

Abstract
Further chemical optimization of the MLSCN/MLPCN probe ML077 (KCC2 IC50 = 537 nM) proved to be challenging as the effort was characterized by steep SAR. However, a multidimensional iterative parallel synthesis approach proved productive. Herein we report the discovery and SAR of an improved novel antagonist (VU0463271) of the neuronal-specific potassium-chloride cotransporter 2 (KCC2), with an IC50 of 61 nM and >100-fold selectivity versus the closely related Na-K-2Cl cotransporter 1 (NKCC1) and no activity in a larger panel of GPCRs, ion channels and transporters.
Keywords: Potassium-chloride co-transporter 2, KCC2, NKCC1, antagonist
Due to their key regulatory roles in CNS physiology, cation-chloride cotransporters, and in particular, the neuronal specific K-Cl cotransporter 2 (KCC2) has recently garnered a great deal of attention1–4. KCC2, identified in 1996,5 modulates inhibitory neurotransmission in both the brain and spinal cord.6–11 However, due to a complete lack of selective and potent pharmacological tools,4 the study of KCC2 has relied on either high doses of furosemide or genetic models (mouse and Drosophila knockout or transgenic zebrafish).1–4,12–15 Based on the necessity of small molecule probes to dissect the role of KCC2, an HTS compatible screen in 384-well plates was developed based on thallium (Tl+ flux) in KCC2-overexpressing HEK293 cells.4,16 Under the auspice of the MLSCN/MLPCN, 234,560 compounds were screened against KCC2, and after obligate counter- and secondary screens, 26 hits were identified as KCC2 antagonists.4 Of these, VU0240511 (1) emerged as an attractive, potent hit (KCC2 IC50 = 568 nM), with >100-fold selectivity versus Na-K-2Cl cotransporter 1 (NKCC1), a critical anti-target as inhibition leads to ototoxic effects (Fig. 1). Upon profiling in an ancillary pharmacology panel of 68 GPCRs, ion channels and transporters, 1 showed significant inhibition (>50% @ 10 µM) of several GPCRs and key ion channels (hERG and L-Type Ca2+ channels). Based on similar issues in the past, we converted the secondary amide in 1 to a tertiary N-Me amide, VU0255011 (2). While 2 displayed a slight improvement in KCC2 potency (KCC2 IC50 = 537 nM) and maintained >100-fold selectivity versus NKCC1, we were gratified to note a cleaner ancillary pharmacology profile (no activities >50% @ 10 µM).4 Thus, 2 was declared an MLSCN/MLPCN probe and given the designation ML077.17 As such, ML077 is freely available upon request,18 and we have supplied ML077 to multiple laboratories around the world. While good data is being generated, there is a need for a more potent KCC2 antagonist. In this Letter, we detail the DMPK characterization of ML077 and the further chemical lead optimization of ML077 en route to a more potent in vitro KCC2 antagonist probe.
Figure 1.

Structures of the KCC2 antagonist HTS hit (1) and the KCC2 antagonist MLPCN probe (2), ML077. Simple conversion to the tertiary N-Me amide eliminated all ancillary pharmacology while maintaining KCC2 potency and selectivity.
In the pilot phase of the Molecular Libraries initiative,17 coined the MLSCN, DMPK profiling of probes was not supported. Now in the production phase, or MLPCN, DMPK profiling is required.18 Thus, prior to further optimization, we profiled ML077 in an effort to assess disposition. In our tier 1 in vitro DMPK screen, compound ML077 displayed no significant P450 inhibition in human liver microsomes (IC50 >30 µM vs. 3A4, 2C9, 1A2 and ~24 µM inhibition of 2D6) and high plasma protein binding with fraction unbound (fu) levels between 1 and 2% in rat and human plasma, respectively. Intrinsic clearance (CLint) determined in rat and human liver microsomes indicated that compound ML077 was rapidly cleared in vitro (rat, CLint = 294 mL/min/kg; human, CLint = 228.9 mL/min/kg). An in vitro to in vivo correlation (IVIVC) was established, as ML077 was found to be a highly cleared compound in rat (CL = 185 mL/min/kg) following intravenous administration (1 mg/kg); the high volume of distribution at steady state (Vss 5.0 L/kg) and super-hepatic clearance produced a relatively short t1/2 (26 min) in vivo. Thus, to deliver an in vivo KCC2 probe, significant improvements in the DMPK profile are required. Note, however, that despite a poor PK, ML077 was used successfully to block KCC2 in spinal cord through intrathecal injection.11
The initial chemical optimization plan for ML077, utilizing multi-dimensional iterative parallel synthesis,19 is detailed in Figure 2, and the synthesis of analogs of ML077 was performed as shown in Scheme 1. Various heteroaryl amines 3 (1° and 2°) are treated with α-chloroacetyl chloride 4 (or α-alkyl substituted variants) to deliver functionalized α-chloroamides 5. Commercially available heteroaryl and heterobiaryl chlorides 6 are treated with thiourea under microwave-assisted conditions to produce the corresponding thiols 7. Finally, reaction of α-chloroamides 5 with thiols 7 in the presence of Cs2CO3 affords a diverse array of analogs 8 of ML077.
