early embryonic hearts initiate cardiac endothelial cytodifferentiation. Dev Biol. 1985;112:414–426. doi: 10.1016/0012-1606(85)90414-2. [DOI] [PubMed] [Google Scholar]
- Kruithof BP, Krawitz SA, Gaussin V. Atrioventricular valve development during late embryonic and postnatal stages involves condensation and extracellular matrix remodeling. Dev Biol. 2007;302:208–217. doi: 10.1016/j.ydbio.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Kruithof BP, van Wijk B, Somi S, Kruithof-de Julio M, Perez Pomares JM, Weesie F, Wessels A, Moorman AF, van den Hoff MJ. BMP and FGF regulate the differentiation of multipotential pericardial mesoderm into the myocardial or epicardial lineage. Dev Biol. 2006;295:507–522. doi: 10.1016/j.ydbio.2006.03.033. [DOI] [PubMed] [Google Scholar]
- Kruithof BPT, Duim SN, Moerkamp AT, Goumans MJ. TGFbeta and BMP signaling in cardiac cushion formation; lessons from mice and chicken. Differentiation. 2012 doi: 10.1016/j.diff.2012.04.003. In Press. [DOI] [PubMed] [Google Scholar]
- Kruzynska-Frejtag A, Machnicki M, Rogers R, Markwald RR, Conway SJ. Periostin (an osteoblast-specific factor) is expressed within the embryonic mouse heart during valve formation. Mech Dev. 2001;103:183–188. doi: 10.1016/s0925-4773(01)00356-2. [DOI] [PubMed] [Google Scholar]
- Kumai M, Nishii K, Nakamura K, Takeda N, Suzuki M, Shibata Y. Loss of connexin45 causes a cushion defect in early cardiogenesis. Development. 2000;127:3501–3512. doi: 10.1242/dev.127.16.3501. [DOI] [PubMed] [Google Scholar]
- Kyndt F, Gueffet JP, Probst V, Jaafar P, Legendre A, Le Bouffant F, Toquet C, Roy E, McGregor L, Lynch SA, Newbury-Ecob R, Tran V, Young I, Trochu JN, Le Marec H, Schott JJ. Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation. 2007;115:40–49. doi: 10.1161/CIRCULATIONAHA.106.622621. [DOI] [PubMed] [Google Scholar]
- Kyndt F, Schott JJ, Trochu JN, Baranger F, Herbert O, Scott V, Fressinaud E, David A, Moisan JP, Bouhour JB, Le Marec H, Benichou B. Mapping of X-linked myxomatous valvular dystrophy to chromosome Xq28. Am J Hum Genet. 1998;62:627–632. doi: 10.1086/301747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YT, Beason KB, Brames GP, Desgrosellier JS, Cleggett MC, Shaw MV, Brown CB, Barnett JV. Activin receptor-like kinase 2 can mediate atrioventricular cushion transformation. Dev Biol. 2000;222:1–11. doi: 10.1006/dbio.2000.9698. [DOI] [PubMed] [Google Scholar]
- Lardeux A, Kyndt F, Lecointe S, Marec HL, Merot J, Schott JJ, Le Tourneau T, Probst V. Filamin-a-related myxomatous mitral valve dystrophy: genetic, echocardiographic and functional aspects. J Cardiovasc Transl Res. 2011;4:748–756. doi: 10.1007/s12265-011-9308-9. [DOI] [PubMed] [Google Scholar]
- Lencinas A, Tavares AL, Barnett JV, Runyan RB. Collagen gel analysis of epithelial-mesenchymal transition in the embryo heart: an in vitro model system for the analysis of tissue interaction, signal transduction, and environmental effects. Birth Defects Res C Embryo Today. 2011;93:298–311. doi: 10.1002/bdrc.20222. [DOI] [PubMed] [Google Scholar]
- Levay AK, Peacock JD, Lu Y, Koch M, Hinton RB, Jr, Kadler KE, Lincoln J. Scleraxis is required for cell lineage differentiation and extracellular matrix remodeling during murine heart valve formation in vivo. Circ Res. 2008;103:948–956. doi: 10.1161/CIRCRESAHA.108.177238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln J, Alfieri CM, Yutzey KE. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev Dyn. 2004;230:239–250. doi: 10.1002/dvdy.20051. [DOI] [PubMed] [Google Scholar]
- Lincoln J, Alfieri CM, Yutzey KE. BMP and FGF regulatory pathways control cell lineage diversification of heart valve precursor cells. Dev Biol. 2006;292:292–302. doi: 10.1016/j.ydbio.2005.12.042. [DOI] [PubMed] [Google Scholar]
