

Abstract

The genetic incorporation of one azide-containing and one keto-containing noncanonical amino acids into a protein at one amber and one ochre mutation sites respectively followed by their orthogonal reactions with one hydroxylamine-containing and one cyclooctyne-containing dyes allows highly efficient one-pot site-specific dual labeling of the protein in a catalyst-free fashion.
Keywords: Förster resonance energy transfer, dual labeling, noncanonical amino acid, amber suppression, ochre suppression
Förster resonance energy transfer (FRET) between a pair of donor and acceptor dyes is an invaluable tool to study dynamic protein conformational changes such as conformation rearrangement and folding/unfolding.[1] The efficiency of energy transfer that is dependent on the distance between the two dyes not only represents the conformational distributions but also reflects their change upon time. Two methods are usually applied to achieve dual labeling of a protein with a FRET pair. One is to genetically fuse two green fluorescent protein (GFP) variants at the N- and C-termini of a protein.[2] The other is to modify two cysteine residues in a protein with a small-molecule FRET pair.[1b] The GFP labeling approach has important advantages such as high labeling specificity and simplicity.[3] However, labeling a protein with two GFP variants is, in general, restricted at two termini. The size of GFP (~27 kDa) is also large enough to potentially interfere with the structures and functions of proteins to which they are fused. The cysteine labeling approach resolves issues such as site and size restrictions associated with the GFP labeling approach. It also has advantages such as the flexibility in choosing small molecule fluorophores and achieving labeling at the single-residue level. The straightforward labeling reactions and the commercial availability of many thiol-reactive dyes have made the cysteine labeling approach a favourable choice especially for the single-molecule FRET analysis.[4] However, the cysteine labeling approach requires mutating all non-targeted cysteine residues, and is therefore not applicable for proteins in which cysteine residues serve critical structural and/or functional roles. In addition, dual labeling of two cysteine residues, in general, leads to heterogeneously labeled proteins due to the lack of selectivity between two cysteine residues. To achieve site-selective labeling of a protein with a small-molecule FRET pair, several other methods have been developed.[5] These methods either work for specific proteins or are complicated so that their general applications are difficult. An alternative dual labeling approach was developed by Schultz and coworkers, in which a cysteine residue and a noncanonical amino acid (NAA), p-acetyl-L-phenylalanine coded by an amber codon were used to site-selectively dually label a protein.[6] Although elegant, this approach still requires mutating non-targeted cysteine residues and thus is not applicable to many proteins.
An ideal dual labeling approach that resolves the issues related to methods discussed above is to install two different bioorthogonal and chemically reactive groups into a protein followed by their selective reactions with corresponding dyes. One way to achieve this goal is to incorporate two different NAAs into a target protein. Recently Chin group and our group independently developed two methods for the genetic incorporation of two different NAAs into one protein.[7] Our double NAA incorporation method relies on the suppression of two stop codons, namely an amber UAG codon and an ochre UAA codon, which is achieved by genetically encoding two orthogonal pairs, an evolved M. jannaschii tyrosyl-tRNA synthetase pair[8] and a wild type or evolved pyrrolysyl-tRNA synthetase pair.[9] Herein, we show that this double NAA incorporation method can be applied to genetically install two different bioorthogonal functional groups into a protein, allowing catalyst-free and site-specific one-pot labeling of the protein with a FRET pair.