Figure 2.

Initial chemical optimization plan for ML077 to improve KCC2 potency.
Scheme 1.

Reagents and conditions: (a) Et3N, CH2Cl2, 0 °C, 65–95%; (b) thiourea, 220 °C, microwave, 15 min, 70–85%; (c) Cs2CO3, CH3CN, rt, 56–80%.
Analogs 8 were screened at both 20 µM and 2 µM concentrations prior to full CRCs in an 86Rb uptake assay. SAR was incredibly steep, with the majority of analogs 8 affording <20% inhibition at 20 µM. Functionalization at any position of the western 6-phenyl moiety of ML077 with small alkyl groups, alkoxy groups or halogens was not tolerated. Similarly, replacements for the pyridazine (pyridines, pyrazines, pyrimidines and thiadiazoles) were also not tolerated, with the exception of two weak, 2-pyridyl-based KCC2 antagonists 9 and 10 (Fig. 3). Moreover, alternative substitutions to the eastern thiazole or alternative heterocycles (pyridines, pyrazine, pyrimidines, etc…) to replace the thiazole were inactive.
Figure 3.

2-Pyridyl-based weak KCC2 antagonists 9 and 10 from first generation libraries.
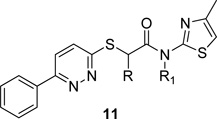
Thus, an unsubstituted 6-phenyl pyridazine, in combination with the 4-methyl thiazole, was required for KCC2 inhibition. Therefore, we focused on incorporating larger alkyl and cycloalkly moieties to replace the tertiary N-Me amide in 2 (Fig. 2), as well as exploring the impact of simple alkyl substitution α-to the amide (Table 1).19 For the latter, we first evaluated the racemic mixture, and then resolved, by chiral SFC, and assayed the single enantiomers.19 This effort was far more productive, affording KCC2 antagonists 11 with improved potency relative to 2, and the first examples of enantioselective KCC2 inhibition. 11a–d employed either N-Me or N-Et tertiary amides with a racemic α-Me or α-Et moiety, and these analogs showed weak KCC2 inhibition (27–57%@2µM). When the steric bulk of the tertiary amide was increased to an N-cyclopropyl moiety, KCC2 inhibitory activity was improved. The racemic α-Me congener 11e and the α-Et derivative 11h displayed IC50’s of 570 nM and 756 nM, respectively, comparable to 2. Chiral SFC resolution of the single enantiomers of 11e afforded 11f, the (+)-enantiomer, and 11g the (−)-enantiomer. Here, the (+)-enantiomer 11f is a potent KCC2 antagonist (IC50 = 152 nM), while the (−)-enantiomer 11g is ~10-fold less active (IC50 = 1900 nM). A similar enantiopreference is observed with the α-ethyl congeners 11i and 11j. These data represent the first example of enantiospecific inhibition of KCC2. Based on the impact of the N-cyclopropyl amide, we then surveyed this modification with an unsubstituted core, affording 11k, the most potent KCC2 antagonist reported to date (IC50 = 61 nM).19 As we further increased the steric bulk of the tertiary amide to N-cyclobutyl, 11m (IC50 = 177 nM), and N-cyclopentyl, 11n (IC50 = 1057 nM), KCC2 potency diminished (Table 1). Based on these data, efforts focused on the further characterization of 11k (VU0463271).
