

Abstract
Mistranslation leads to elevated mutagenesis and replication arrest, both of which are hypothesized to result from the presence of mixed populations of wild type and mistranslated versions of DNA polymerase III subunit proteins. Consistent with this possibility, expression of missense alleles of dnaQ (which codes for the proofreading subunit ε in wild type (dnaQ+) cells is shown to lead to SOS induction as well as mutagenesis. Exposure to sublethal concentrations of streptomycin, an aminoglycoside antibiotic known to promote mistranslation, also leads to SOS induction.
Keywords: DNA replication, SOS induction, Mutagenesis, Mistranslation, Streptomycin, Mutator
1. INTRODUCTION
Protein synthesis has relatively low fidelity, such that in normal cells amino acids are misincorporated at rates estimated to be in the range of 10−3 to 10−5 [1–5]. Since ribosomal mutants with increased translational fidelity (“restrictive” mutations), as well as decreased fidelity (ribosomal ambiguity or Ram mutations) can be isolated, the observed normal error rates are thought to be growth-optimized, balancing the costs of mistranslation against the costs of more stringent error-proofing [6]. At error-rates of 10−4, 10% of proteins consisting of 1,000 amino acids will contain at least one error, and 50% ribosomes (7,132 amino acids) will contain one or more errors. Similarly, at background mistranslation rates of 10−4, 61% of DNA polymerase III holoenzyme assemblies[(αεθ)3τ3δδ’χψ(β2)3 =9, 523 amino acids] will have at least one amino acid error. Whether this built-in protein heterogeneity has significant biological consequences has long been debated (e.g., [2, 7, 8]), and is an area of renewed interest [9, 10].
Elevated mistranslation does have a significant impact on DNA replication as demonstrated by a mutator phenotype (translational stress-induced mutagenesis, TSM) conferred by mutations in genes specifying tRNAs [11–14], tRNA-modifying enzymes [15], and ribosomal proteins that control fidelity [16], or by exposure to sublethal concentrations of streptomycin, an antibiotic that enhances mistranslation. It has been hypothesized that occasional recruitment of mistranslated dominant-negative ε (proofreading) or α (polymerase) subunits can account for the TSM phenotype [13, 17]. TSM requires RecA-dependent homologous recombination functions for the full manifestation of the mutator effect [12, 18–20]. Homologous recombination functions were proposed to be required to rescue cells from replication arrest that accompanies error-prone DNA synthesis by corrupted DNA polymerase [17]. In accordance with this hypothesis, SOS functions were shown to be spontaneously induced in TSM-constitutive (mutA) cells [17]. Here, in a proof-of-principle experiment, we show that expression of missense dnaQ (ε subunit) alleles in dnaQ+ cells is sufficient to trigger SOS induction as measured by a prophage induction assay. We also find that exposure to sublethal concentrations of streptomycin, an antibiotic known to promote mistranslation, also leads to SOS induction.
2. MATERIALS AND METHODS
Strains
The E. coli SIVET (selectable in vivo expression technology) strain SG104 (MG1655 H-19B ΔN::kan cro-tnpR168 ΔOP::bla galK::resC-tet-resC) was derived from the SIVET strain K10449 [21] as described [17].
Plasmids
Plasmids pSGQ, pSG12G, pSG103G and pSG108G, bearing, respectively, wild type E. coli dnaQ allele, dnaQ(D12G), dnaQ(D103G) and dnaQ(D108G) were constructed in a low copy number vector plasmid as follows. A 1482 bp Nco I-Nsi I fragment containing the lacIq gene along with its Ptrc promoter from plasmid pSE380 [22] was cloned into the multicloning site of the pSC101-derived low copy number vector pMW119 [23] to obtain the vector plasmid pTM100. Plasmid pSGQ (dnaQ+ AmpR SpcR) was constructed by cloning a Nco I-Hind III 981 bp PCR fragment bearing a dnaQ+ allele, as well as a Sac I-Sph I 1180 bp fragment bearing the aad9+ allele (conferring spectinomycin resistance) from the shuttle plasmid pFW5 [24] into pTM100. Plasmids pSGQ12G, pSGQ103G and pSGQ108G were similarly constructed using the corresponding alleles of dnaQ, generously provided as pBR329-based clones by Jeffrey H. Miller (UCLA). Plasmids pSGQ(E14A) through pSGQ(E199A) series were derived by site-directed mutagenesis from pSGQ by inverse PCR following the primer design strategy described by Zheng et al. [25]. Similarly mutD5 (T15I) and dnaQ49 (V96G) alleles were constructed by inverse PCR in the same vector on the basis of sequence information from the literature [26]. Primers used for construction of the dnaQ alleles used in this study are listed in Table 1. All alleles were verified by DNA sequence analysis using primer 5’-GCT GTC AAA CAT CAG GAG C.
