

SUMMARY
Epigenetic regulation may involve heritable chromatin states but how chromatin features can be inherited through DNA replication is incompletely understood. We address this question using cell free replication of chromatin. Previously, we showed that a Polycomb Group complex, PRC1, remains continuously associated with chromatin through DNA replication. Here we investigate the mechanism of persistence. We find that a single PRC1 subunit, Posterior Sex Combs (PSC) can reconstitute persistence through DNA replication. PSC binds nucleosomes and self-interacts, bridging nucleosomes into a stable, oligomeric structure. Within these structures, individual PSC-chromatin contacts are dynamic. Stable association of PSC with chromatin, including through DNA replication, depends on PSC-PSC interactions. Our data suggest labile individual PSC-chromatin contacts allow passage of the DNA replication machinery while PSC-PSC interactions prevent PSC from dissociating, allowing it to rebind to replicated chromatin. This mechanism may allow inheritance of chromatin proteins including PRC1 through DNA replication to maintain chromatin states.
Keywords: chromatin, DNA replication, Polycomb, epigenetic, cell free system
INTRODUCTION
Epigenetics, including the inheritance of regulatory information across cell generations without changing genetic information, is believed to be central to differentiation and is increasingly implicated in disease (Feinberg, 2007). Two unsolved questions in epigenetics are: what is the nature of inherited information, and what are the mechanisms of its transmission? Chromatin features, including histone modifications, histone variants, remodeled nucleosomes, chromatin folding, and chromatin binding proteins, are widely believed to function as epigenetic information (Margueron and Reinberg, 2010; Probst et al., 2009). Chromatin is an attractive candidate to be a carrier of epigenetic information because it is directly coupled to the DNA sequence it regulates (Kaufman and Rando, 2010). Although mechanisms for inheritance of DNA methylation and some histone modifications through DNA replication have been described, the behavior of most chromatin proteins and associated chromatin features during DNA replication and mitosis are incompletely understood (Margueron and Reinberg, 2010).
The Polycomb Group (PcG) proteins, initially described in Drosophila melanogaster (Lewis, 1978; Struhl, 1981), are implicated in epigenetic silencing of gene expression that persist through development. A long-standing model for how PcG proteins mediate stable silencing is that they create unique chromatin structures that are inherited through cell generations (Paro, 1990). Extensive work has described multiple PcG protein complexes and their effects on chromatin and gene expression (Muller and Verrijzer, 2009; Simon and Kingston, 2009). However, precisely how these mechanisms are used by PcG proteins to maintain gene silencing across cell divisions, is still not clear.
Understanding how chromatin proteins like the PcG behave during DNA replication is central to epigenetics. Because DNA replication occurs rapidly and only once during each cell cycle, it is difficult to study the behavior of proteins at specific genes during DNA replication in cells, and perturbations of DNA replication trigger global cellular responses. We are using cell free replication of chromatin to study how passage of the DNA replication machinery affects PcG protein-chromatin interactions. Using a reconstituted system makes it possible to isolate and manipulate mechanistic events during DNA replication. By analogy, in vitro transcription systems, although they recapitulate only part of gene regulation, continue to be valuable in defining steps in transcription and regulatory mechanisms (e.g. (Cheng et al., 2012; Jishage et al., 2012; Shukla et al., 2011) ).
The system we use to study replication of PcG protein bound chromatin is based on the SV40 virus (reviewed in (Fanning and Zhao, 2009)). The SV40 large T-Antigen (TAg) binds to plasmids containing the SV40 origin sequence). TAg serves as both the origin recognition factor, and the replicative helicase (Dean et al., 1987; Stahl et al., 1986), functions that require many proteins in eukaryotic cells. TAg binds to and unwinds the origin DNA in an ATP-dependent manner and, in the presence of cytoplasmic extracts from mammalian cells, recruits DNA polymerase and associated factors to establish bidirectional DNA replication forks that replicate the plasmid template (Li and Kelly, 1984; Stillman and Gluzman, 1985; Wobbe et al., 1985). Because it bypasses the complex biochemical steps involved in replication origin selection and initiation, the SV40 system cannot be used to study how these events might influence PcG proteins. However, once replication is initiated in this system, it is thought to largely recapitulate chromosomal DNA replication mechanisms. The SV40 system was used to identify many of the components of the DNA replication fork (Waga et al., 1994; Waga and Stillman, 1994; Wobbe et al., 1987), and also chromatin assembly factor 1, which assembles nucleosomes onto newly replicated DNA (Smith and Stillman, 1989; Stillman, 1986).
We previously used the SV40 cell free replication system to study PcG proteins and found that a PRC1 core complex (PCC) consisting of three subunits, PSC, Polycomb (Pc), and dRING (Francis et al., 2001)) persists on chromatin or DNA through DNA replication in vitro (Francis et al., 2009). Corroborating this result, we found that a key PcG protein in PRC1, PSC, is found on newly replicated chromatin in Drosophila tissue culture cells (Francis et al., 2009). Here, we investigate the mechanism by which PCC is maintained on chromatin through DNA replication.
RESULTS
PCC is transferred to both leading and lagging strands
If persistence through replication is important for maintaining gene expression states, PCC should transfer to both leading and lagging strands, even though their replication has mechanistic differences that might affect binding of PcG proteins. In the SV40 system, bidirectional replication forks replicate circular plasmids. Therefore, each copy is replicated partially as leading and partially as lagging strand so that analysis of intact plasmids would not reveal a strand bias.
To test whether PCC preferentially segregates to one strand we carried out replication reactions in vitro in the presence of 32P-labelled dATP and isolated replicated (radiolabelled) DNA associated with PCC. We asked whether it contains replicated DNA from both strands by hybridizing it to strand specific probes (Fig. 1). This experiment is possible because the SV40 origin of replication and thus the direction of replication is known. DNA replicated in either direction isolated from control reactions hybridized to both strands, as expected. Replicated DNA which was bound to PCC also hybridized to both strands, independent of the direction of replication. Thus PCC is transferred to both newly replicated strands.
Figure 1. PCC is transferred to both leading and lagging strands during DNA replication.
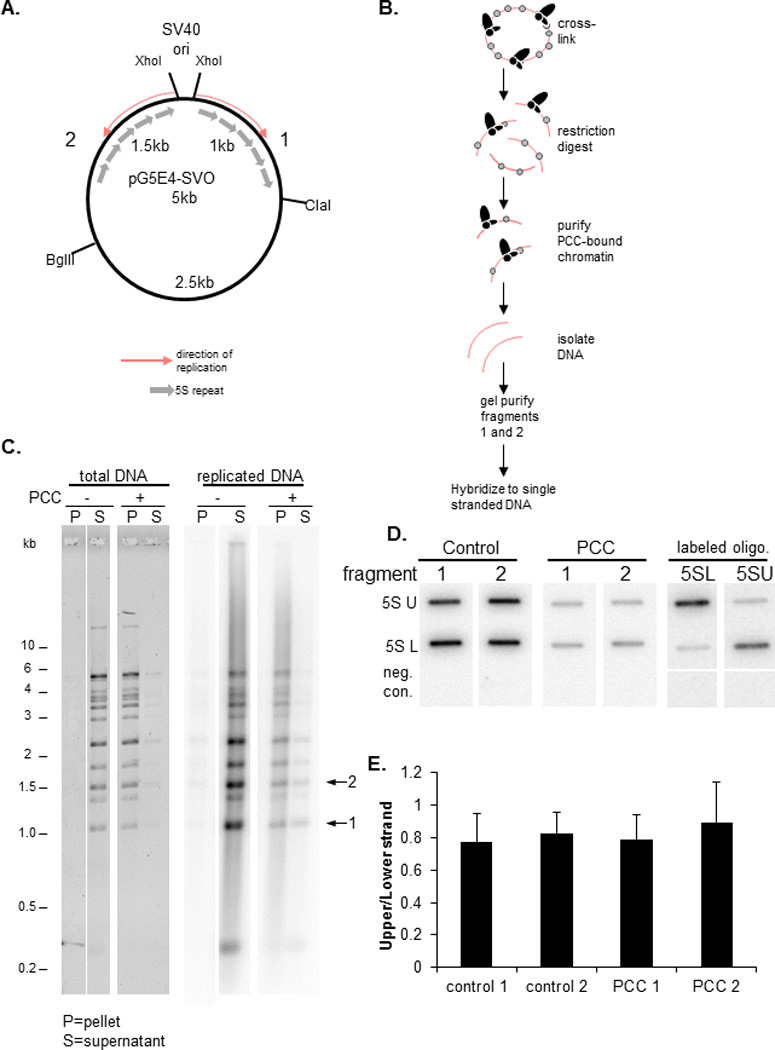
A) Schematic of plasmid used for DNA replication. Fragments 1 and 2 were used for analysis of leading and lagging strand association with PCC. Both fragments contain repeats of the 5S rDNA sequence and are replicated in opposite directions so that the leading and lagging strands are opposite between the two. B) Schematic of isolation of PCC-bound fragments 1 and 2. After replication, reactions were cross-linked with formaldehyde to preserve PCC-chromatin interactions. Chromatin templates were purified by sucrose gradient sedimentation, dialyzed, and digested with restriction enzymes to release fragments 1 and 2 and streptavidin-coated beads used to isolate PCC-bound chromatin fragments. Supernatant from the streptavidin pulldown was analyzed for control reactions which lacked PCC. Purified DNA was separated by agarose gel electrophoresis and fragments 1 and 2 isolated and used to probe blots of single stranded DNA corresponding to each strand. C) Gel of purified DNA from PcG bound or unbound restriction digested chromatin from DNA replication reactions. Left panel shows SYBR gold stain of total DNA and right panel a phosphorimager scan of replicated DNA. Arrows indicate the fragments 1 and 2. Cross-linked chromatin is not completely digested so that there are several partial digestion products. D) Phosphorimager scan of hybridization of PCC-bound or control replicated DNA hybridized to slot blots with immobilized strand-specific probes. Right panels are blots probed with a labeled oligonucleotide corresponding to the upper or lower strand to confirm the purity of the single stranded DNA and the specificity of hybridization conditions. Negative control is a sequence not present in the plasmid. U=upper strand; L=lower strand E) Summary graph of the average ratio of upper to lower strand for blots probed with fragments 1 or 2. Error bars in this and all subsequent graphs are standard deviation.