- Lincoln J, Yutzey KE. Molecular and developmental mechanisms of congenital heart valve disease. Birth Defects Res A Clin Mol Teratol. 2011;91:526–534. doi: 10.1002/bdra.20799. [DOI] [PubMed] [Google Scholar]
- Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. 2007;171:1407–1418. doi: 10.2353/ajpath.2007.070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna-Zurita L, Prados B, Grego-Bessa J, Luxan G, del Monte G, Benguria A, Adams RH, Perez-Pomares JM, de la Pompa JL. Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J Clin Invest. 120:3493–3507. doi: 10.1172/JCI42666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Lu MF, Schwartz RJ, Martin JF. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development. 2005;132:5601–5611. doi: 10.1242/dev.02156. [DOI] [PubMed] [Google Scholar]
- MacGrogan D, Luna-Zurita L, de la Pompa JL. Notch signaling in cardiac valve development and disease. Birth Defects Res A Clin Mol Teratol. 91:449–459. doi: 10.1002/bdra.20815. [DOI] [PubMed] [Google Scholar]
- Markwald RR, Fitzharris TP, Bolender DL, Bernanke DH. Sturctural analysis of cell:matrix association during the morphogenesis of atrioventricular cushion tissue. Dev Biol. 1979;69:634–654. doi: 10.1016/0012-1606(79)90317-8. [DOI] [PubMed] [Google Scholar]
- Markwald RR, Fitzharris TP, Manasek FJ. Structural development of endocardial cushions. Am J Anat. 1977;148:85–119. doi: 10.1002/aja.1001480108. [DOI] [PubMed] [Google Scholar]
- Markwald RR, Fitzharris TP, Smith WN. Sturctural analysis of endocardial cytodifferentiation. Dev Biol. 1975;42:160–180. doi: 10.1016/0012-1606(75)90321-8. [DOI] [PubMed] [Google Scholar]
- Markwald RR, Funderburg FM. Use of 6-diazo-5-oxo-L-norleucine to study interaction between myocardial glycoconjugate secretion and endothelial activation in the early embryonic chick heart. Dev Biol. 1983;99:395–407. doi: 10.1016/0012-1606(83)90289-0. [DOI] [PubMed] [Google Scholar]
- Markwald RR, Norris RA, Moreno-Rodriguez R, Levine RA. Developmental basis of adult cardiovascular diseases: valvular heart diseases. Ann N Y Acad Sci. 2011;1188:177–183. doi: 10.1111/j.1749-6632.2009.05098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwald RR, Smith WN. Distribution of mucosubstances in the developing rat heart. J Histochem Cytochem. 1972;20:896–907. doi: 10.1177/20.11.896. [DOI] [PubMed] [Google Scholar]
- McBrien A, Chaudhari M, Crossland DS, Aspey H, Heads-Baister A, Griselli M, O’Sullivan J, Hasan A. Single-centre experience of 101 paediatric and adult Ross procedures: mid-term results. Interact Cardiovasc Thorac Surg. 2012 doi: 10.1093/icvts/ivr149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messas E, Bel A, Szymanski C, Cohen I, Touchot B, Handschumacher MD, Desnos M, Carpentier A, Menasche P, Hagege AA, Levine RA. Relief of mitral leaflet tethering following chronic myocardial infarction by chordal cutting diminishes left ventricular remodeling. Circ Cardiovasc Imaging. 2012;3:679–686. doi: 10.1161/CIRCIMAGING.109.931840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel JB. Anoikis in the cardiovascular system: known and unknown extracellular mediators. Arterioscler Thromb Vasc Biol. 2003;23:2146–2154. doi: 10.1161/01.ATV.0000099882.52647.E4. [DOI] [PubMed] [Google Scholar]
- Milewicz DM, Guo DC, Tran-Fadulu V, Lafont AL, Papke CL, Inamoto S, Kwartler CS, Pannu H. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- Millan FA, Denhez F, Kondaiah P, Akhurst RJ. Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development. 1991;111:131–143. doi: 10.1242/dev.111.1.131. [DOI] [PubMed] [Google Scholar]
- Mjaatvedt CH, Kern CB, Norris RA, Fairey S, Cave CL. Normal distribution of melanocytes in the mouse heart. Anat Rec A Discov Mol Cell Evol Biol. 2005;285:748–757. doi: 10.1002/ar.a.20210. [DOI] [PubMed] [Google Scholar]
- Mulholland DL, Gotlieb AI. Cell biology of valvular interstitial cells. Can J Cardiol. 1996;12:231–236. [PubMed] [Google Scholar]
