

Abstract
Judicious modifications to the structure of the previously reported HCV NS5A inhibitor1, resulted in more potent anti-HCV compounds with similar and in some cases improved toxicity profiles. The synthesis of nineteen new NS5A inhibitors is reported along with their ability to block HCV replication in an HCV 1b replicon system. For the most potent compounds chemical stability, stability in liver microsomes and inhibition of relevantCYP450 enzymes is also presented.
Keywords: NS5A, Antiviral, HCV
Hepatitis C virus (HCV) is the leading cause of chronic liver disease, liver transplantation and is the most common blood-borne infection in developed countries.1 The prevalence of HCV infection ranges from 1–5% in most developed countries and is estimated to be 5-fold greater than HIV infection.1 Until recently, the only approved drugs for the treatment of HCV were pegylated interferon-α (IFN) and ribavirin (RBV), both of which are poorly tolerated and have limited efficacy, with less than 50% response rates among individuals infected with the most common virus genotype (1b).2 The FDA approval, in May 2011, of the protease inhibitors (PI) boceprevir (Victrelis) and telaprevir (Incivek) which created a lot of excitement however, their real impact on standard of care (SOC) remains unclear as sustained virologic response (SVR) for genotype 1 HCV is still only about 70 to 80% when administered with IFN and RBV.3
Efforts toward improved HCV treatment include the development of antiviral agents that inhibit HCV via new targets such as the viral NS5A protein. Despite significant gaps in the understanding of the role of NS5A in the virus replication cycle, it is known that it plays an integral role in a number of host and/or pathogen events such as RNA replication, assembly of viral particles and host immune response.4 NS5A phosphokinase is phosphorylated by cellular protein kinases and the phosphorylation sites are conserved among the various HCV genotypes.5,6Recently, the NS5A inhibitor BMS-790052 (Figure 1) was discovered and displayed in vitro potency ((EC50) of ≤ 50 pM)7 that translated to rapid viral load decline (≈ 3.3 log10) after a single 100 mg dose with no signs of toxicity.8 While compound 1 has established NS5A as a valid target for effective clinical treatment of HCV, it also provided a foundation for the design of second-generation inhibitors that could be more resistant to selection of mutant virus with reduced potential cytotoxic liabilities. This lead us to dissect the symmetrical inhibitor 1 into four major structural zones: zone I (biphenyl linker zone) has already been studied extensively by other research groups; zone II (imidazole portion) despite claims of small modifications no significant exploration of this area has been disclosed; zone III (proline moiety), minor modifications such as fluorination or cyclopropanation have been reported; zone IV (amino acid and its “cap”) has been extensively studied.8,9
Figure 1.

Chemical structure of BMS-790052
In consideration of novelty and retention of potency, we initially chose to focus on both zones II and III. Indeed, the lack of significant exploration of zone II along with the ease of halogenation of the imidazole ring, which would allow for the efficient preparation of more complex structures using transition metal catalyzed cross coupling reactions, made zone II ideal for investigation. Similarly, zone III has not been extensively explored while readily available proline analogs would allow for extensive modifications from well known organic transformations. Herein we wish to report our preliminary structure activity relationship (SAR) efforts for zones II and III.
The biphenyl core of the targeted compounds was prepared according to general Scheme 1. Diketone 1 was first converted to the α, α′-dibromodiketone using Br2 in CH2Cl2 then coupled with N-Boc protected proline derivatives A10 in the presence of DIPEA. Refluxing the corresponding ester intermediates with ammonium acetate in toluene allowed for the formation of the imidazole ring and lead to the versatile Boc protected intermediates 3a-c in 63% to 75%yields.
Scheme 1.

Reagents and conditions: (a) Br2, CH2Cl2, rt, overnight; (b) (i) A, DIPEA, rt, 5 h; (ii) NH4OAc, toluene, 100-110 °C, 15-20 h.