Table 1.
Partially overlapping inverse PCR Primers used for mutant dnaQ allele constructiona
| Allele | Forward Primer | Reverse Primer | Site |
|---|---|---|---|
| E14A | 5’ CTC GAT ACC GcA ACC ACC GGT ATG AAC CAG ATT GGT GC |
5’ GGT GGT TgC GGT ATC GAG AAC GAT CTG GCG TGT AAT TGC |
None |
| E26A | 5’ GCG CAC TAT GcC GGC CAC AAG ATC ATT GAG ATT G |
5’ GAT CTT GTG GCC GgC ATA GTG CGC ACC AAT CTG |
Nae I |
| E32A | 5’ CAA GAT CAT TGc GAT TGG CGC CGT TGA AGT GGT G |
5’ CGG CGC CAA TCg CAA TGA TCT TGT GGC CTT CAT AG |
Hae II |
| E37A | 5’ GAG ATT GGC GCC GTT GcA GTG GTG AAC CGT CGC CTG |
5’ GTT CAC CAC TgC AAC GGC GCC AAT CTC AAT GAT CTT G |
Hae II |
| E71A | 5’ GCC GAT GcA TTT TTT CTA GAT AAG CCC ACG TTT G |
5’ CTT ATC TAG AAA AAA TgC ATC GGC AAT ACC ATG |
Xba I |
| E81A | 5’ CAC GTT TGC TGc AGT AGC CGA TGA GTT CAT GG |
5’ CAT CGG CTA CTg CAG CAA ACG TGG GCT TAT CG |
Pst I |
| E94A | 5’ CGG CGC CGc GTT GGT GAT CCA TAA C | 5’ ACC AAC gCG GCG CCG CGA ATA TAG TC |
Kas I |
| E110A | 5’ GGA CTA TGc ATT TTC GTT GCT TAA GCG C |
5’ GCA ACG AAA ATg CAT AGT CCA TAA AGC C |
Nsi I |
| E173A | 5’ GAT CCT TGC GGc CGT TTA TCT GGC GAT GAC CG |
5’ CAG ATA AAC GgC CGC AAG GAT CTG GGC ATC GAG |
Nae I |
| E199A | 5’ CAA CAA GGC GcC GCA ACA ATT CAG CGC ATT GTA CGT CAG G |
5’ CAA TGC GCT GAA TTG TTG CGg CGC CTT GTT GCT GTT GTG |
Kas I |
|
T15I (mutD5) |
5’ ATC GTT CTA GAT ACC GAA AtC ACC GGT ATG AAC CAG |
5’ CCG GTG aTT TCG GTA TCT AGA ACG ATC TGG CGT G |
Xba I |
|
V96G (dnaQ49) |
5’ CGG CGC CGA GTT GGg GAT CCA TAA CGC AGC G |
5’ ATG GAT CcC CAA CTC GGC GCC GCG AAT ATA G |
Kas I |
Sequence change leading to the amino acid substitution is indicated by the lowercase letter in each forward and reverse primer set. Silent sequence change(s) for creating the indicated restriction site is (are) shown by underlined letters in each primer set. Sequence overlap within each set of primers is shown in boldface.