- Nakamura T, Colbert MC, Robbins J. Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ Res. 2006;98:1547–1554. doi: 10.1161/01.RES.0000227505.19472.69. [DOI] [PubMed] [Google Scholar]
- Nishii K, Kumai M, Egashira K, Miwa T, Hashizume K, Miyano Y, Shibata Y. Mice lacking connexin45 conditionally in cardiac myocytes display embryonic lethality similar to that of germline knockout mice without endocardial cushion defect. Cell Commun Adhes. 2003;10:365–369. doi: 10.1080/cac.10.4-6.365.369. [DOI] [PubMed] [Google Scholar]
- Nishii K, Kumai M, Shibata Y. Regulation of the epithelial-mesenchymal transformation through gap junction channels in heart development. Trends Cardiovasc Med. 2001;11:213–218. doi: 10.1016/s1050-1738(01)00103-7. [DOI] [PubMed] [Google Scholar]
- Niu Z, Iyer D, Conway SJ, Martin JF, Ivey K, Srivastava D, Nordheim A, Schwartz RJ. Serum response factor orchestrates nascent sarcomerogenesis and silences the biomineralization gene program in the heart. Proc Natl Acad Sci U S A. 2008;105:17824–17829. doi: 10.1073/pnas.0805491105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris RA, Kern CB, Wessels A, Wirrig EE, Markwald RR, Mjaatvedt CH. Detection of betaig-H3, a TGFbeta induced gene, during cardiac development and its complementary pattern with periostin. Anat Embryol (Berl) 2005;210:13–23. doi: 10.1007/s00429-005-0010-z. [DOI] [PubMed] [Google Scholar]
- Norris RA, Moreno-Rodriguez R, Hoffman S, Markwald RR. The many facets of the matricelluar protein periostin during cardiac development, remodeling, and pathophysiology. J Cell Commun Signal. 2009a;3:275–286. doi: 10.1007/s12079-009-0063-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris RA, Moreno-Rodriguez RA, Sugi Y, Hoffman S, Amos J, Hart MM, Potts JD, Goodwin RL, Markwald RR. Periostin regulates atrioventricular valve maturation. Developmental Biology. 2008;316:200–213. doi: 10.1016/j.ydbio.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris RA, Potts JD, Yost MJ, Junor L, Brooks T, Tan H, Hoffman S, Hart MM, Kern MJ, Damon B, Markwald RR, Goodwin RL. Periostin promotes a fibroblastic lineage pathway in atrioventricular valve progenitor cells. Dev Dyn. 2009b;238:1052–1063. doi: 10.1002/dvdy.21933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosthoek PW, Wenink AC, Macedo AJ, Gittenberger-de Groot AC. The parachute-like asymmetric mitral valve and its two papillary muscles. J Thorac Cardiovasc Surg. 1997;114:9–15. doi: 10.1016/S0022-5223(97)70111-9. [DOI] [PubMed] [Google Scholar]
- Oosthoek PW, Wenink AC, Vrolijk BC, Wisse LJ, DeRuiter MC, Poelmann RE, Gittenberger-de Groot AC. Development of the atrioventricular valve tension apparatus in the human heart. Anat Embryol (Berl) 1998a;198:317–329. doi: 10.1007/s004290050187. [DOI] [PubMed] [Google Scholar]
- Oosthoek PW, Wenink AC, Wisse LJ, Gittenberger-de Groot AC. Development of the papillary muscles of the mitral valve: morphogenetic background of parachute-like asymmetric mitral valves and other mitral valve anomalies. J Thorac Cardiovasc Surg. 1998b;116:36–46. doi: 10.1016/S0022-5223(98)70240-5. [DOI] [PubMed] [Google Scholar]
- Padala M, Hutchison RA, Croft LR, Jimenez JH, Gorman RC, Gorman JH, 3rd, Sacks MS, Yoganathan AP. Saddle shape of the mitral annulus reduces systolic strains on the P2 segment of the posterior mitral leaflet. Ann Thorac Surg. 2009;88:1499–1504. doi: 10.1016/j.athoracsur.2009.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock JD, Lu Y, Koch M, Kadler KE, Lincoln J. Temporal and spatial expression of collagens during murine atrioventricular heart valve development and maintenance. Dev Dyn. 2008;237:3051–3058. doi: 10.1002/dvdy.21719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pomares JM, Phelps A, Sedmerova M, Carmona R, Gonzalez-Iriarte M, Munoz-Chapuli R, Wessels A. Experimental studies on the spatiotemporal expression of WT1 and RALDH2 in the embryonic avian heart: a model for the regulation of myocardial and valvuloseptal development by epicardially derived cells (EPDCs) Dev Biol. 2002;247:307–326. doi: 10.1006/dbio.2002.0706. [DOI] [PubMed] [Google Scholar]
- Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol. 2005;243:287–335. doi: 10.1016/S0074-7696(05)43005-3. [DOI] [PubMed] [Google Scholar]
- Phillips HM, Hildreth V, Peat JD, Murdoch JN, Kobayashi K, Chaudhry B, Henderson DJ. Non-cell-autonomous roles for the planar cell polarity gene Vangl2 in development of the coronary circulation. Circ Res. 2008;102:615–623. doi: 10.1161/CIRCRESAHA.107.160861. [DOI] [PubMed] [Google Scholar]
- Phillips HM, Murdoch JN, Chaudhry B, Copp AJ, Henderson DJ. Vangl2 acts via RhoA signaling to regulate polarized cell movements during development of the proximal outflow tract. Circ Res. 2005;96:292–299. doi: 10.1161/01.RES.0000154912.08695.88. [DOI] [PubMed] [Google Scholar]
- Phillips HM, Rhee HJ, Murdoch JN, Hildreth V, Peat JD, Anderson RH, Copp AJ, Chaudhry B, Henderson DJ. Disruption of planar cell polarity signaling results in congenital heart defects and cardiomyopathy attributable to early cardiomyocyte disorganization. Circ Res. 2007;101:137–145. doi: 10.1161/CIRCRESAHA.106.142406. [DOI] [PubMed] [Google Scholar]
- Poelmann RE, Gittenberger-de Groot AC, Hierck BP. The development of the heart and microcirculation: role of shear stress. Med Biol Eng Comput. 2008a;46:479–484. doi: 10.1007/s11517-008-0304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelmann RE, Van der Heiden K, Gittenberger-de Groot A, Hierck BP. Deciphering the endothelial shear stress sensor. Circulation. 2008b;117:1124–1126. doi: 10.1161/CIRCULATIONAHA.107.753889. [DOI] [PubMed] [Google Scholar]
- Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J Heart Valve Dis. 2004;13:841–847. [PubMed] [Google Scholar]
- Rajamannan NM, Gersh B, Bonow RO. Calcific aortic stenosis: from bench to the bedside--emerging clinical and cellular concepts. Heart. 2003;89:801–805. doi: 10.1136/heart.89.7.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie J, Jimenez J, He Z, Sacks MS, Yoganathan AP. The material properties of the native porcine mitral valve chordae tendineae: an in vitro investigation. J Biomech. 2006;39:1129–1135. doi: 10.1016/j.jbiomech.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Rivera-Feliciano J, Lee KH, Kong SW, Rajagopal S, Ma Q, Springer Z, Izumo S, Tabin CJ, Pu WT. Development of heart valves requires Gata4 expression in endothelial-derived cells. Development. 2006;133:3607–3618. doi: 10.1242/dev.02519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Ho PM, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Rosamond WD, Sorlie PD, Stafford RS, Turan TN, Turner MB, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross DN. Homograft replacement of the aortic valve. Lancet. 1962;2:487. doi: 10.1016/s0140-6736(62)90345-8. [DOI] [PubMed] [Google Scholar]
- Runyan RB, Markwald RR. Invasion of mesenchyme into three-dimensional collagen gels: a regional and temporal analysis of interaction in embryonic heart tissue. Dev Biol. 1983;95:108–114. doi: 10.1016/0012-1606(83)90010-6. [DOI] [PubMed] [Google Scholar]
- Sacks MS, Enomoto Y, Graybill JR, Merryman WD, Zeeshan A, Yoganathan AP, Levy RJ, Gorman RC, Gorman JH., 3rd In-vivo dynamic deformation of the mitral valve anterior leaflet. Ann Thorac Surg. 2006;82:1369–1377. doi: 10.1016/j.athoracsur.2006.03.117. [DOI] [PubMed] [Google Scholar]
- Sacks MS, He Z, Baijens L, Wanant S, Shah P, Sugimoto H, Yoganathan AP. Surface strains in the anterior leaflet of the functioning mitral valve. Ann Biomed Eng. 2002;30:1281–1290. doi: 10.1114/1.1529194. [DOI] [PubMed] [Google Scholar]