PRC1 component PSC persists on chromatin through DNA replication
Previous work indicates that the central chromatin modifying protein in PRC1 is PSC. PSC has many of the activities of the whole complex, including tight binding to DNA and chromatin, inhibition of chromatin remodeling and transcription, and chromatin compaction (Francis et al., 2004; Francis et al., 2001; King et al., 2002). These activities are conserved in PRC1 through evolution (Beh et al., 2012; Grau et al., 2011), and PSC that lacks these activities but can assemble into PRC1 has a lethal phenotype in flies (King et al., 2005; Wu and Howe, 1995). We therefore tested if PSC alone can remain associated with chromatin through DNA replication. PSC was bound to chromatin templates that were replicated and sedimented through sucrose gradients to separate PSC-bound and unbound chromatin. Replicated chromatin from these reactions sediments near the bottom of gradients, indicating that it is bound by PSC (Fig. 2A–C). If PSC is added with the replication extract (“W.E.”), templates sediment as unbound plasmids, indicating that these concentrations of PSC do not bind chromatin in the presence of replication extracts. This was also observed wtih PCC (Francis et al., 2009). Thus, we conclude that PSC persists on chromatin templates through DNA replication, since PSC molecules that are released into solution will not re-bind in extracts. Addition of competitor chromatin to replication reactions to capture any released PSC also does not reduce binding of PSC to replicated chromatin (Fig. S1). Like PCC, PSC inhibits DNA replication in vitro (Francis et al., 2009) so that fewer products are observed. The replication products are full length (demonstrated by denaturing agarose gels, Fig. 2B), suggesting inhibition occurs at the level of initiation.
Figure 2. PcG protein Posterior Sex Combs is sufficient for persistence through DNA replication.
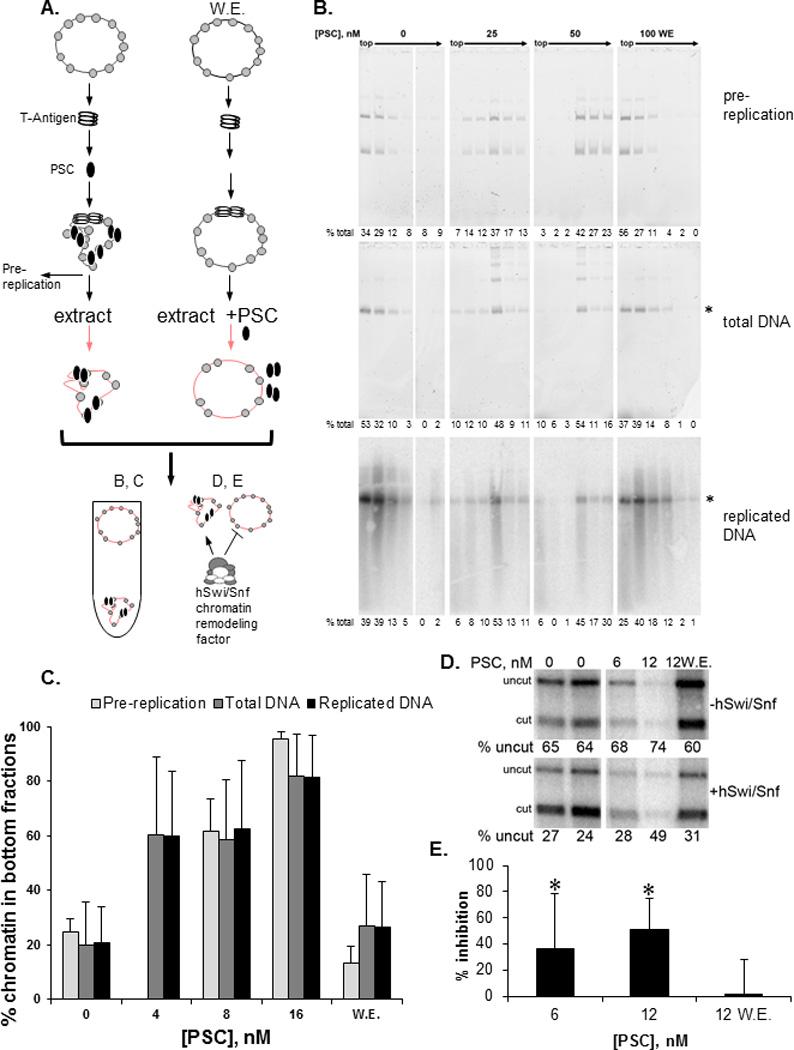
A) Schematic of SV40 replication experiments. Plasmids were incubated with SV40 T-Antigen, followed by PSC; replication was started by addition of extract. PSC was added with replication extracts in reactions labeled “W.E.” (with extract). Replication products were separated by sucrose gradient sedimentation (B, C) or used as substrates for chromatin remodeling reactions (D, E). B) Sucrose gradient fractions from a replication experiment carried out with PSC. Top panel: SYBR gold stained native gel of DNA in fractions from a gradient to analyze PSC binding to chromatin prior to replication; Middle panel: SYBR gold stained denaturing gel of total DNA from fractions of a gradient separating replication reactions; Bottom panel: phosphorimager scan of the middle gel showing radiolabeled replication products which are predominantly full length (asterisks marks full length plasmid). C) Summary of sucrose gradient sedimentation of replication reactions. Bars show the average % of chromatin present in the bottom three fractions of the gradient. D) Example of chromatin remodeling assay on replication fractions. Panels show phosphorimager scan of replicated DNA incubated with (bottom) or without (top) hSwi/Snf. Chromatin remodeling mediated by hSwi/Snf increases restriction enzyme digestion; a decrease in digestion reflects inhibition of chromatin remodeling. E) Summary of inhibition of chromatin remodeling by PSC after DNA replication. Asterisks indicate that inhibition is significantly higher than when PSC is added with the extracts by student’s one-tailed t-test (p<0.05). Note that similar amounts of DNA are present in each lane but the differences in signal reflect inhibition of DNA replication by PSC, similar to what was observed previously for PCC (Francis et al., 2009); denaturing gels (B) indicate that replication products are full length so that inhibition likely occurs at the level of initiation. See also Figure S1.
To determine if the PSC that remains on chromatin is functional, we asked if PSC-bound, replicated templates are refractory to ATP-dependent chromatin remodeling, a hallmark of PRC1-bound chromatin (Shao et al., 1999). When PSC is bound to chromatin prior to DNA replication, but not when it is added with the replication extract, chromatin remodeling of replication products is inhibited (Fig. 2D, E). We conclude that persistence through DNA replication can be reconstituted with PSC alone, and thus that PSC can be used to dissect how PCC persists through DNA replication.