Table 1.
Structures and activities of analogs 11.
 | ||||
|---|---|---|---|---|
| Cmpd | R | R1 | % Inhib. @ 2 µM |
IC50 (nM) |
| 2 | H | Me | 82 | 537 |
| 11a | (±)-Me | Me | 46 | ND |
| 11b | (±)-Et | Me | 27 | ND |
| 11c | (±)-Et | Et | 45 | ND |
| 11d | (±)-Me | Et | 57 | ND |
| 11e | (±)-Me | 87 | 570 | |
| 11f | a(±)-Me | ND | 152 | |
| 11g | a(−)-Me | ND | 1,900 | |
| 11h | (±)-Et | 84 | 756 | |
| 11i | a(+)-Et | ND | 385 | |
| 11j | a(−)-Et | 40 | ND | |
| 11k | H | ND | 61 | |
| 11m | H | ND | 177 | |
| 11n | H | ND | 1057 | |
ND: not determined.
enantiomers separated by chiral SFC and (+) or (−) rotation noted,19 absolute stereochemistry is unknown.
Figure 4 displays the full dose-response curves for both 2 (ML077) and 11k (VU0463271) for both KCC2 and NKCC1, our standard anti-target in 86Rb uptake assays.4 Here, both 2 (IC50 = 537 nM) and 11k (IC50 = 61 nM) are potent KCC2 antagonists, but only display weak, partial inhibition of NKCC1 function at concentrations up to 100 µM. In the Lead Profiling screen at Ricerca (68 GPCRs, ion channels and transporter radioligand binding assays),21 11k displayed no significant activities (no inhibition >50%@10 µM). Thus, 11k is ~9-times more potent than ML077, and maintains an excellent ancillary pharmacology profile. In our tier 1 in vitro DMPK screen, compound 11k displayed only very P450 inhibition in human liver microsomes (IC50 >30 µM vs. 3A4, 2C9, 2D6 and ~20 µM inhibition of 1A2). Plasma protein binding, with fraction unbound (fu) levels, could not be determined for 11k, as it was found to be unstable in rat plasma (6% remaining after 1 hour at 37 °C). Intrinsic clearance (CLint) determined in human liver microsomes indicated that compound 11k was rapidly cleared in vitro, and was found to be a moderate-to-high clearance compound in rat (CL = 57 mL/min/kg) following intravenous administration (1 mg/kg); the low volume of distribution at steady state (Vss 0.4 L/kg), coupled with moderate-to-high clearance produced a relatively short t1/2 (9 min) in vivo. Thus, to deliver an in vivo KCC2 probe, significant improvements in the DMPK profile are still required. However, 11k represents a significant improvement in terms of an in vitro KCC2 antagonist probe relative to ML077.
Figure 4.

Dose-response curves for 2 (ML077) and 11k (VU0463271). A) Full dose-response curves for 2 and 11k on KCC2 function in an 86Rb uptake assay, highlighting the increased potency of 11k versus the original probe ML077 (2). B) Full dose-response curves for 2 and 11k on NKCC1 function in an 86Rb uptake assay, with both displaying weak partial inhibition at concentrations up to 100 µM.
Based on the steep SAR and poor in vivo PK, we elected to revisit our HTS hits in an effort to develop an in vivo KCC2 antagonist probe. Very few hits from the HTS showed reasonable KCC2 potency in conjunction with NKCC1 selectivity; however, dihydropyrimidine dione 12 (IC50 = 640 nM) and [1,2,3]triazolo[4,5-d]pyrimidine 13 (IC50 = 1.2 µM) represented acceptable starting points (Fig. 5). Similar multidimensional iterative parallel synthesis approaches for the lead optimization of 12 and 13 led to inactive compounds once again. Recognition of the structural similarities between 12, 2 and 11k led us to generate chimeras, replacing the dihydropyrimidine dione of 12 with the 4-methyl thiazole moiety found in 2 and 11k and a number of other azaheterocycles. However, all the chimeras were inactive or very weak inhibitors (IC50’s >20 µM) of KCC2.
Figure 5.