- Sacks MS, Yoganathan AP. Heart valve function: a biomechanical perspective. Philos Trans R Soc Lond B Biol Sci. 2007;362:1369–1391. doi: 10.1098/rstb.2007.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandbo N, Dulin N. Actin cytoskeleton in myofibroblast differentiation: ultrastructure defining form and driving function. Transl Res. 2011;158:181–196. doi: 10.1016/j.trsl.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JA, Jackson LF, Lee DC, Camenisch TD. Form and function of developing heart valves: coordination by extracellular matrix and growth factor signaling. J Mol Med. 2003;81:392–403. doi: 10.1007/s00109-003-0456-5. [DOI] [PubMed] [Google Scholar]
- Sepich DS, Calmelet C, Kiskowski M, Solnica-Krezel L. Initiation of convergence and extension movements of lateral mesoderm during zebrafish gastrulation. Dev Dyn. 2005;234:279–292. doi: 10.1002/dvdy.20507. [DOI] [PubMed] [Google Scholar]
- Shelton EL, Yutzey KE. Tbx20 regulation of endocardial cushion cell proliferation and extracellular matrix gene expression. Dev Biol. 2007;302:376–388. doi: 10.1016/j.ydbio.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton EL, Yutzey KE. Twist1 function in endocardial cushion cell proliferation, migration, and differentiation during heart valve development. Dev Biol. 2008;317:282–295. doi: 10.1016/j.ydbio.2008.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snarr BS, Kern CB, Wessels A. Origin and fate of cardiac mesenchyme. Dev Dyn. 2008;237:2804–2819. doi: 10.1002/dvdy.21725. [DOI] [PubMed] [Google Scholar]
- Snarr BS, Wirrig EE, Phelps AL, Trusk TC, Wessels A. A spatiotemporal evaluation of the contribution of the dorsal mesenchymal protrusion to cardiac development. Dev Dyn. 2007;236:1287–1294. doi: 10.1002/dvdy.21074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider P, Hinton RB, Moreno-Rodriguez RA, Wang J, Rogers R, Lindsley A, Li F, Ingram DA, Menick D, Field L, Firulli AB, Molkentin JD, Markwald R, Conway SJ. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ Res. 2008;102:752–760. doi: 10.1161/CIRCRESAHA.107.159517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Fassler R, Mishina Y, Jiao K, Baldwin HS. Essential functions of Alk3 during AV cushion morphogenesis in mouse embryonic hearts. Dev Biol. 2007;301:276–286. doi: 10.1016/j.ydbio.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Stein I. Short P-R interval, prolonged QRS complex (Wolff, Parkinson, White syndrome); report of fourteen cases and a review of the literature. Ann Intern Med. 1946;24:60–71. doi: 10.7326/0003-4819-24-1-60. [DOI] [PubMed] [Google Scholar]
- Sugi Y, Ito N, Szebenyi G, Myers K, Fallon JF, Mikawa T, Markwald RR. Fibroblast growth factor (FGF)-4 can induce proliferation of cardiac cushion mesenchymal cells during early valve leaflet formation. Dev Biol. 2003;258:252–263. doi: 10.1016/s0012-1606(03)00099-x. [DOI] [PubMed] [Google Scholar]
- Sugi Y, Yamamura H, Okagawa H, Markwald RR. Bone morphogenetic protein-2 can mediate myocardial regulation of atrioventricular cushion mesenchymal cell formation in mice. Dev Biol. 2004;269:505–518. doi: 10.1016/j.ydbio.2004.01.045. [DOI] [PubMed] [Google Scholar]
- Tan H, Junor L, Price RL, Norris RA, Potts JD, Goodwin RL. Expression and deposition of fibrous extracellular matrix proteins in cardiac valves during chick development. Microsc Microanal. 2011;17:91–100. doi: 10.1017/S1431927610094365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao G, Levay AK, Gridley T, Lincoln J. Mmp15 is a direct target of Snai1 during endothelial to mesenchymal transformation and endocardial cushion development. Dev Biol. 2011;359:209–221. doi: 10.1016/j.ydbio.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua-Belmonte JC, de la Pompa JL. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004;18:99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkatchenko TV, Moreno-Rodriguez RA, Conway SJ, Molkentin JD, Markwald RR, Tkatchenko AV. Lack of periostin leads to suppression of Notch1 signaling and calcific aortic valve disease. Physiol Genomics. 2009;39:160–168. doi: 10.1152/physiolgenomics.00078.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend TA, Robinson JY, Deig CR, Hill CR, Misfeldt A, Blobe GC, Barnett JV. BMP-2 and TGFbeta2 shared pathways regulate endocardial cell transformation. Cells Tissues Organs. 194:1–12. doi: 10.1159/000322035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend TA, Robinson JY, How T, DeLaughter DM, Blobe GC, Barnett JV. Endocardial cell epithelial-mesenchymal transformation requires Type III TGFbeta receptor interaction with GIPC. Cell Signal. 24:247–256. doi: 10.1016/j.cellsig.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend TA, Wrana JL, Davis GE, Barnett JV. Transforming growth factor-beta-stimulated endocardial cell transformation is dependent on Par6c regulation of RhoA. J Biol Chem. 2008;283:13834–13841. doi: 10.1074/jbc.M710607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Akker NM, Lie-Venema H, Maas S, Eralp I, DeRuiter MC, Poelmann RE, Gittenberger-De Groot AC. Platelet-derived growth factors in the developing avian heart and maturating coronary vasculature. Dev Dyn. 2005;233:1579–1588. doi: 10.1002/dvdy.20476. [DOI] [PubMed] [Google Scholar]
- Van den Akker NM, Winkel LC, Nisancioglu MH, Maas S, Wisse LJ, Armulik A, Poelmann RE, Lie-Venema H, Betsholtz C, Gittenberger-de Groot AC. PDGF-B signaling is important for murine cardiac development: its role in developing atrioventricular valves, coronaries, and cardiac innervation. Dev Dyn. 2008;237:494–503. doi: 10.1002/dvdy.21436. [DOI] [PubMed] [Google Scholar]
- Visconti RP, Ebihara Y, LaRue AC, Fleming PA, McQuinn TC, Masuya M, Minamiguchi H, Markwald RR, Ogawa M, Drake CJ. An in vivo analysis of hematopoietic stem cell potential: hematopoietic origin of cardiac valve interstitial cells. Circ Res. 2006;98:690–696. doi: 10.1161/01.RES.0000207384.81818.d4. [DOI] [PubMed] [Google Scholar]
- Wessels A, Markman MW, Vermeulen JL, Anderson RH, Moorman AF, Lamers WH. The development of the atrioventricular junction in the human heart. Circ Res. 1996;78:110–117. doi: 10.1161/01.res.78.1.110. [DOI] [PubMed] [Google Scholar]
- Wessels A, Perez-Pomares JM. The epicardium and epicardially derived cells (EPDCs) as cardiac stem cells. Anat Rec A Discov Mol Cell Evol Biol. 2004;276:43–57. doi: 10.1002/ar.a.10129. [DOI] [PubMed] [Google Scholar]
- Wessels A, Sedmera D. Developmental anatomy of the heart: a tale of mice and man. Physiol Genomics. 2003;15:165–176. doi: 10.1152/physiolgenomics.00033.2003. [DOI] [PubMed] [Google Scholar]
- Wessels A, van den Hoff MJB, Adamo M, Lockhart M, Sauls K, Briggs LE, RAN, Phelps AL, van Wijk B, Perez-Pomares JM, Dettman RW, Burch BE. Epicardially-derived Fibroblasts and their Contribution to the Parietal Leaflets of the Atrioventricular Valves in the Murine Heart. Developmental Biology. 2012 doi: 10.1016/j.ydbio.2012.04.020. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirrig EE, Yutzey KE. Transcriptional regulation of heart valve development and disease. Cardiovasc Pathol. 2011;20:162–167. doi: 10.1016/j.carpath.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin HC, Shekhar A, McQuinn TC, Butcher JT. Hemodynamic patterning of the avian atrioventricular valve. Dev Dyn. 2011;240:23–35. doi: 10.1002/dvdy.22512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, von Gise A, Ma Q, Hu YW, Pu WT. Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev Biol. 2010;338:251–261. doi: 10.1016/j.ydbio.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuberbuhler JR, Allwork SP, Anderson RH. The spectrum of Ebstein’s anomaly of the tricuspid valve. J Thorac Cardiovasc Surg. 1979;77:202–211. [PubMed] [Google Scholar]