The SIVET prophage induction assay is based on the conversion of chloramphenicol-sensitive (CmS) cells to a chloramphenicol-resistant (CmR) phenotype upon SOS induction [21], and was carried out as described previously [17, 21] and summarized below. Briefly, the E. coli SG104 (an MG1655-derived SIVET strain [17]) cells carrying a pSC101-derived plasmid bearing an allele of dnaQ (see Table 2) were grown overnight at 37°C in LB medium supplemented with tetracycline (5 µg/ml), kanamycin (20 µg/ml) and ampicillin (30 µg/ml). Five ml of fresh LB medium supplemented with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was inoculated with 0.1 ml of a 1:500,000 dilution of the overnight culture (estimated to have approximately 1,000 cells) and the cells were allowed to grow overnight at 37°C (or 28°C where noted) with aeration. To determine SIVET induction (ratio of CmR and CmS cells), the cultures were plated on LB agar supplemented with kanamycin (20 µg/ml), ampicillin (30 µg/ml), and chloramphenicol (10 µg/ml). To select all SIVET lysogens, the cultures were plated on LB agar containing kanamycin (20 µg/ml) and ampicillin (30 µg/ml). Colonies were counted after incubating the plates for 48 h at 37°C. SIVET induction results (CmR/CmS ratios) were averaged from 3 to 5 replicate cultures. Experiments using minimal medium A [27] were carried out as above except that 0.1 mM IPTG was used.
Table 2.
Effect of plasmid-borne dnaQ alleles on SIVET induction and RifR mutant frequency in LB mediuma
| SIVET Induction (CmR/CmS) | Mutation Frequency (RifR/RifS) | |||
|---|---|---|---|---|
| dnaQ allele | Frequency (SD) | Ratio | Frequency (SD) | Ratio |
| on plasmid | [× 10−4] | (dnaQ/WT) | [× 10−7] | (dnaQ/WT) |
| Wild Type(dnaQ+) | 2.4 (0.71) | 1 | 0.85 (0.34) | 1 |
| D12G | 29 (7.7) | 12 | 1,441 (195) | 1,695 |
| D103G | 27 (6) | 11 | 942 (150) | 1,108 |
| D108G | 2 (0.64) | <1 | ND | ND |
| E14A | 57.4 (19.7) | 24 | 4,845 (1435) | 5,700 |
| E26A | 3.5 (1.3) | 2 | 0.33 (0.12) | <1 |
| E32A | 15.2 (9) | 6 | 1.1 (0.49) | 1 |
| E37A | 2.7 (1.3) | 1 | 0.67 (0.4) | <1 |
| E71A | 1.5 (0.39) | <1 | 0.14 (0.05) | <1 |
| E81A | 2.4 (2.4) | 1 | 0.67 (0.48) | <1 |
| E94A | 2.1 (1.4) | <1 | 0.08 (0.03) | <1 |
| E110A | 2.3 (0.74) | 1 | 0.15 (0.06) | <1 |
| E173A | 2.8 (1.4) | 1 | 0.87 (0.25) | 1 |
| E199A | 2.1 (1.4) | 1 | 0.16 (0.03) | <1 |
| V96G (dnaQ49) | 1.4 (0.32) | <1 | 17 (12) | 20 |
| T15I (mutD5) | 2.8 (0.7) | 1 | 265 (27) | 312 |
SIVET induction (CmR/CmS ratio) was assayed as described in the text after growth at 37°C for all alleles except for dnaQ49 and mutD5, which were grown at 28°C. SD, Standard deviation; WT here refers to basal SIVET induction and mutation frequency values shown in the first row of data; ND, not determined. Data were averaged from 3–5 replicate cultures.
To determine the effect of exposure to streptomycin on SIVET induction, 5 ml of LB medium containing either no streptomycin (mock-treatment) or 5 µg/ml streptomycin (5 replicate cultures for each condition) was inoculated with approximately 1,000 cells from a fresh overnight culture of SG104 grown in the presence of kanamycin, ampicillin and tetracycline as described above. Cells were allowed to grow overnight at 37°C with aeration before plating to determine CmR/CmS ratios as described above.
For determining rifampicin-resistant (RifR) mutant frequencies, 5 ml of fresh LB medium was inoculated with 1% volume of an overnight culture of SG104 cells bearing a pSC101-derived plasmid, and the cells were grown for 18 h at 37°C with aeration. To determine the number of RifR mutants, 0.1 ml aliquots of the culture (or appropriately diluted culture in the case of mutator strains) were plated on LB agar plates supplemented with 100 µg/ml rifampicin, followed by incubation at 37°C for 48 h. Viable cell count was determined by plating appropriate dilutions of the same culture on LB agar. Average number of RifR mutants (3 to 5 replicate cultures) was divided by the average number of total viable cells to derive the mutant frequency.