Structures of the KCC2 antagonist HTS hits 12 and 13, with selectivity versus NKCC1, and the multi-dimensional iterative parallel synthesis approach for their chemical optimization.
In summary, we have further optimized the KCC2 antagonist probe ML077, by application of a multi-dimensional iterative parallel synthesis approach, to afford 11k (VU0463271), the most potent (KCC2 IC50 = 61 nM) and selective (>100-fold versus NKCC1 and inactive in an ancillary pharmacology panel of 68 GPCRs, ion channels and transporters) KCC2 antagonist reported to date. While possessing a favorable in vitro DMPK profile, 11k is a highly cleared, short half-life compound not suitable as an in vivo probe. Attempts to optimize additional HTS hits, including chimeras with 11k, were not productive. Overall, the SAR of all the KCC2 antagonist series was extremely steep. At present, the MLPCN screening deck has more than doubled since the KCC2 screen was originally performed, and we are actively pursuing a rescreen to identify more tractable hits with the potential to develop an in vivo probe to compliment our in vitro KCC2 probe 11k. As both ML077 and 11k were developed for the MLPCN, they are freely available upon request. Additional refinements and studies are in progress and will be reported in due course.
Acknowledgments
The authors acknowledge Emily Days and Christopher Farmer of the Vanderbilt High Throughput Screening facility and the legacy MLSCN Screening Center for GPCRS, ion channels and transporters. This work was supported by grants from the NIH. Vanderbilt is a Specialized Chemistry Center within the Molecular Libraries Probe Centers Network (U54MH84659).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Woo N-S, Lu J, England R, McClellan R, Dufour S, Moint DB, Deutch AY, Lovinger DM, Delpire E. Hippocampus. 2002;12:258–268. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- 2.Zhu L, Polley N, Mathews GC, Delpire E. Epilepsy Res. 2008;79:201–212. doi: 10.1016/j.eplepsyres.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgado C, Pinto-Ribeiro F, Tavares I. Neurosci. Lett. 2008;438:102–106. doi: 10.1016/j.neulet.2008.04.032. [DOI] [PubMed] [Google Scholar]
- 4.Delpire E, Days E, Lewis LM, Mi D, Kim K, Lindsley CW, Weaver CD. Proc. Natl. Acad. Sci. USA. 2009;106:5383–5388. doi: 10.1073/pnas.0812756106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Payne JA, Stevenson TJ, Donaldson LF. J. Biol. Chem. 1996;271:16245–16252. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- 6.Nomura H, Sakai A, Umino M, Suzuki H. Neurosci, Res. 2006;56:435–440. doi: 10.1016/j.neures.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 7.Jolivalt CG, Lee CA, Ramos KM, Calcutt NA. Pain. 2008;140:48–57. doi: 10.1016/j.pain.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miletic G, Miletic V. Pain. 2008;137:532–539. doi: 10.1016/j.pain.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang W, Liu L-Y, Xu TL. Neuroscience. 2008;152:502–510. doi: 10.1016/j.neuroscience.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 10.Cramer SW, Baggott C, Cain J, Tilghman J, Allcock B, Miranpuri G, Rajpal S, Sun D, Resnicj D. Mol. Pain. 2008;4:36. doi: 10.1186/1744-8069-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Austin TM, Delpire E. Anesth. Analg. 2011;113:1509–1515. doi: 10.1213/ANE.0b013e31822e0a5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hubner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyl K, Jentsch TJ. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- 13.Zhu L, Lovinger D, Delpire E. J. Neurophysiol. 2005;93:1557–1568. doi: 10.1152/jn.00616.2004. [DOI] [PubMed] [Google Scholar]