PSC and PCC can bridge nucleosomes
Electron microscopy indicates that PSC and PCC compact chromatin, bringing nucleosomes into clusters while leaving intervening loops of DNA (Francis et al., 2004). To test whether PSC holds nucleosomes together in solution, PSC-bound chromatin templates were digested with micrococcal nuclease (MNase) to remove linker DNA, and sedimented through sucrose gradients (Fig. 3). 60–80% of the generated mononucleosomes sediment near the bottom of the gradient, unlike free mononucleosomes suggesting PSC forms bridges between nucleosomes on the plasmid. PCC also bridges nucleosomes (Fig. 3B, C).
Figure 3. PSC and PCC bridge nucleosomes.
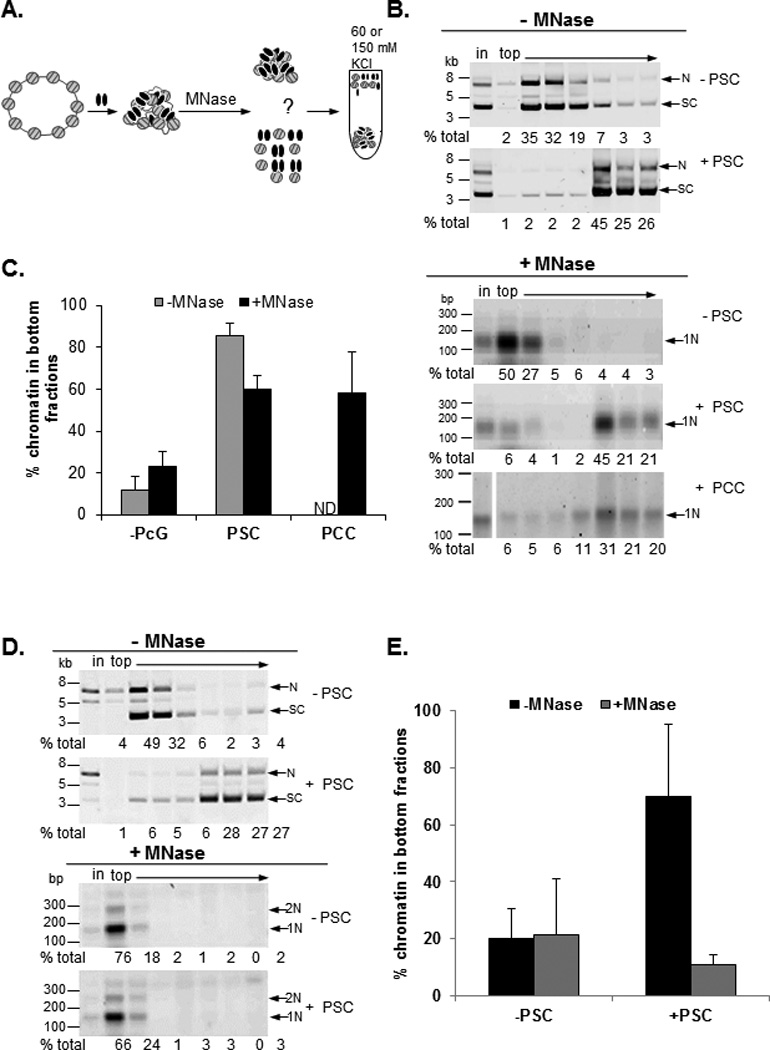
A) Schematic of nucleosome bridging experiments. B) Example of nucleosome bridging assay. Each panel shows SYBR gold stained DNA from sucrose fractions of undigested (−MNase) or digested (+MNase) plasmid alone or incubated with PSC or PCC as indicated. N=nicked; SC=supercoiled forms of plasmid; 1N=mononucleosome; in=input. C) Summary graph of average % of chromatin in the bottom three fractions of sucrose gradients. D) Representative and E) summary of nucleosome bridging assay(s) sedimented through gradient with 150 mM KCl. See also Figure S2.
Bridged nucleosomes lacking linker DNA bind less stably to PSC
To determine whether the MNase treated bridged complexes are less stable than intact, PcG bound chromatin, binding reactions and MNase digestion were carried out with PSC under our standard (60 mM KCl) conditions but reactions were sedimented through sucrose gradients containing 150 mM KCl. Sedimentation of PSC-bound plasmids was unaffected by the elevated KCl. In contrast, the mononucleosomes from PSC-bound, MNase digested chromatin sediment near the top of the gradient (Fig. 3D, E). Streptavidin pulldown experiments with biotin tagged PSC demonstrate that PSC does not remain bound to mononucleosomes after sedimentation through sucrose gradients in 150 mM KCl (Fig. S2). These data indicate that the bridged species lacking linker DNA is less stable than intact bridged chromatin. They also demonstrate that nucleosome bridging does not reflect formation of insoluble aggregates since it is reversed by elevated KCl. These experiments suggest that PSC-nucleosome interactions are dynamic. On plasmid templates, nucleosomes can be transiently released and recaptured because of their high local concentration, while released mononucleosomes will diffuse away rapidly, making rebinding unlikely. Removal of linker DNA with MNase likely also reduces stability of binding of PSC to nucleosomes (Lo and Francis, 2010).
PSC can bridge nucleosomes in trans
We wondered if nucleosome bridging occurs only between adjacent nucleosomes, or can occur among widely separated nucleosomes or even in trans. We asked whether PSC can create nucleosome clusters, analogous to bridged complexes, from mononucleosome substrates (Fig. 4). Various concentrations of PSC were bound to a mixture of mononucleosomes labelled with biotin or Cy3 on the 5’ end of the DNA (5nM total mononucleosomes) (Fig. 4A, S3A). Binding reactions were fractionated by sucrose gradient sedimentation. Without PSC, mononucleosomes sediment near the top of the gradient. As PSC is increased from 2.5 to 40 nM, the template sediments first in the middle and finally near the bottom of the gradient (Fig. 4B, C). To determine whether PSC-mononucleosome complexes contain bridged nucleosomes, biotinylated nucleosomes were isolated from the peak gradient fractions using streptavidin- coated beads. If complexes contain bridged nucleosomes, then Cy3-labelled nucleosomes should co-purify with biotinylated ones. Cy3-labelled mononucleosomes were not isolated with biotinylated ones from the top (unbound) or middle (first PSC-nucleosome complex) fractions of gradients but were enriched on streptavidin-coated beads incubated with fractions near the bottom of the gradients (Fig. 4B, D). In this experiment, Cy3-labelled nucleosomes are present at 4-fold higher levels than biotinylated ones, yet close to 80% of the Cy3-labelled nucleosomes are isolated with the biotinlyated ones. This suggests that PSC-induced nucleosome clusters contain at least 3–4 nucleosomes (if PSC binds with a 1:1 stoichiometry with nucleosomes and forms bridges between only two nucleosomes then the maximum possible association of Cy3-labelled mononucleosomes would be 25%). Thus, low concentrations of PSC bind mononucleosomes but do not bring them together, while higher concentrations bring nucleosomes together in trans. Previously we found that PSC binds mononucleosomes but inhibits their remodeling only at high concentrations (Francis et al., 2001; Lo and Francis, 2010), which coincide with concentrations at which we observe bridging of mononucleosomes. In contrast, an array of even 2 nucleosomes is efficiently inhibited by PSC (Lo and Francis, 2010). This can be explained if bridging of nucleosomes is required to inhibit chromatin remodeling, since this reaction would be intramolecular for nucleosomal arrays but intermolecular for mononucleosomes and thus less efficient. Thus, inhibition of chromatin remodeling is correlated with nucleosome bridging.
Figure 4. PSC can bridge nucleosomes in trans.