3. RESULTS AND DISCUSSION
E. coli DNA polymerase III holoenzyme is a multimer of 10 distinct polypeptides organized into three polymerase cores (αεθ), a clamp loader (τ3-δδ’χψ) and three processivity rings (β2) [28, 29]. The trimeric polymerase core (αεθ) is responsible for DNA synthesis (α subunit) and proofreading (ε subunit). In addition to its proofreading 3’–5’ exonuclease activity, ε plays an important structural role in the core such that in its absence, DNA synthesis becomes less processive in addition to being highly error-prone [30–32]. The function of the θ subunit is not known, but a structural role in stabilizing ε is inferred [33].
To determine whether coexpression of missense mutant ε subunit protein in the presence of wild type ε can trigger SOS induction, we used prophage induction, a surrogate for SOS induction, as an assay. In the so-called SIVET assay, SOS derepression leads to the expression of a transposase that acts at a distal site to convert cells from a chloramphenicol-sensitive to chloramphenicol-resistant (CmR) phenotype [21], and is thus suitable for detecting transient SOS induction occurring in a fraction of cells [17]. We have previously used this sensitive assay to demonstrate that cells expressing the mutA tRNA (which leads to aspartate→glycine mistranslation) suffer SOS induction presumably due to the occasional recruitment of corrupted pol-III cores (αεθ in which the ε has a missense error) to the replication fork [17]. In the current experiments, we asked if direct expression of missense mutant ε proteins in wild type (dnaQ+) cells triggers both mutagenesis and SOS induction. Because mutant tRNAs causing D→G (aspartate→glycine) and E→A (glutamate→alanine) missense substitutions are known to confer relatively strong mutator phenotypes [11–13], we chose to look at D→G and E→A dnaQ missense alleles in this study.
Table 2 shows that E14A, D12G, and D103G, the strongest three of the tested mutator dnaQ alleles, also show the highest SIVET induction (24-, 12- and 11-fold, respectively). D12G and E14A affect two essential and highly conserved amino acids in the Exo I motif of ε (Fig. 1), whereas the D103G affects the Exo II motif. The strongest mutator allele (E14A) is also the strongest SIVET inducer, and alleles that did not elevate mutagenesis did not elevate SIVET induction, consistent with a linkage between error-prone DNA synthesis and replication arrest. An exception is provided by the E32A allele, which shows moderate SIVET induction (6-fold) with no increase in mutagenesis. Interestingly, the data show that E14A is a stronger mutator under these conditions than mutD5 (T15I), the best known dominant-negative dnaQ allele that specifies a mutant protein that retains about 2% of the nuclease activity of wild type ε [34]. Increased mutagenesis by E14A may be due to a total loss of editing nuclease activity due to its critical role at the catalytic center [35, 36], or due to other factors such as stability leading ultimately to an increased ability to displace the wild type protein from polymerase cores. SIVET induction in cells expressing the mutD5 allele was modest, even though a significant >300-fold elevation in mutagenesis was observed. Because we have used a co-expression system in which a mutant dnaQ allele is expressed in the background of a wild type allele, the absence of an effect for a majority of the E→A alleles shown in Table 2 by itself does not necessarily imply that the corresponding mutant proteins have normal proofreading function because they could be recessive or unstable.
Fig. 1.
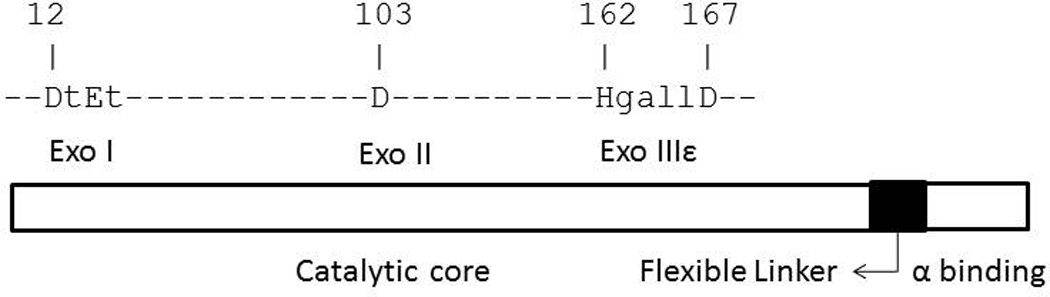
Domain structure of ε protein showing exonuclease motifs, with uppercase letters showing highly conserved amino acid residues.