Exploration of 4-substituted imidazole derivatives, as shown in Scheme 2, marked the beginning of our synthetic efforts and initial SAR studies. Bromination of the 4-position of the imidazoles of 3a was accomplished using N-bromosuccinimide (NBS) in dichloromethane (52% yield). After removal of Boc protection with 6N HCl, the resulting amine was coupled with N-methylcarbamate protected valine using N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) and N-hydroxybenzotriazole (HOBt) in the presence of N, N-diisopropylethylamine (DIPEA). The palladium catalyzed cross coupling reaction of 5a under Stille conditions11 resulted in 4-aryl and 4-heteroaryl compounds 6-8 in moderate yields providing the first in this zone II series of aryl substituted imidazoles.
Scheme 2.

Reagents and conditions: (a) X = Br, I: NXS, CH2Cl2, rt, 30 min; X = Cl: NCS, 40 °C, 15 h; X = F, NFSI, NaHCO3, CH2Cl2/acetone (1:1), 50 °C. (b) 6 N HCl, CH3OH, 50 °C, 5 h; (c) HOOC-CH[CH(CH3)2]-NHCOOCH3, EDC, HOBt, DIPEA, 0 °C – rt, 15 h; (d) R-Sn(Bu)3, Pd(PPh3)2Cl2, dioxane, 80 °C, overnight.
Introduction of thiophene, furan or phenyl at the 4-position of the imidazole lead to a noticeable decrease of potency toward HCV replication (EC50 of 100, 26 and 200 pM respectively) when compared to 1. Evaluation of the brominated intermediate 5a for anti-replication activity versus HCV proved surprising as it displayed an EC50 of 5.9 pM and EC90 of 14 pM; thus compound 5a was equipotent to BMS-790052 at the EC50 and EC90. This unexpected result prompted us to investigate the influence of other halogens at the 4-position of the imidazole ring. Iodine and chlorine were thus introduced on the imidazole ring of compound 3a using N-iodosuccinimide (NIS) and N-chlorosuccinimide (NCS) respectively while fluorine was introduced by careful reaction with N-fluorobenzenesulfonimide (NFSI) and NaHCO3. It is noteworthy that this fluorination reaction leads to the formation of both the difluorinated derivative 4d and the monofluorinated derivative 4e. As above, simple Boc deprotection under acidic conditions followed by the coupling of N-methylcarbamate protected valine in the presence of HOBt and DIPEA resulted in the dihalogeneated compounds 5b-d and monofluoro derivative 5e. Remarkably the bis-iodo, bis-chloro and bis-fluoro compounds 5b, 5c and 5d exhibited an EC50 of 6.4 ± 3.0, 5.5 ± 1.9 and 6.8 ± 3.3 pM respectively which, as in the case of the bis-bromo compound, are equipotent to the reference compound 1. The EC90 values for these three were all close to 10 pM representing a modest improvement relative to the reference compound 1. In addition, all of the bis-halogen compounds 5c and 5d exhibited increased toxicity in primary human lymphocytes, although they were non-toxic at concentrations up to 100 μM in Vero cells. These results suggest that a small modification in this region (Zone II) such as introduction of a halogen is well tolerated but introduction of larger groups such as thiophene, furane and phenyl are detrimental to the HCV replicon activity.
Zone III modifications (Scheme 3) included introduction of various triazoles using copper catalyzed alkyne azide cycloadditions.12 Azido intermediates 3b and 3c were first deprotected under acidic conditions then coupled with N-methylcarbamate protected valine in presence of HOBt and DIPEA to give compounds 9-10 in respectively 64 and 59% yields. Finally, treatment of 9-10 with various alkenes in the presence of CuSO4 and sodium ascorbate afforded the 1,4-triazolo derivatives 11-14 in good yields. Like for Zone II, introduction of bulky modifications such as 1,4-substituted triazoles on the proline moiety (Zone III) considerably decreased potencies. On the other hand, introduction of a small group such an azido group seems to be well tolerated. Indeed, (4-R)-azido compound 10 with an EC50 = 9.5 pM appeared as potent as reference compound 1 without displaying any signs of toxicity up to 100 μM in PBM, CEM and Vero cells. Interestingly, (4-S)-azido compound 9 appeared slightly less active (EC50 = 23 pM) and showed toxicities in PBM and CEM cells (IC50 = 20.3 and 18.4 μM respectively).
Scheme 3.