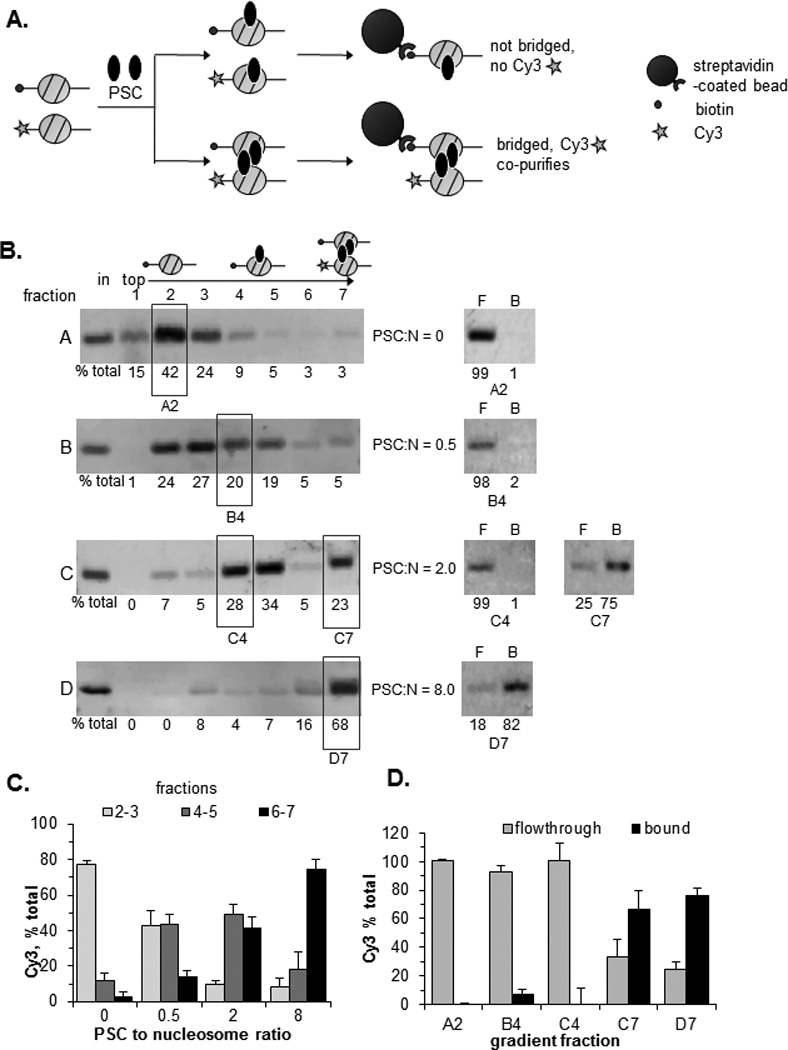
A) Schematic of assay to test PSC bridging of mononucleosomes. Mononucleosome bridging was quantified by measuring the amount of Cy3-labelled nucleosomal DNA that co-purifies with the biotinylated nucleosomes. B) Left panels show Cy3-labelled nucleosomal DNA in sucrose gradient fractions of mononucleosome-PSC mixtures. The distribution of labelled mononucleosomal DNAs was also confirmed with SYBR gold staining, and with a Cy5 label on the biotinylated DNA; both fragments have identical distributions in the gradient (not shown). Right panels show streptavidin pulldown results from the fractions indicated with boxes. F=flow-through; B= bound. PSC:N is ratio of PSC to nucleosomes. C) Summary of migration of Cy3-labelled nucleosomes in sucrose gradients. D) Co-purification of Cy3-labelled mononucleosomes with biotinylated nucleosomes. Gels show Cy3-labelled DNA in the flow through and bound to streptavidin-coated beads. Fraction numbers correspond to gradients in B. See also Figure S3.
The experiment above demonstrated that PSC can bridge nucleosomes in trans, but only at high ratios of PSC to nucleosomes, while bridging of chromatin occurs at ratios of about 1:1 (Fig. 2). To distinguish a requirement for a high concentration of PSC from a high PSC to mononucleosomes ratio to observe bridging in trans, a mononucleosome mixing experiment was carried out with both nucleosomes and PSC at 40 nM (i.e. a 1:1 ratio) (Fig. S3B). PSC bridges mononucleosomes under these conditions indicating that trans nucleosome interactions do not require high ratios of PSC to nucleosomes, but high concentrations of PSC. The ability of PSC to bridge non-adjacent nucleosomes could allow it to bridge chromatin in front of and behind replication forks, or to form or stabilize chromatin loops among PcG protein bound sites as observed in vivo (Lanzuolo et al., 2007).
Nucleosome bridging depends on chromatin binding and self-interaction
PSC is a 1603 amino acid protein which has conserved domains in its N-terminal region that are important for its assembly into PRC1, and a large, intrinsically disordered C-terminal region which is required for both its activities on chromatin and its in vivo function (Fig. 5A) (Beh et al., 2012; Emmons et al., 2009; Francis et al., 2004; King et al., 2005). We previously showed that PSC self-interacts (Lo and Francis, 2010), and a simple mechanism for nucleosome bridging would be interaction among nucleosome bound PSCs. To test this model, we analyzed a series of PSC truncations to determine which sequences in PSC are required for nucleosome bridging. We first measured chromatin binding for each PSC truncation using sucrose gradient sedimentation. PSC1–572 does not alter chromatin sedimentation, suggesting it does not bind stably to chromatin, which is consistent with previous analyses (King et al., 2005). All of the other truncated versions of PSC bind chromatin, but two, PSC456–909 and PSCΔ761–1355, require 4-fold higher protein concentrations for binding (Fig. 5B, grey bars, S4A). Reduced chromatin binding correlates with a decrease in DNA binding affinity (Fig. S4B). PSC1–909, PSC456–1603, and PSCΔ761–1355 bridges nucleosomes at concentrations sufficient for binding while PSC456–909 does not (Fig. 5B, black bars, S4A). Thus, chromatin binding alone does not predict bridging activity since PSC456–909 and PSCΔ761–1355. Previous analysis of these truncations indicates that PSC1–572 does not inhibit chromatin remodeling, while PSC456–909 does so weakly on nucleosomal arrays and not on mononucleosome substrates (King et al., 2005; Lo and Francis, 2010). PSC1–572 can assemble into PRC1, but cannot replace PSC function in vivo while PSC1–909, which bridges nucleosomes, supports gene silencing and viability (King et al., 2005; Wu and Howe, 1995).
Figure 5. Chromatin binding and self-interaction are important for nucleosome bridging and persistence through DNA replication.
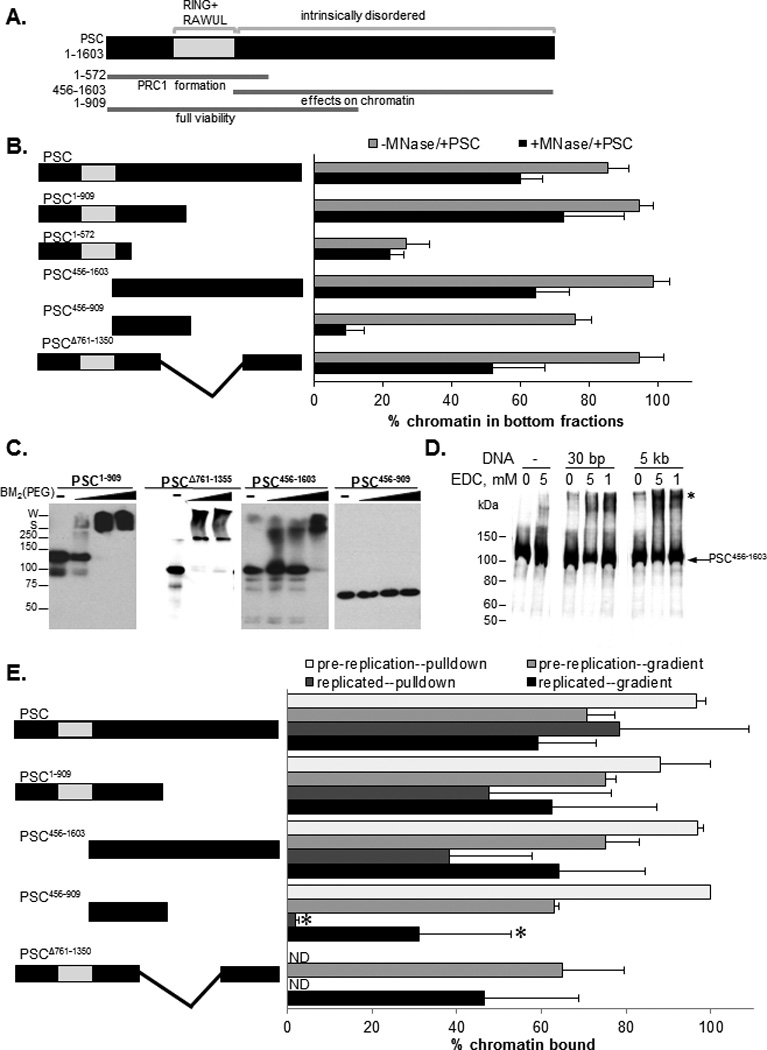
A) Schematic diagrams of PSC. Functional regions are based previous data and conserved domains (Brunk et al., 1991; Sanchez-Pulido et al., 2008) are in grey. B) Summary of bridging assays with truncated PSC. Grey and black bars show average % of chromatin in the bottom three fractions of the gradient without and with MNase digestion, indicative of chromatin binding and nucleosome bridging, respectively. C) Representative Western blot analysis of protein cross-linking of truncated PSC with BM2(PEG). Note that all samples in a set were run on the same gel, but different sets are from different gels so that markers are approximate. S=stacking gel border; W=well. D) Western blot analysis of the effect of DNA (30 bp oligonucleotide or 5 kb plasmid) on cross-linking of PSC456–1603 by EDC; asterisk indicates cross-linked products. E) Summary of replication experiments carried out with truncated PSC proteins. Graph shows average % of chromatin that is in the bottom three fractions of sucrose gradients or isolated on streptavidin coated beads using biotinylated PSC truncations of binding or replication reactions. Proteins were used at 50–100 nM except for PSC456–909 and PSCΔ761–1355 which were used at 200 nM. Asterisks indicates statistically significant difference between pre-replication and replicated chromatin by t-test (P<0.05). See also Figure S4.