We also found that dnaQ49 (V96G), a recessive allele known to have a strong mutator phenotype at 37°C but a moderate mutator effect at a lower temperature [37], confers a moderate mutator phenotype (20-fold; Table 2) at 28°C, indicating that the mutant ε protein must participate in replication to an appreciable extent in the presence of wild type ε at the lower temperature. As expected for a recessive allele, at 37°C dnaQ49 (V96G) barely elevated mutagenesis (<4 fold) in the dnaQ+ background, and had no effect on SIVET induction (data not shown).
The linkage between mutagenesis and SOS (SIVET) induction raises the question whether SOS polymerases IV and V make a contribution to mutagenesis. While such a contribution cannot be ruled out, the dominant contributor to mutagenesis is likely to be defective DNA polymerase III proofreading as evidenced by data on mutD5 (significant mutagenesis without SIVET induction), and E32A (SIVET induction without mutagenesis). Thus defective DNA synthesis (DNA elongation) leading to SIVET induction, and defective proofreading leading to mutagenesis, may be separable, but apparently occur together in cells expressing D12G, D103G or E14A alleles. The data in Tables 2 are consistent with the hypothesis that elevated mistranslation, by increasing the levels of dominant-negative missense mutant versions of critical DNA polymerase subunit proteins, leads to increased mutagenesis as well as replication arrest [17].
Defective DNA synthesis by a corrupted DNA polymerase III was previously proposed to lead to an accumulation of single-stranded DNA (ssDNA) gaps at replication forks, which in turn led to double-stranded breaks and arrested replication [17]. If so, slower growth in minimal medium might ameliorate the replication defect by allowing more time for the repair of ssDNA gaps, as previously proposed to explain the rich medium-sensitivity of polA cells defective for DNA polymerase I [38]. Table 3 shows that in minimal medium, expression of D12G or D103G alleles continues to confer a robust mutator phenotype (albeit not as strong as in rich medium), but very weak SIVET induction.
Table 3.
Effect of plasmid-borne dnaQ alleles on SIVET induction and RifR mutant frequency in minimal mediuma
| SIVET Induction (CmR/CmS) | Mutation Frequency (RifR/RifS) | |||
|---|---|---|---|---|
| dnaQ allele | Frequency (SD) | Ratio | Frequency (SD) | Ratio |
| on plasmid | [× 10−4] | (dnaQ/WT) | [× 10−7] | (dnaQ/WT) |
| Wild Type (dnaQ+) | 1.6 (0.4) | 1 | 0.92 (0.81) | 1 |
| D12G | 2.6 (1.1) | <2 | 418 (78) | 454 |
| D103G | 2.8 (0.94) | <2 | 426 (56) | 463 |
SIVET induction was assayed after growth at 37°C in minimal medium A as described in the text. SD, Standard deviation; WT here refers to basal SIVET induction and mutation frequency values shown in the first row of data; Data were averaged from 3–5 replicate cultures.
The aminoglycoside streptomycin binds to ribosomes, leading initially to codon misreading, and later to a complete inhibition of protein synthesis. In E. coli, aminoglycoside exposure leads to rapid cell killing, and lethality is linked to mistranslation rather than inhibition of protein synthesis per se because non-aminoglycoside inhibitors of protein synthesis such as chloramphenicol or tetracycline are bacteriostatic rather than bactericidal. Since mistranslation can corrupt DNA replication and lead to replication arrest, it is interesting to ask if exposure to sublethal concentrations of streptomycin also leads to SIVET induction. Table 4 shows that exposure to 5 µg/ml streptomycin does lead to appreciable (5-fold) SIVET induction, consistent with the possibility that mistranslation in streptomycin-exposed cells may lead to replication arrest. Recent findings suggest that oxygen radicals make a contribution to aminoglycoside lethality [39, 40], and cells defective for alkB and other DNA repair genes are peculiarly sensitive to an aminoglycoside [41], suggesting an additional possible mechanism for SIVET induction. AlkB is capable of efficiently repairing ssDNA lesions, acting on a variety of alkylated bases as well as cyclic base lesions induced by lipid peroxidation [42–44]. Since mistranslation leads to misreplication, and replication arrest [17], it is possible that generation of ssDNA gaps by mistranslationally corrupted DNA polymerase is critical to aminoglycoside lethality. In this scenario, cell killing results from overwhelming the cell’s capacity to repair oxidative, as well as other types of damage processes that target ssDNA gaps generated by a corrupted replication apparatus.