Reagents and conditions: (a) 6 N HCl, CH3OH, 50 °C, 5 h (b) HOOC-CH[CH(CH3)2]-NHCOOCH3, EDC, HOBt, DIPEA, 0 °C, rt, overnight. (c) alkyne, sodium ascorbate, CuSO4.5H2O, t-BuOH/H2O (1:1), rt, overnight.
Based on these results, a combination of our best modifications in both zone II and III was also studied. Thus, (4-S)- and (4-azido compounds 9 and 10 were halogenated at the R)- azido compounds 9 and 10 were halogenated at the 4-position of the imidazoles rings by using either NBS or NCS in CH2Cl2 (Scheme 4) and all the synthesized compounds displayed an EC50 ≤ 10 pM. Interestingly, compound 17 showed an EC50 of 2.6 pM with no toxicity toward Vero cells up to 100 μM.
Scheme 4.

Reagents and conditions: (a) X = Br: NBS, CH2Cl2, rt, 0.5-1 h; X = Cl: NCS, CH2Cl2, 50 °C, 4-24 h.
Among all the molecules synthesized, the most promising compounds 5a-d were further investigated. First of all, their stability at pH 3 and 7 in phosphate buffer was evaluated. After 48 h at 20°C, LC/MS analysis at time 0, 1, 8, 24 and 48 h revealed no evidence of degradation of these compounds. Next stability in human liver microsomes was established (Table 2). Compounds 5a-d (1 μM) were incubated with human liver microsomes in potassium phosphate buffer and 1 mg/mL microsomal protein. Samples were collected at 5, 15, 30 and 60 min, analyzed by LC-MS/MS and half-life (t1/2) and intrinsic clearance (CLint) for each compound were calculated based on the elimination rate constant (k). Interestingly, compounds 5b and 5d show higher metabolic stability and therefore lower clearance (CLint< 8.6) compared with BMS-790052.
Table 2. Half-life and intrinsic clearance values in human liver microsomes of compounds 5a-d and BMS-790052.
| Cmpd | Elimination rate constant (k) | t1/2 (min) | CLint (μL/min/mg protein) |
|---|---|---|---|
| 5a | 0.01 | 67.3 | 10.3 |
| 5b | 0.0058 | 119.5 | 5.8 |
| 5c | 0.018 | 38.5 | 18.0 |
| 5d | 0.0056 | 123.8 | 5.6 |
| BMS-790052 | 0.012 | 56.3 | 12.3 |
Drug-drug interactions have become an important issue in modern health care. It is now apparent that many drug-drug interactions can be explained by alterations in the metabolic lenzymes that are present in the liver and other extra-hepatic tissues. Many of the major pharmacokinetic interactions between drugs are due to hepatic cytochrome P450 (CYP450) enzymes being affected by one or more of the drugs present in a given individual. Furthermore, drug interactions can be a result of inhibition or induction of CYP450 enzymes. The potential drug-drug interaction liabilities of these compounds were investigated by a CYP450 reversible inhibition assay. Compounds 5b and 5d did not appear to inhibit the major CYP450 enzymes at the IC90 level and had a favorable CYP profile, which is suggestive of no potential drug-drug interactions (Table 3). Due to the large therapeutic window, the inhibition observed for 3A4 (in the micromolar range) is expected not to be clinically relevant at therapeutic doses.
Table 3. Cytochrome P450 inhibition data for compounds 5b, 5d and BMS-790052a.
| CYP1A2 (μM) | CYP3A4 (μM) | CYP2D6 (μM) | CYP2C9 (μM) | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Cmpd | IC50 | IC90 | IC50 | IC90 | IC50 | IC90 | IC50 | IC90 |
| 5b | > 100 | > 100 | 13.8 ± 11.2 | > 100 | > 100 | > 100 | 81.6 ± 26.1 | > 100 |
| 5d | > 100 | > 100 | 3.1 ± 2.3 | > 100 | > 100 | > 100 | > 100 | > 100 |
| BMS-790052 | > 100 | > 100 | 7.2 ± 1.7 | > 100 | > 100 | > 100 | 59.5 ± 13.6 | > 100 |
Mean ± SD of at least two independent assays; IC50 = 50% inhibitory concentration. The following positive controls were used (IC50, μM): α -naphthoflavone for 1A2 (> 93% at 3 μM); ketoconazole for 3A4 (> 93% at 10 μM); PH-053 (proprietary) for 2D6 (> 70% at 100 μM); sulfaphenazole for 2C9 (> 90% at 10 μM).