To determine if self-interaction can explain the difference in bridging activity among PSC truncations that bind chromatin we used chemical cross-linking to test PSC truncations for self-interaction. PSC1–909 and PSCΔ761–1355 formed high molecular weight cross-linked species with two different cross-linkers, indicating that they self-interact. PSC456–1603 also formed cross-linked species but at higher cross-linker concentrations (Fig. 5C, S5C). Cross-linking of PSC456–1603 is stimulated by DNA (Fig. 5D), which is consistent with a previous report of DNA-induced interactions among PCCs (Mohd-Sarip et al., 2006). DNA stimulation of cross-linking may reflect binding of multiple proteins to single DNAs, although 30 base pair oligonucleotides added in excess of protein stimulate protein cross-linking similarly to plasmids, arguing against this possibility. PSC456–909, which is impaired for nucleosome bridging did not form high molecular weight species when incubated with either cross-linker under these conditions. We verified these self-interaction results with PSC truncations using co-immunoprecipitation assays (Fig. S5D, E). We also tested the two weak chromatin binding truncations in competition binding assays to determine if self-interaction has functional consequences for chromatin binding. Self-interaction should stabilize chromatin binding, and comparison of PSCΔ761–1335, and PSC456–909, demonstrates that this is the case (Fig. S4F, G). Thus, self-association can compensate for weakened DNA binding activity (as is the case for PSCΔ761–1335), but bridging is not observed without self-association (as observed with PSC456–909).
Nucleosome bridging activity is correlated with persistence through DNA replication
PSC truncations were tested for their ability to remain associated with chromatin through DNA replication. PSC truncations that can bridge nucleosomes persist through DNA replication (Fig. 5E; S4 H–O) while PSC456–909 is impaired for persistence through DNA replication. At concentrations of PSC456–909 10-fold higher than used for PSC, nucleosome bridging, persistence through DNA replication, and DNA-stimulated self-interaction are observed (data not shown). Thus, this protein may harbor the core activities of PSC but is severely impaired for functions other than chromatin binding under our assay conditions (See also Supplemental Discussion).
Although the findings from the sucrose gradient sedimentation experiments described above are reproducible, the defect observed for PSC456–909 is more subtle than that observed in the bridging assay. We therefore repeated this experiment with biotinylated versions of the key proteins and streptavidin beads to capture proteins and associated chromatin templates. Using this protocol, PSC and the tested truncations, including PSC456–909 bind in the absence of extracts (“pre-replication”). After the replication reaction, however, essentially no binding of PSC456–909 is observed when either total or replicated templates are analyzed (Fig. 5E; S4P, Q). In contrast, PSC, PSC1–909 and PSC456–1603 are all bound to replicated chromatin. We conclude that nucleosome bridging activity is correlated with persistence through DNA replication and that both correlate with chromatin binding and self-association.
PSC-chromatin contacts are dynamic
A central question in how PCC and PSC can persist through DNA replication using their nucleosome bridging activity is whether the proteins lose contact with chromatin during DNA replication. Intuitively, it seems that chromatin must be transiently released while the underlying DNA is copied. Yet our data indicates that PSC and PCC are not released from the template. This suggests either that PSC and PCC do not ever lose contact with the chromatin or that they transiently release the chromatin, but are held near the template and subsequently rebind. We carried out a series of experiments to address this issue.
To test if PCC contacts with chromatin are dynamic on the time scale of these experiments we carried out competition binding experiments with an average of 2 or 4 complexes per chromatinized plasmid. We reasoned that the stable binding of PCC observed previously could reflect cooperative interactions among complexes rather than stable binding of individual complexes. If this is correct, then PCC should bind more stably when multiple copies of the complex are present on the same template and nucleosomes are bridged. Both ratios of PCC allowed capture of the majority of plasmids with streptavidin-coated beads (using biotinylated PCC), and competitor effectively blocks binding (Fig. 6A–C; S5A–C). However, when competitor is added after binding, to sequester PCC that is released from chromatin, less chromatin is captured by low than high ratios of PCC. This experiment suggests bridging by PCC stabilizes binding. In the course of our studies, we noticed that PCC binds tightly to ssDNA (Fig. S5), and we used ssDNA as competitor in these experiments. This result is interesting because all replication forks contain ssDNA.
Figure 6. PSC and PCC-chromatin contacts are dynamic and replication extracts contain components that compete for their binding.
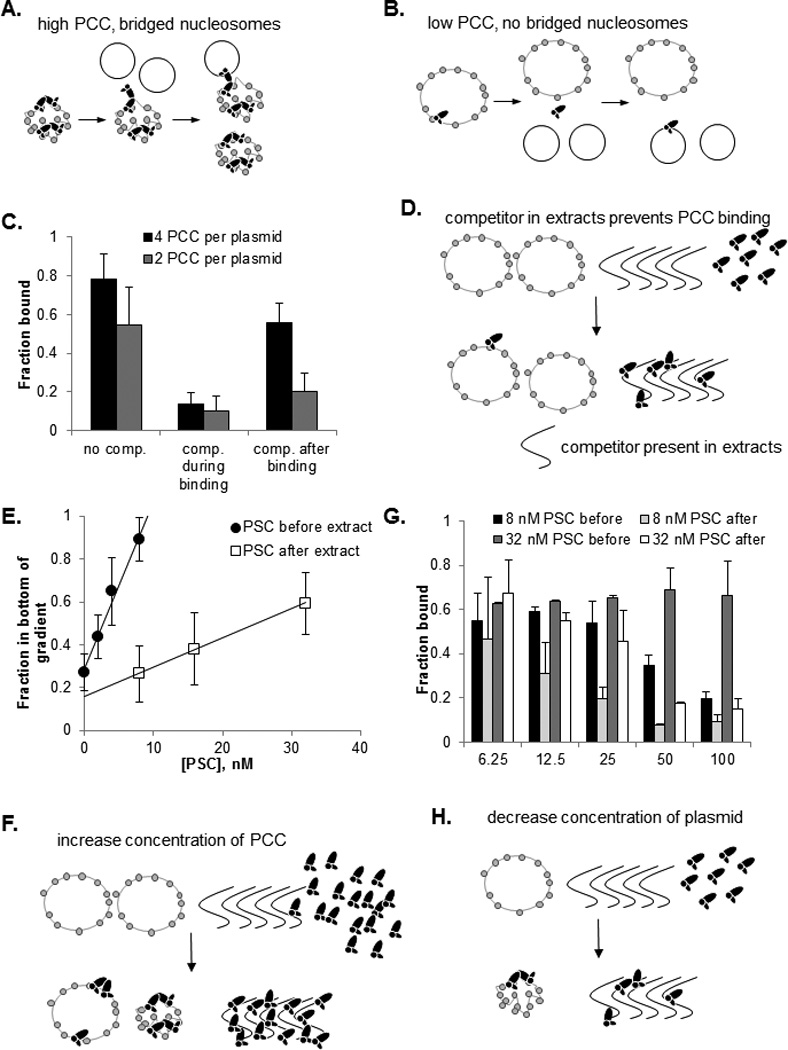
A, B) Schematic diagram demonstrating the predicted behavior of low versus high ratios of PCC to plasmid in competition assays. Note that for high concentrations of PCC (A), transient loss of PCC-chromatin contacts can result in rebinding to the same template (bottom) or bridging between template and competitor (top). C) Summary of competition experiments. See also Figure S5. D) Schematic diagram of competition between factors in extract (which may include RNA that is present in the extracts and binds PSC and PCC, not shown) and chromatin for PCC or PSC binding. E) Summary of PSC titration experiments in which PSC was added before or after extract. Points were fit with a linear regression (R2 ≥0.99). The ratio of the slopes in the absence versus presence of extracts is 5.7. F) Schematic diagram explaining the effect of raising PSC or PCC concentration in binding reactions. G) Summary of template titration experiments in extracts. H) Schematic diagram of the effect of decreasing chromatin template in binding experiments in extracts. F) See also Figure S6.