Table 4.
Effect of exposure to sublethal concentrations of streptomycin on SIVET induction
| SIVET Induction (CmR/CmS) |
|||
|---|---|---|---|
| Experimenta | Streptomycin (µg/ml) | Frequency(SD) [× 10−4] | Ratio (Str+/Str−) |
| Experiment 1 | 0 | 1.3 (0.57) | - |
| 5 | 7.8 (6.5) | 6 | |
| Experiment 2 | 0 | 0.84 (0.18) | - |
| 5 | 4.1 (5.9) | 5 | |
Data represent averages from five replicate cultures in each experiment. SIVET induction was assayed as described in the text. SD, standard deviation; Str, streptomycin.
ACKNOWLEDGMENTS
Authors are grateful to Drs. David I. Friedman and Jeffrey H. Miller for strains. This work was supported in part by USPHS grant GM58253.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST: Authors declare that there are no conflicts of interest.
REFERENCES
- 1.Bouadloun F, Donner D, Kurland CG. Codon-specific missense errors in vivo. EMBO J. 1983;2:1351–1356. doi: 10.1002/j.1460-2075.1983.tb01591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drummond DA, Wilke CO. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet. 2009;10:715–724. doi: 10.1038/nrg2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edelmann P, Gallant J. Mistranslation in E. coli. Cell. 1977;10:131–137. doi: 10.1016/0092-8674(77)90147-7. [DOI] [PubMed] [Google Scholar]
- 4.Ellis N, Gallant J. An estimate of the global error frequency in translation. Mol. Gen. Genet. 1982;188:169–172. doi: 10.1007/BF00332670. [DOI] [PubMed] [Google Scholar]
- 5.Kramer EB, Farabaugh PJ. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. Rna. 2007;13:87–96. doi: 10.1261/rna.294907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurland CG, Hughes D, Ehrenberg M. Limitations of translational accuracy. In: Neidhardt FC, Curtiss RI, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaecter M, Umbarger HE, editors. Escherichia coli and Salmonella: Cellular and Molecular Biology. Washington, DC: ASM Press; 1996. pp. 979–1004. [Google Scholar]
- 7.Orgel LE. The maintenance of the accuracy of protein synthesis and its relevance to ageing: a correction. Proc. Natl. Acad. Sci. U. S. A. 1970;67:1476. doi: 10.1073/pnas.67.3.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallant J, Kurland C, Parker J, Holliday R, Rosenberger R. The error catastrophe theory of aging. Point counterpoint. Exp Gerontol. 1997;32:333–346. doi: 10.1016/s0531-5565(96)00030-7. [DOI] [PubMed] [Google Scholar]
- 9.Reynolds NM, Lazazzera BA, Ibba M. Cellular mechanisms that control mistranslation. Nat Rev Microbiol. 2010;8:849–856. doi: 10.1038/nrmicro2472. [DOI] [PubMed] [Google Scholar]
- 10.Schimmel P. Mistranslation and its control by tRNA synthetases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011;366:2965–2971. doi: 10.1098/rstb.2011.0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorazi R, Lingutla JJ, Humayun MZ. Expression of mutant alanine tRNAs increases spontaneous mutagenesis in Escherichia coli. Mol. Microbiol. 2002;44:131–142. doi: 10.1046/j.1365-2958.2002.02847.x. [DOI] [PubMed] [Google Scholar]
- 12.Murphy HS, Humayun MZ. Escherichia coli cells expressing a mutant glyV (glycine tRNA) gene have a UVM-constitutive phenotype: implications for mechanisms underlying the mutA or mutC mutator effect. J. Bacteriol. 1997;179:7507–7514. doi: 10.1128/jb.179.23.7507-7514.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slupska MM, Baikalov C, Lloyd R, Miller JH. Mutator tRNAs are encoded by the Escherichia coli mutator genes mutA and mutC: a novel pathway for mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 1996;93:4380–4385. doi: 10.1073/pnas.93.9.