Initial resistant virus selection studies using 5b and BMS-790052 were performed and mutations identified by population sequencing. Mutations observed for BMS-790052 included L31V, which was previously reported,13 in addition to several unreported mutations, which we will disclose in full detail in subsequent publications. Compound 5b selected for several different mutations including Y93H, which was previously reported as an NS5A resistance mutation.13
In conclusion, extremely potent new NS5A inhibitors (picomolar activity) with a wide therapeutic window (> 104) are disclosed. Among all the compounds prepared 5b and 5d exhibited improved liver microsome stability compared to reference compound BMS-790052 and no inhibition of major human CYP enzymes was exhibited at therapeutically relevant concentrations. In addition, these two halogenated compounds displayed improved stability toward human liver microsomes versus BMS-790052 while maintaining comparable anti-HCV replication activity. Finally, further mutation selection studies are currently being investigated in our laboratory and will be the subject of future publications.
Table 1. Chemical Structure, Anti-HCV Activity and Cytotoxicity of Compounds 5-19.
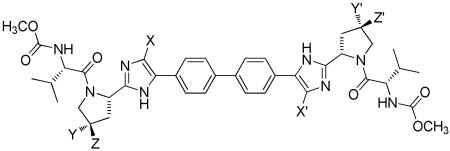
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Cmpd | X | X′ | Y | Y′ | Z | Z′ | HCV Replicon (pM) | Cytotoxicity, CC50 (μM)* | |||
|
| |||||||||||
| EC50 | EC90 | PBM | CEM | Vero | |||||||
| BMS-790052 | H | H | H | H | H | H | 4.2 ± 1.5 | 20 ± 7.5 | 19 | 9.6 | 21 |
| 5b | Cl | Cl | H | H | H | H | 6.4 ± 3.0 | 11.4 ± 4.8 | 2.8 | 12 | ≥ 100 |
| 5a | Br | Br | H | H | H | H | 5.9 ± 3.9 | 14 ± 8.3 | 2.9 | 5.7 | 84 |
| 5c | I | I | H | H | H | H | 5.5 ± 1.9 | 11 ± 2.2 | 4.7 | 35 | > 100 |
| 5d | F | F | H | H | H | H | 6.8 ± 3.3 | 21.2 ± 11.0 | 19 | 3.0 | > 100 |
| 5e | H | F | H | H | H | H | 14.4 ±4.0 | 52.5 ±11.1 | 16 | 2.8 | 83 |
| 6 | Ph | Ph | H | H | H | H | 200 | 700 | > 100 | 37.4 | > 100 |
| 7 |
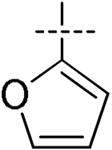
|
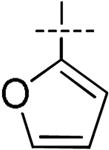
|
H | H | H | H | 26 | 97 | 49.2 | 2.9 | >100 |
| 8 |
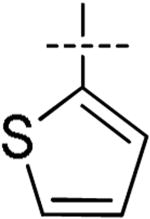
|
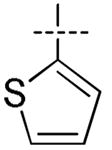
|
H | H | H | H | 100 | 300 | 44.6 | > 100 | > 100 |
| 9 | H | H | N3 | N3 | H | H | 23 | 35.2 | 20.3 | 18.4 | > 100 |
| 10 | H | H | H | H | N3 | N3 | 18.4 ± 12.6 | 54.2 ± 34.1 | > 100 | ≥ 100 | > 100 |
| 11 | H | H | H | H |
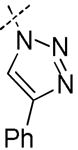
|
|
2700 | 9200 | > 100 | > 100 | > 100 |
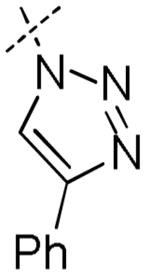
| 12 | H | H | H | H |
|
|
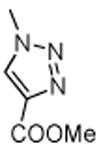
11.8 | 42.8 | 6.6 | 10.7 | > 100 |
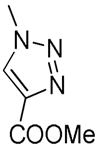
| 13 | H | H |
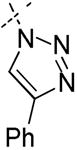