The finding that PCC-chromatin interactions are dynamic, but stabilized by bridging provides a possible explanation for how PCC can both be retained on templates and release chromatin transiently during replication. If this is correct, it further predicts that PCC and PSC must rebind to chromatin after passage of the replication fork. This raises an apparent contradiction, since our data indicate that PCC and PSC do not bind chromatin in replication extracts under conditions where persistence is observed. Indeed, this feature of the system is critical to the conclusion that the proteins are not released into solution during replication. A possible explanation is that if PCCs are tethered to the template through interactions with other, chromatin bound PCCs, their local concentration near the template will be higher than in the extracts which may allow rebinding to chromatin. To investigate the prediction that high concentrations of PSC (or PCC) can bind chromatin in extracts, we titrated PSC into chromatin binding reactions before or after adding replication extracts (Fig. 6D,E; S6A). We find that PSC can bind chromatin when added after extract, but 5–6 times higher concentrations are required than when PSC is pre-bound in buffer. Similar results were observed in a smaller number of experiments with PCC (not shown).
To determine if the reduction in PSC binding to chromatin in extracts is due to competition from factors in the extract, or inactivation of PSC binding in extracts, we titrated template in reactions where PSC was bound before or after addition of extracts and measured binding by streptavidin pulldown of biotinylated PSC and bound chromatin. Our rationaleis that if extracts contain components that compete for PSC binding, then less PSC is available for binding in extracts than in buffer. Decreasing the amount of chromatin template under these conditions should increase the fraction of template that is bound (since both PCC/PSC and chromatin are above their Kd for binding (Francis et al., 2009)). In contrast, if PSC binds chromatin less well in extract because its affinity is reduced (so that it is no longer above its Kd for chromatin), then the fraction bound will remain the same. We find that as template is titrated down, a higher fraction is bound (Fig. 6 G,H; S6 B). This is consistent with a model in which the extracts do not affect the affinity of PSC for chromatin but instead compete for binding to it, thus decreasing the amount of PSC available for binding. From these experiments, we conclude that PSC and PCC-chromatin interactions are dynamic, but stabilized by interactions with other PSC/PCCs on the template (nucleosome bridging). In extract, PSC and PCC binding is attenuated, likely because of competitor factors present in the extracts. However, at elevated concentrations, which should be present near chromatin templates where PSC or PCC are tethered through PSC-PSC interactions, PSC and PCC can bind chromatin in extracts.
DISCUSSION
A Nucleosome bridging model for persistence through DNA replication
We suggest the following model for persistence through DNA replication (Fig. 7). Each PcG complex binds both to chromatin and at least one other PcG complex, forming a stable structure on chromatin in which nucleosomes are bridged. This structure is refractory to chromatin remodeling, and may also be important for inhibition of transcription. Within the bridged configuration individual PcG-chromatin contacts are dynamic (Fig. 6), allowing segments of DNA to be released from PcG proteins so they can be replicated (we do not know if the replicative helicase or other components of the replication fork promote disruption of PcG-chromatin contacts or if the intrinsic dynamics of PSC-chromatin interactions are sufficient). When a complex breaks contact with chromatin during DNA replication, PSC-PSC interactions with another complex bound further away from the replication fork prevent it from dissociating and instead allow it to rebind to newly replicated chromatin as it becomes available. This model can explain how the PcG complex can persist through DNA replication, but still allow the replication machinery access to the DNA. The labile nature of individual PcG-chromatin contacts allows these contacts to be rearranged so that the complexes can transfer from in front of to behind the replication fork. These dynamic contacts may also explain why chromatin bound by PCC is still accessible to restriction enzymes and DNaseI (Francis et al., 2001). The unstructured nature of the region of PSC that is important for these activities may be flexible, allowing bridges to form across variable distances in chromatin. Thus, individually dynamic PCCs form a stable chromatin structure through PSC-PSC interactions that can withstand passage of a DNA replication fork.
Figure 7. Bridging model for persistence of PcG complexes through DNA replication in vitro.
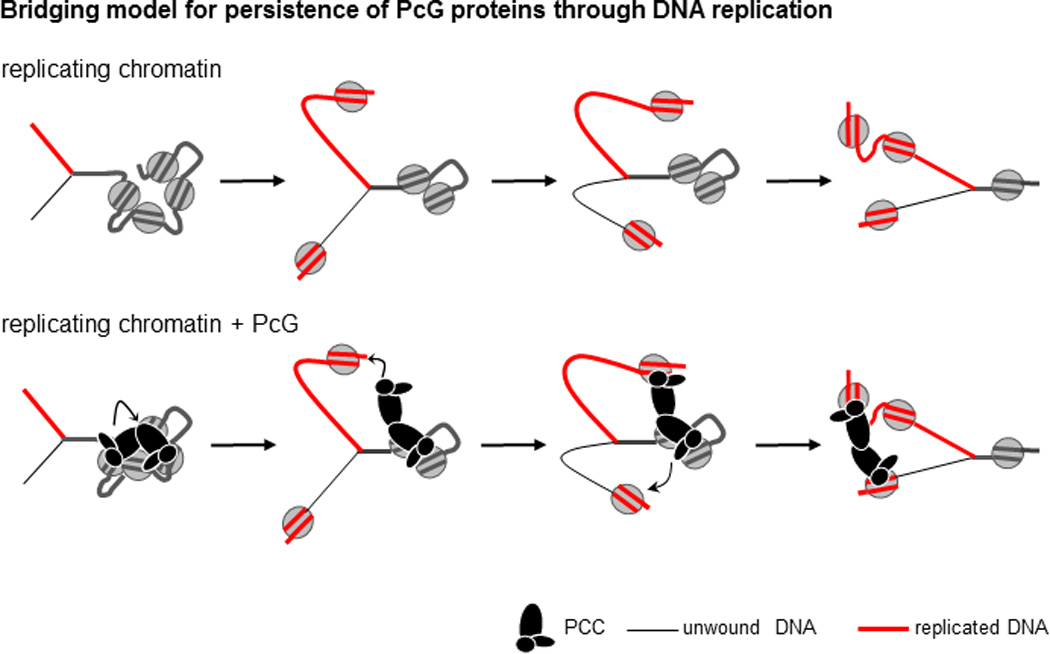
Top scheme shows a replication fork moving through chromatin; lower panel shows the same steps with PcG complexes transferring from unreplicated to replicated chromatin. Curved arrows indicate hypothetical mechanistic steps. The bridging model proposes that a combination of PcG-PcG and PcG-chromatin interactions allows dynamic rearrangement of chromatin contacts without loss of PcG complexes. See text for details.
Sites that bind PcG proteins in vivo can interact over large distances (Bantignies and Cavalli, 2011; Lanzuolo et al., 2007). Although PcG proteins may not be primarily responsible for forming these contacts (Comet et al., 2011; Li et al., 2011), nucleosome bridging could stabilize them. A previous study described trans interactions among nucleosomal arrays mediated by PRC1 (Lavigne et al., 2004). In these experiments, PRC1-bound nucleosomal arrays which were immobilized on beads could recruit additional arrays from solution without addition of new PRC1. This activity is likely related to the nucleosome bridging activity described here, but is distinct since it occurs with slower kinetics and cannot be carried out by PSC alone.
Does nucleosome bridging occur in vivo?