- 14.Hekmat-Scafe DS, Lundy MY, Ranga R, Tanouye MA. J. Neurosci. 2006;26:8943–8954. doi: 10.1523/JNEUROSCI.4998-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reynolds A, Brustein E, Liao M, Mercado A, Babilonia E, Mount DB, Drapeau P. J. Neurosci. 2008;28:1588–1597. doi: 10.1523/JNEUROSCI.3791-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weaver CD, Harden D, Dworetzky SI, Robertson B, Knox RJ. J. Biomol. Screen. 2004;9:671–677. doi: 10.1177/1087057104268749. [DOI] [PubMed] [Google Scholar]
- 17.For information on the MLI or MLPCN please see: http://mli.nih.gov/mli/mlpcn/
- 18.To request your free sample of ML077 or 11k, please email: craig.lindsley@vanderbilt.edu
- 19.Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver CD, Lindsley CW. J. Combi. Chem. 2008;10:345–354. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]
- 20.Experimental details and characterization data for compounds in Table 1. Synthesis of 11k: To a solution of cyclopropyl-(4-methyl-thiazol-2-yl)-amine (160 mg, 1 mmol) (117 mg, 1 mmol, 82.5 µL) and chloroacetyl chloride (117 mg, 1 mmol, 82.5 µL) in CH2Cl2 (3.4 mL, 0.3 M) at 0 °C was added Et3N (104 mg, 1 mmol, 144 µL). After 30 min, complete consumption of starting material as followed by liquid chromatography-mass spectrometry (LC-MS) was observed. Saturated, aqueous solution of NH4Cl was added. The aqueous layer was extracted with CH2Cl2 (× 2). The combined organic layers were dried (MgSO4), filtered, and concentrated in vacuo. The residue was taken to the next step without further purification. To a solution of 6-phenylpyridazine-3-thiol (214 mg, 1.14 mmol) and 2-chloro-N-cyclopropyl-N-(4-methylthiazol-2-yl)acetamide (~1 mmol) in CH3CN (5 mL, 0.2 M) at ambient temperature was added Cs2CO3 (506 mg, 1.55 mmol). After 18 h the reaction mixture was diluted with EtOAc and washed with H2O (× 2). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. The residue was purified by flash chromatography (Hexane/EtOAc, 4:1) to provide desired product as a yellow oil (216 mg, 56% over two steps). 1H NMR (400 MHz, CDCl3): δ 7.99 (d, J = 1.6 Hz, 1 H), 7.97 (s, 1 H), 7.64 (d, J = 9.2 Hz, 1 H), 7.48-7.45 (m, 4 H), 6.59 (s, 1 H), 4.80 (s, 2 H), 3.31-3.28 (m, 1 H), 2.38 (s, 3 H), 1.28 (m, 2 H), 1.01 (m, 2 H); 13C NMR (100 MHz, CDCl3): δ 169.4, 160.2, 159.0, 156.5, 147.5, 135.9, 129.8, 128.9, 126.6, 126.2, 123.4, 110.3, 34.8, 30.5, 17.4, 11.3; HRMS (ESI) calcd for C19H19N4OS2 [M+H+] 383.1000 found 383.1000. Characterization of 11e–g 1H NMR (400 MHz, CDCl3): δ 8.01 (d, J = 2.0 Hz, 1 H), 7.99 (d, J = 1.6 Hz, 1 H), 7.65 (d, J = 9.2 Hz, 1 H), 7.51-7.48 (m, 3 H), 7.37 (d, J = 9.2 Hz, 1 H), 6.60 (s, 1 H), 6.05 (bs, 1 H), 3.33 (bs, 1 H), 2.36 (s, 3 H), 1.77 (d, J = 6.8 Hz, 3 H), 1.24-1.18 (m, 2 H), 1.10-1.05 (m, 1 H), 0.90-0.86 (m, 1 H); 13C NMR (100 MHz, CDCl3): δ 173.7, 160.3, 159.1, 156.6, 147.6, 135.8, 129.9, 129.0, 128.9, 127.7, 126.6, 126.2, 123.5, 110.5, 40.86, 30.68, 18.59, 17.44; HRMS (ESI) calcd for C20H21N4OS2 [M+H+] 397.1154 found 397.1157. Peak A [α]D20 +290.3° (c = 0.033, CHCl3) SFC preffered column: chiralpak ia RT = 2.23 min; Peak A [α]D20 −296.0° (c = 0.036, CHCl3) SFC preferred column: chiralpak ia RT = 2.79 min. Experimental details for the KCC2-overexpressing HEK293 cells and for 86Rb uptake experiments can be found in reference 4.
- 21.For full information on the targets in the Lead Profiling Screen at Ricerca, please see: www.ricerca.com