|
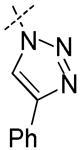
|
H | H | 757 102 | > 104 | 96 | > 100 | > 100 |
| 14 | H | H |
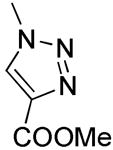
|
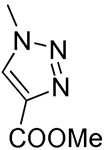
|
H | H | > 104 | > 104 | > 100 | > 100 | > 100 |
| 15 | Cl | Cl | H | H | N3 | N3 | 4.7 | 9.6 | 7.8 | 3.8 | > 100 |
| 16 | Cl | H | H | H | N3 | N3 | 6.3 | 29.5 | 9.7 | 31 | > 100 |
| 17 | Br | Br | H | H | N3 | N3 | 2.6 | 21.6 | 19.8 | 10.8 | > 100 |
| 18 | Cl | Cl | N3 | N3 | H | H | 11.2 | 74.6 | 3.3 | 4.0 | 98.1 |
| 19 | Br | Br | N3 | N3 | H | H | 8.3 | 55.3 | 3.4 | 4.5 | > 100 |
All compounds had a CC50 of > 1 μM in Huh-7 cells
Acknowledgments
This work was supported in part by NIH grant 5P30-AI-50409 (CFAR), 5R01-AI-071846 and by the Department of Veterans Affairs. Dr. Schinazi is the founder and a major shareholder of RFS Pharma, LLC. Emory received no funding from RFS Pharma, LLC to perform this work and vice versa.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Liang JT, Hoofnagle JH. Hepatitis C, Biomedical Research Reports. Academic Press; London, UK: 2000. pp. 185–203. [Google Scholar]
- 2.Bobeck DR, Schinazi RF, Coats SJ. Antivir Ther. 2010;15:935. doi: 10.3851/IMP1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheridan C. Nature Biotech. 2011;29:553. doi: 10.1038/nbt0711-553. [DOI] [PubMed] [Google Scholar]
- 4.Schinazi RF, Bassit L, Gavegnano CJ. Viral Hepatitis. 2010;17:77. doi: 10.1111/j.1365-2893.2009.01246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katze MG, Kwieciszewski B, Goodlett DR, Blakely CM, Neddermann P, Tan SL, Aebersold R. Virology. 2000;278501 doi: 10.1006/viro.2000.0662. [DOI] [PubMed] [Google Scholar]
- 6.Kim J, Lee D, Choe J. Biochem Biophys Res Commun. 1999;257777 doi: 10.1006/bbrc.1999.0460. [DOI] [PubMed] [Google Scholar]
- 7.Fridell RA, Qiu D, Wang C, Valera L, Gao M. Antimicrob Agents Chemother. 2010;54:3641. doi: 10.1128/AAC.00556-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR, 2nd, Lemm JA, Wang C, Knipe JO, Chien C, Colono RJ, Grasela DM, Meanwell NA, Hamann LG. Nature. 2010;465:96. doi: 10.1038/nature08960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Sun JH, Gao M, O'Boyle DR, II, Lemm JA, Roberts SB, Belema M, Meanwell NA. WO 2012009394 A2 20120119 PCT Int Appl. 2012; (b) Lopez OD, St Laurent DR, Goodrich J, Romine JLee, Serrano-Wu M, Yang FK, Kakarla R, Yang XJ, Qiu YP, Snyder LB. US 20110294819 A1 20111201 US Pat Appl Publ. 2011; (c) Belema M, Romine JL, Nguyen VN, Wang G, Lopez OD, St Laurent DR, Chen Q, Bender JA, Bender JA, Yang Z, Hewawasam P, Xu NN, Meanwell NA, Easter JA, Su BN, Smith MJ. US 20110237636 A1 20110929 US Pat Appl Publ. 2011; d) Belema M, Hewawasam P. US 20110237636 A1 20110929 US Pat Appl Publ. 2011; (d) Bender