A direct test of this question has so far not been possible. If PRC1 persists on chromatin through DNA replication in vivo as shown here in vitro, PSC may be diluted by two-fold on replicated chromatin until additional PcG proteins are recruited. We carried out Chromatin Immunoprecipitation (ChIP) using Drosophila S2 cells at different stages in the cell cycle that were isolated by centrifugal elutriation (Fig. S7). The PSC to chromatin ratio shows a trend towards decreasing as the cell cycle progresses from G1 through S and into G2 at five PREs. PSC association with 3 of the 5 PREs is statistically significantly decreased in G2 vs. G1. Thus, PSC levels at some genes are reduced in G2 vs. G1, but not to the extent expected if PSC was segregated to newly replicated chromatin and no additional PSC added in S-phase. These data do not directly test our in vitro model, but are consistent with its predictions, as is the requirement for the region of PSC that mediates nucleosome bridging for maintaining silencing in flies (King et al., 2005; Wu and Howe, 1995) and the binding of PSC to newly replicated chromatin in cells (Francis et al., 2009). A recent report analyzing several PcG proteins (Pc, PHO, E(Z), and the modification H3K27me3) through S-phase using cells synchronized with hydroxyurea reported an increase in PcG protein binding at PREs prior to their replication (Lanzuolo et al., 2011). The difference observed by these authors is larger than what we observe in cells fractionated by elutriation although both data sets suggest that PcG levels at PREs are higher before than after DNA replication, consistent with segregation of PcG proteins, and with cell-cycle modulated recruitment of PcG proteins (Aoto et al., 2008; Hernandez-Munoz et al., 2005; Lanzuolo et al., 2011).
Are PcG proteins directed to newly replicated chromatin?
One question not answered by the experiments carried out to date is whether or how PSC or PCC might be specifically directed to newly replicated chromatin, as opposed to transferring to a more distal location in front of the fork. In our in vitro system where replication is carried out on plasmid templates, the possible distance the complexes can travel is limited, and ultimately they end up on newly replicated DNA since the whole template is replicated. If complexes make many transfers during the course of the reaction, these would not be observed. In vivo, if complexes transfer in either direction in an unconstrained manner, they will not remain located near target sites, and persistence through DNA replication would not facilitate maintenance of gene expression states. PCC binds tightly to ssDNA and it is possible that this contributes to recognition of replicating DNA, thereby promoting transfer to newly replicated chromatin. In support of interactions between PSC and ssDNA, the single stranded DNA binding protein RPA is enriched in purifications of PSC from Drosophila S2 cells (A. Wani, unpublished data). More experiments will be needed to test this idea, and more generally to determine whether PSC recognized features of the replication fork or newly replicated chromatin. In vivo, PcG proteins are targeted by DNA binding proteins, and their affinity for chromatin is also influenced by histone modifications. Either or both of these factors could also contribute to retaining complexes at specific sites, which can be tested in future experiments.
In summary, we describe a bridging mechanism by which PcG proteins can be transferred from unreplicated to replicated chromatin that mayallow PcG proteins to provide a continuous memory of silencing. The combination of chromatin binding and self-association is found in other chromatin proteins (Canzio et al., 2011; McBryant et al., 2006), raising the possibility that the mechanism described here may also be used by other epigenetic regulators.
EXPERIMENTAL PROCEDURES
Complete experimental protocols are available in the Extended Experimental procedures
Experiments were repeated at least three times with at least two different preparations of template and proteins unless indicated.
Proteins
Drosophila PcG proteins, human Swi/Snf, SV40 large T-Antigen and histones, were prepared as described (Francis et al., 2001; Landford, 1988). Active concentrations for DNA binding were measured for PSC and PCC (Francis et al., 2001); assuming proteins bind DNA as monomers, the active fraction is 0.2–0.3. All stated concentrations for PSC and PCC are active, except Fig. 5 in which total concentrations are used to compare active and inactive proteins.
Templates
Chromatin templates were prepared with plasmid pG5E4-SVO (Francis et al., 2009).
Replication of chromatin in SV40 cell-free system and chromatin remodeling
Replication of chromatin in the SV40 cell-free system using TAg and HeLa cell S100 extracts and chromatin remodeling of replication products, were carried out essentially as described (Francis et al., 2009).
Nucleosome bridging assay
Chromatinized plasmids or linear nucleosomal arrays were used at 40 or 80nM nucleosomes with 1–2 molar ratio of active protein to nucleosomes for PSC, PCC, PSC1–909 or PSC456–1603 and up to 5 molar ratio of total protein to nucleosomes for PSC1–572 or PSC456–909. Linker DNA was digested at 30°C for 5–10 min by the addition of 1U MNase (USB) 10 mM CaCl2, and 0.5 µg of a 29-bp dsDNA. Reactions were stopped with 15 mM EDTA and centrifuged through sucrose gradients. DNA from each fraction was resolved in agarose gels and visualized with SYBR gold stain (Invitrogen) on a Typhoon Imager. Mononucleosome bridging was carried out under similar conditions without MNase digestion.
Protein cross linking
Protein cross-linking was carried out with EDC, a zero length, amine-carboxyl cross-linker, and BM2(PEG), which cross-links sulfhydryls up to 14.7 Å as described (Lo and Francis, 2010).
Competition binding assay
Chromatin binding was carried out with biotinylated PCC and circular single stranded DNA as competitor. Competitor was added either during the initial binding step or after PCC was bound to chromatin. Reactions were cross-linked with formaldehyde to preserve binding during pull-down with streptavidin coated beads.
Analysis of binding to leading vs. lagging strand
See legend of Fig. 1 and Extended Experimental Procedures.
Supplementary Material
HIGHLIGHTS.
Mechanisms for inheritance of chromatin states are poorly understood
- PRC1 component PSC alone persists on chromatin through DNA replication in vitro
-
◦Self-interacting PSC can bridge nucleosomes to form stable structures
-
◦Nucleosome bridging allows rearrangement of chromatin contacts without PSC release
-
◦This mechanism may allow transfer of PRC1 from unreplicated to replicated chromatin
-
◦
Acknowledgements
We thank Dr. William Forrester for advice on elutriation. We thank Kyle McElroy and Drs. Sharad Ramanathan, Alex Schier, Erin O’Shea, Robert Kingston and Catherine Dulac for comments on the manuscript. This work was supported by a research grant to N.J.F. from the National Institutes of Health (GM78456).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aoto T, Saitoh N, Sakamoto Y, Watanabe S, Nakao M. Polycomb group protein-associated chromatin is reproduced in post-mitotic G1 phase and is required for S phase progression. J Biol Chem. 2008;283:18905–18915. doi: 10.1074/jbc.M709322200. [DOI] [PubMed] [Google Scholar]
- Bantignies F, Cavalli G. Polycomb group proteins: repression in 3D. Trends Genet. 2011 doi: 10.1016/j.tig.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Beh LY, Colwell LJ, Francis NJ. A core subunit of Polycomb repressive complex 1 is broadly conserved in function but not primary sequence. PNAS. 2012;109 doi: 10.1073/pnas.1118678109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunk BP, Martin EC, Adler PN. Drosophila genes Posterior Sex Combs and Suppressor two of zeste encode proteins with homology to the murine bmi-1 oncogene. Nature. 1991;353:351–353. doi: 10.1038/353351a0. [DOI] [PubMed] [Google Scholar]