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slupska MM, King AG, Lu LI, Lin RH, Mao EF, Lackey CA, Chiang JH, Baikalov C, Miller JH. Examination of the role of DNA polymerase proofreading in the mutator effect of miscoding tRNAs. J Bacteriol. 1998;180:5712–5717. doi: 10.1128/jb.180.21.5712-5717.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao J, Leung HE, Winkler ME. The miaA mutator phenotype of Escherichia coli K-12 requires recombination functions. J. Bacteriol. 2001;183:1796–1800. doi: 10.1128/JB.183.5.1796-1800.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balashov S, Humayun MZ. Escherichia coli Cells Bearing a Ribosomal Ambiguity Mutation in rpsD Have a Mutator Phenotype That Correlates with Increased Mistranslation. J. Bacteriol. 2003;185:5015–5018. doi: 10.1128/JB.185.16.5015-5018.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Al Mamun AA, Gautam S, Humayun MZ. Hypermutagenesis in mutA cells is mediated by mistranslational corruption of polymerase, and is accompanied by replication fork collapse. Mol. Microbiol. 2006;62:1752–1763. doi: 10.1111/j.1365-2958.2006.05490.x. [DOI] [PubMed] [Google Scholar]
- 18.Al Mamun AA, Rahman MS, Humayun MZ. Escherichia coli cells bearing mutA, a mutant glyV tRNA gene, express a recA-dependent error-prone DNA replication activity. Mol. Microbiol. 1999;33:732–740. doi: 10.1046/j.1365-2958.1999.01520.x. [DOI] [PubMed] [Google Scholar]
- 19.Ren L, Al Mamun AA, Humayun MZ. The mutA mistranslator tRNA-induced mutator phenotype requires recA and recB genes, but not the derepression of lexA-regulated functions. Mol. Microbiol. 1999;32:607–615. doi: 10.1046/j.1365-2958.1999.01378.x. [DOI] [PubMed] [Google Scholar]
- 20.Balashov S, Humayun MZ. Mistranslation Induced by Streptomycin Provokes a RecABC/RuvABC-dependent Mutator Phenotype in Escherichia coli Cells. J. Mol. Biol. 2002;315:513–527. doi: 10.1006/jmbi.2001.5273. [DOI] [PubMed] [Google Scholar]
- 21.Livny J, Friedman DI. Characterizing spontaneous induction of Stx encoding phages using a selectable reporter system. Mol. Microbiol. 2004;51:1691–1704. doi: 10.1111/j.1365-2958.2003.03934.x. [DOI] [PubMed] [Google Scholar]
- 22.Brosius J. Laboratory methods: Superlinkers in cloning and expression vectors. DNA. 1989;8:759–777. doi: 10.1089/dna.1989.8.759. [DOI] [PubMed] [Google Scholar]
- 23.Al Mamun AA, Tominaga A, Enomoto M. Detection and characterization of the flagellar master operon in the four Shigella subgroups. J. Bacteriol. 1996;178:3722–3726. doi: 10.1128/jb.178.13.3722-3726.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Podbielski A, Spellerberg B, Woischnik M, Pohl B, Lutticken R. Novel series of plasmid vectors for gene inactivation and expression analysis in group A streptococci (GAS) Gene. 1996;177:137–147. doi: 10.1016/0378-1119(96)84178-3. [DOI] [PubMed] [Google Scholar]
- 25.Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004;32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jonczyk P, Nowicka A, Fijalkowska IJ, Schaaper RM, Ciesla Z. In vivo protein interactions within the Escherichia coli DNA polymerase III core. J. Bacteriol. 1998;180:1563–1566. doi: 10.1128/jb.180.6.1563-1566.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller JH. A short course in Bacterial Genetics: A laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor, N. Y: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- 28.McInerney P, Johnson A, Katz F, O'Donnell M. Characterization of a triple DNA polymerase replisome. Mol Cell. 2007;27:527–538. doi: 10.1016/j.molcel.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 29.Georgescu RE, Kurth I, O'Donnell ME. Single-molecule studies reveal the function of a third polymerase in the replisome. Nat Struct Mol Biol. 2012;19:113–116. doi: 10.1038/nsmb.