JA, Hewawasam P, Kadow JF, Lopez OD, Meanwell NA, Nguyen VN, Romine JL, Snyder LB, St Laurent DR, Wang G, Xu NN, Belema M. WO 2010117635 A1 20101014 PCT Int Appl. 2010; (e) Belema M, Nguyen VN, Serrano-Wu M, St Laurent DR, Qiu YP, Ding M, Meanwell NA, Snyder LB. US 20100080772 A1 20100401 US Pat Appl Publ. 2010; (f) Bachand C, Belema M, Deon DH, Good AC, Goodrich J, Hamann LG, James CA, Langley DR, Lavoie R, Lopez OD, Martel A, Meanwell NA, Nguyen VN, Romine JL, Ruediger EH, Snyder LB, St Lauurent DR, Yang FK, Wang G. WO 2008144380 A1 20081127 PCT Int Appl. 2008; (g) Milbank JBJ, Tran TD, Wakenhut F. WO 2011154871 A1 20111215 PCT Int Appl. 2011; (h) Vandyck K, Verschueren WG, Raboisson PJMB. WO 2012013643 A1 20120202 PCT Int Appl. 2012; (i) Li LP, Zhong M. WO 2011156543 A2 20111215 PCT Int Appl. 2011; (j) Qiu YL, Wang C, Ying L, Peng XW, Or YS. WO 2010091413 A1 20100812 PCT Int Appl. 2010; (k) Li LP, Zhong M. WO 2011150243 A1 20111201 PCT Int Appl. 2011; (l) Kim S, Gao Q, Yang FK. US 20100158862 A1 20100624 US Pat Appl Publ. 2010; (m) Bachand C, Belema M, Deon DH, Good AC, Godrich J, James CA, Lavoie R, Lopez OD, Martel A, Meanwell NA, Nguyen VN, Romine JL, Ruediger EH, Snyder LB, St Laurent DR, Yang FK, Langley DR, Wang G, Hamann LG. WO 2009102318 A1 20090820 PCT Int Appl. 2009; (n) Bachand C, Belema M, Deon DH, Good AC, Goodrich J, James CA, Lavoie R, Lopez OD, Martel A, Meanwell NA, Nguyen VN, Romine JL, Ruediger EH, Snyder LB, St Laurent DR, Yang FK, Langley DR, Hamann LG. WO 2008021936 A2 20080221 PCT Int Appl. 2008; (o) Belema M, Romine JL, Nguyen VN, Wang G, Lopez OD, St Laurent DR, Chen Q, Bender JA, Yang Z, Hewawasam P, Xu NN, Meanwell NA, Easter JA, Su BN, Smith MJ. WO 2011075439 A1 20110623 PCT Int Appl. 2011; (p) Corte JR, Fang T, Decicco CP, Pinto DJP, Rossi KA, Hu ZL, Jeon Y, Quan ML, Smallheer JM, Wang YF, Yang W. WO 2011100401 A1 20110818 PCT Int Appl. 2011
- 10.Webb TR, Eigenbrot CJ. Org Chem. 1991;56:3009. [Google Scholar]
- 11.Shrestha S, Bhattarai BR, Lee KH, Cho H. Bioorg Med Chem. 2007;15:6535. doi: 10.1016/j.bmc.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 12.For reviews on the CuAAC see; (a) Hein JE, Fokin VV. Chem Soc Rev. 2010;39:1302. doi: 10.1039/b904091a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Aragao-Leoneti V, Campo VL, Gomes AS, Field RA, Carvalho I. Tetrahedron. 2010;66:9475. [Google Scholar]; (c) Amblard F, Cho JH, Schinazi RF. Chem Rev. 2009;109:4207. doi: 10.1021/cr9001462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemm J, O'Boyle D, II, Liu M, Nower PT, Colonno R, Deshpande MS, Snyder LB, Martin SW, St Laurent DR, Serrano-Wu MH, Romine JL, Meanwell NA. Virol. 2010;84:482. doi: 10.1128/JVI.01360-09. [DOI] [PMC free article] [PubMed] [Google Scholar]