- Canzio D, Chang EY, Shankar S, Kuchenbecker KM, Simon MD, Madhani HD, Narlikar GJ, Al-Sady B. Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol Cell. 2011;41:67–81. doi: 10.1016/j.molcel.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B, Li T, Rahl PB, Adamson TE, Loudas NB, Guo J, Varzavand K, Cooper JJ, Hu X, Gnatt A, et al. Functional association of Gdown1 with RNA polymerase II poised on human genes. Mol Cell. 2012;45:38–50. doi: 10.1016/j.molcel.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comet I, Schuettengruber B, Sexton T, Cavalli G. A chromatin insulator driving three-dimensional Polycomb response element (PRE) contacts and Polycomb association with the chromatin fiber. Proc Natl Acad Sci U S A. 2011;108:2294–2299. doi: 10.1073/pnas.1002059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean FB, Bullock P, Murakami Y, Wobbe CR, Weissbach L, Hurwitz J. Simian virus 40 (SV40) DNA replication: SV40 large T antigen unwinds DNA containing the SV40 origin of replication. Proc Natl Acad Sci U S A. 1987;84:16–20. doi: 10.1073/pnas.84.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmons RB, Genetti H, Filandrinos S, Lokere J, Wu CT. Molecular genetic analysis of Suppressor 2 of zeste identifies key functional domains. Genetics. 2009;182:999–1013. doi: 10.1534/genetics.108.097360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning E, Zhao K. SV40 DNA replication: from the A gene to a nanomachine. Virology. 2009;384:352–359. doi: 10.1016/j.virol.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- Francis NJ, Follmer NE, Simon MD, Aghia G, Butler JD. Polycomb Proteins Remain Bound to Chromatin and DNA during DNA Replication In Vitro. Cell. 2009:110–122. doi: 10.1016/j.cell.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis NJ, Kingston RE, Woodcock CL. Chromatin compaction by a Polycomb Group Protein Complex. Science. 2004;306:1574–1577. doi: 10.1126/science.1100576. [DOI] [PubMed] [Google Scholar]
- Francis NJ, Saurin AJ, Shao Z, Kingston RE. Reconstitution of a functional core Polycomb repressive complex. Mol Cell. 2001;8:545–556. doi: 10.1016/s1097-2765(01)00316-1. [DOI] [PubMed] [Google Scholar]
- Grau DJ, Chapman BA, Garlick JD, Borowsky M, Francis NJ, Kingston RE. Compaction of chromatin by diverse Polycomb group proteins requires localized regions of high charge. Genes Dev. 2011;25:2210–2221. doi: 10.1101/gad.17288211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Munoz I, Taghavi P, Kuijl C, Neefjes J, van Lohuizen M. Association of BMI1 with polycomb bodies is dynamic and requires PRC2/EZH2 and the maintenance DNA methyltransferase DNMT1. Mol Cell Biol. 2005;25:11047–11058. doi: 10.1128/MCB.25.24.11047-11058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jishage M, Malik S, Wagner U, Uberheide B, Ishihama Y, Hu X, Chait BT, Gnatt A, Ren B, Roeder RG. Transcriptional regulation by Pol II(G) involving mediator and competitive interactions of Gdown1 and TFIIF with Pol II. Mol Cell. 2012;45:51–63. doi: 10.1016/j.molcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman PD, Rando OJ. Chromatin as a potential carrier of heritable information. Curr Opin Cell Biol. 2010;22:284–290. doi: 10.1016/j.ceb.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King IF, Emmons RB, Francis NJ, Wild B, Muller J, Kingston RE, Wu CT. Analysis of a polycomb group protein defines regions that link repressive activity on nucleosomal templates to in vivo function. Mol Cell Biol. 2005;25:6578–6591. doi: 10.1128/MCB.25.15.6578-6591.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King IFG, Francis NJ, Kingston RE. Native and recombinant Polycomb-Group complexes establish a selective block to template accessibility to repress transcription in vitro. Mol Cell Biol. 2002;22:7919–7928. doi: 10.1128/MCB.22.22.7919-7928.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landford RE. Expression of simian virus 40 T antigen in insect cells using a baculovirus expression vector. Virology. 1988;167:72–81. doi: 10.1016/0042-6822(88)90055-4. [DOI] [PubMed] [Google Scholar]
- Lanzuolo C, Lo Sardo F, Diamantini A, Orlando V. PcG complexes set the stage for epigenetic inheritance of gene silencing in early S phase before replication. PLoS Genet. 2011;7:e1002370. doi: 10.1371/journal.pgen.1002370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzuolo C, Roure V, Dekker J, Bantignies F, Orlando V. Polycomb response elements mediate the formation of chromosome higher-order structures in the bithorax complex. Nat Cell Biol. 2007;9:1167–1174. doi: 10.1038/ncb1637. [DOI] [PubMed] [Google Scholar]
- Lavigne M, Francis NJ, King IF, Kingston RE. Propagation of silencing: recruitment and repression of naive chromatin in trans by Polycomb repressed chromatin. Mol Cell. 2004;13:415–425. doi: 10.1016/s1097-2765(04)00006-1. [DOI] [PubMed] [Google Scholar]
- Lewis EB. A gene complex controlling segmentation in Drosophila. Nature. 1978;276:565–570. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- Li HB, Muller M, Bahechar IA, Kyrchanova O, Ohno K, Georgiev P, Pirrotta V. Insulators, not Polycomb response elements, are required for long-range interactions between Polycomb targets in Drosophila melanogaster. Mol Cell Biol. 2011;31:616–625. doi: 10.1128/MCB.00849-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Kelly TJ. Simian virus 40 DNA replication in vitro. Proc Natl Acad Sci USA. 1984;81:6973–6977. doi: 10.1073/pnas.81.22.6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SM, Francis NJ. Inhibition of chromatin remodeling by polycomb group protein posterior sex combs is mechanistically distinct from nucleosome binding. Biochemistry. 2010;49:9438–9448. doi: 10.1021/bi100532a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet. 2010;11:285–296. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBryant SJ, Adams VH, Hansen JC. Chromatin architectural proteins. Chromosome Res. 2006;14:39–51. doi: 10.1007/s10577-006-1025-x. [DOI] [PubMed] [Google Scholar]
- Mohd-Sarip A, van der Knaap JA, Wyman C, Kanaar R, Schedl P, Verrijzer CP. Architecture of a polycomb nucleoprotein complex. Mol Cell. 2006;24:91–100. doi: 10.1016/j.molcel.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Muller J, Verrijzer P. Biochemical mechanisms of gene regulation by polycomb group protein complexes. Curr Opin Genet Dev. 2009;19:150–158. doi: 10.1016/j.gde.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Paro R. Imprinting a determined state into the chromatin of Drosophila. TIG. 1990;6:416–421. doi: 10.1016/0168-9525(90)90303-n. [DOI] [PubMed] [Google Scholar]
- Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol. 2009;10:192–206. doi: 10.1038/nrm2640. [DOI] [PubMed] [Google Scholar]
- Sanchez-Pulido L, Devos D, Sung ZR, Calonje M. RAWUL: a new ubiquitin-like domain in PRC1 ring finger proteins that unveils putative plant and worm PRC1 orthologs. BMC Genomics. 2008;9:308. doi: 10.1186/1471-2164-9-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Raible F, Mollaaghababa R, Guyon JR, Wu C-t, Bender W, Kingston RE. Stabilization of chromatin structure by PRC1, a polycomb complex. Cell. 1999;98:37–46. doi: 10.1016/S0092-8674(00)80604-2. [DOI] [PubMed] [Google Scholar]
- Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–79. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- Smith S, Stillman BW. Purification and characterization of CAF-1, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell. 1989;58:15–25. doi: 10.1016/0092-8674(89)90398-x. [DOI] [PubMed] [Google Scholar]
- Stahl H, Droge P, Knippers R. DNA helicase activity of SV40 large tumor antigen. Embo J. 1986;5:1939–1944. doi: 10.1002/j.1460-2075.1986.tb04447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillman B. Chromatin assembly during SV40 DNA replication in vitro. Cell. 1986;45:555–565. doi: 10.1016/0092-8674(86)90287-4. [DOI] [PubMed] [Google Scholar]
- Stillman BW, Gluzman Y. Replication and supercoiling of Simian Virus 40 DNA in cell extracts from human cells. Mol Cell Biol. 1985;5:2051–2060. doi: 10.1128/mcb.5.8.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G. A gene product required for correct initiation of segmental determination in Drosophila. Nature. 1981;293:36–41. doi: 10.1038/293036a0. [DOI] [PubMed] [Google Scholar]
- Waga S, Bauer G, Stillman B. Reconstitution of complete SV40 DNA replication with purified replication factors. J Biol Chem. 1994;269:10923–10934. [PubMed] [Google Scholar]
- Waga S, Stillman B. Anatomy of a DNA replication fork revealed by reconstitution of SV40 DNA replication in vitro. Nature. 1994;369:207–212. doi: 10.1038/369207a0. [DOI] [PubMed] [Google Scholar]
- Wobbe CR, Dean F, Weissbach L, Hurwitz J. In vitro replication of duplex circular DNA containing the simian virus 40 origin site. Proc Natl Acad Sci U S A. 1985;82:5710–5714. doi: 10.1073/pnas.82.17.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobbe CR, Weissbach L, Borowiec JA, Dean FB, Murakami Y, Bullock P, Hurwitz J. Replication of simian virus 40 origin-containing DNA in vitro with purified proteins. Proc Natl Acad Sci U S A. 1987;84:1834–1838. doi: 10.1073/pnas.84.7.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CT, Howe M. A genetic analysis of the Suppressor 2 of zeste complex of Drosophila melanogaster. Genetics. 1995;140:139–181. doi: 10.1093/genetics/140.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.