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maki H, Kornberg A. Proofreading by DNA polymerase III of Escherichia coli depends on cooperative interaction of the polymerase and exonuclease subunits. Proc. Natl. Acad. Sci. U. S. A. 1987;84:4389–4392. doi: 10.1073/pnas.84.13.4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim DR, McHenry CS. In vivo assembly of overproduced DNA polymerase III. Overproduction, purification, and characterization of the alpha, alpha-epsilon, and alpha-epsilon-theta subunits. J. Biol. Chem. 1996;271:20681–20689. doi: 10.1074/jbc.271.34.20681. [DOI] [PubMed] [Google Scholar]
- 32.Stefan A, Reggiani L, Cianchetta S, Radeghieri A, Gonzalez Vara y Rodriguez A, Hochkoeppler A. Silencing of the gene coding for the epsilon subunit of DNA polymerase III slows down the growth rate of Escherichia coli populations. FEBS Lett. 2003;546:295–299. doi: 10.1016/s0014-5793(03)00604-5. [DOI] [PubMed] [Google Scholar]
- 33.Ozawa K, Jergic S, Park AY, Dixon NE, Otting G. The proofreading exonuclease subunit epsilon of Escherichia coli DNA polymerase III is tethered to the polymerase subunit alpha via a flexible linker. Nucleic Acids Res. 2008;36:5074–5082. doi: 10.1093/nar/gkn489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lehtinen DA, Perrino FW. Dysfunctional proofreading in the Escherichia coli DNA polymerase III core. Biochem. J. 2004;384:337–348. doi: 10.1042/BJ20040660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cisneros GA, Perera L, Schaaper RM, Pedersen LC, London RE, Pedersen LG, Darden TA. Reaction Mechanism of the epsilon Subunit of E. coli DNA Polymerase III: Insights into Active Site Metal Coordination and Catalytically Significant Residues. J Am Chem Soc. 2009 doi: 10.1021/ja8082818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perrino FW, Harvey S, McNeill SM. Two functional domains of the epsilon subunit of DNA polymerase III. Biochemistry. 1999;38:16001–16009. doi: 10.1021/bi991429+. [DOI] [PubMed] [Google Scholar]
- 37.Isbell RJ, Fowler RG. Temperature-dependent mutational specificity of an Escherichia coli mutator, dnaQ49, defective in 3'---5' exonuclease (proofreading) activity. Mutat. Res. 1989;213:149–156. doi: 10.1016/0027-5107(89)90146-2. [DOI] [PubMed] [Google Scholar]
- 38.Joyce CM, Grindley ND. Method for determining whether a gene of Escherichia coli is essential: application to the polA gene. J. Bacteriol. 1984;158:636–643. doi: 10.1128/jb.158.2.636-643.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 40.Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell. 2008;135:679–690. doi: 10.1016/j.cell.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang TM, Yuan J, Nguyen A, Becket E, Yang H, Miller JH. The Aminoglycoside Antibiotic Kanamycin Damages DNA bases in Escherichia coli: Caffeine potentiates the DNA-damaging Effects of Kanamycin while Suppressing Cell Killing by Ciprofloxacin in Escherichia coli and Bacillus anthracis, Antimicrob. Agents Chemother. 2012 doi: 10.1128/AAC.00066-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dinglay S, Trewick SC, Lindahl T, Sedgwick B. Defective processing of methylated single-stranded DNA by E. coli AlkB mutants. Genes Dev. 2000;14:2097–2105. [PMC free article] [PubMed] [Google Scholar]
- 43.Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature. 2002;419:174–178. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- 44.Delaney JC, Smeester L, Wong C, Frick LE, Taghizadeh K, Wishnok JS, Drennan CL, Samson LD, Essigmann JM. AlkB reverses etheno DNA lesions caused by lipid oxidation in vitro and in vivo. Nat Struct Mol Biol. 2005;12:855–860. doi: 10.1038/nsmb996. [DOI] [PubMed] [Google Scholar]