In our previous work, we showed that p-azido-phenylalanine and Nε-propargyloxycarbonyl-lysine (1 and 2 in Scheme 1) could be genetically incorporated into one protein in E. coli. We applied this approach to recombinantly synthesize glutamine binding protein (QBP) with 1 and 2 installed at its 3rd and 141st amino acid positions (QBP(1+2)) respectively and attempted to label QBP(1+2) specifically with a FRET pair (4 and 5 in Scheme 1) using two sequential copper(I)-catalyzed azide-alkyne Huisgen cycloaddition (CuAAC) reactions. Under an optimized reaction condition, we achieved close to 80% labeling of QBP(1+2) with 4 and 5 (please see the supporting information for the detailed labeling procedures and characterizations of the labeled protein). However, several issues arose from this labeling strategy. First, both two NAAs and two dyes are cross-reactive. Two labeling reactions of QBP(1+2) had to be carried out separately to avoid a cross reaction between two dyes and an excessive dye had to be provided during the first labeling step to avoid a cross reaction between 1 and 2 from two QBP(1+2) molecules. Second, CuAAC reactions induce protein aggregation and oxidation. During labeling QBP(1+2), we noticed a significant amount of protein aggregated. The finally dually labeled protein also showed an unexpected peak in its ESI-MS spectrum that is 18 Da more than the expected molecular weight (Supplementary Figure 3), indicating protein oxidation.
Scheme 1.
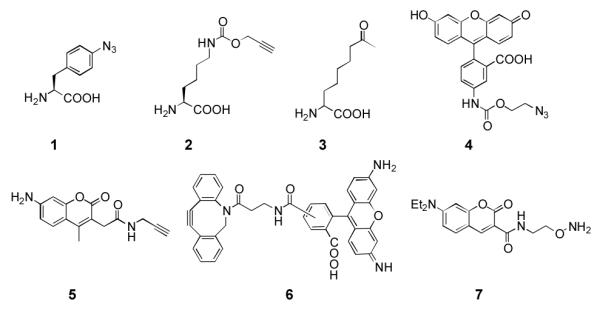
NAAs and dyes used in this study.
With these observed issues associated with CuAAC labeling reactions of QBP(1+2), we next sought to incorporate two other different NAAs into QBP for its straightforward labeling with a FRET pair. We reasoned that two NAAs with two bioorthogonal functional groups that undergo catalyst-free reactions and do not cross-react with each other would allow dual labeling of a protein incorporated with these two NAAs in a one-pot and catalyst-free fashion. This one-pot and catalyst-free dual-labeling strategy will not only simplify the labeling process but also avoid potential protein aggregation and oxidation problems caused by transition metal catalysts. Two NAAs that meet this requirement and can be potentially incorporated into a protein using our double NAA incorporation method are 1 and 2-amino-8-oxononanoic acid (3 in Scheme 1)[10]. These two NAAs undergo a copper-free azide-cyclooctyne click reaction and an oxime formation reaction, respectively. Although the rate of an azide-cyclooctyne click reaction is typically slower than a CuAAC reaction,[11] presumably there is no protein aggregation due to the lack of a transition metal catalyst so that the reaction time could be prolonged to achieve high labeling efficiency. To demonstrate labeling reactions of 1 and 3 are indeed orthogonal to each other, superfolder green fluorescent protein (sfGFP) with either 1 or 3 at its 2nd amino acid position was recombinantly synthesized and labeling reactions of the two purified proteins (sfGFP-1 and sfGFP-3) with both dyes 6 and 7 were then tested. To express sfGFP−1, E. coli BL21 cells were transformed with two compatible plasmids that carried genes coding an evolved MjTyRS (AzFRS) specific for 1, , and sfGFP with an amber mutation at its 2nd amino acid position and grown in 2YT medium supplemented with 1 mM 1; to express sfGFP-3, E. coli BL21 cells were transformed with two compatible plasmids that carried genes coding an evolved M mazei PylRS (AcKRS) specific for Nε-acetyl-lysine, ,[12] and sfGFP with an amber mutation at its 2nd amino acid position and grown in 2YT medium supplemented with 2 mM 3. Genetic incorporation of 3 at an amber mutation site mediated by an AcKRS- pair was demonstrated previously.[10] As shown in Figure 1, sfGFP-1 specifically reacted with 6 but not 7 and sfGFP-3 was selectively labeled with 7 but not 6, proving that an azide-cyclooctyne click reaction and an oxime formation reaction are indeed orthogonal to each other.
Figure 1.
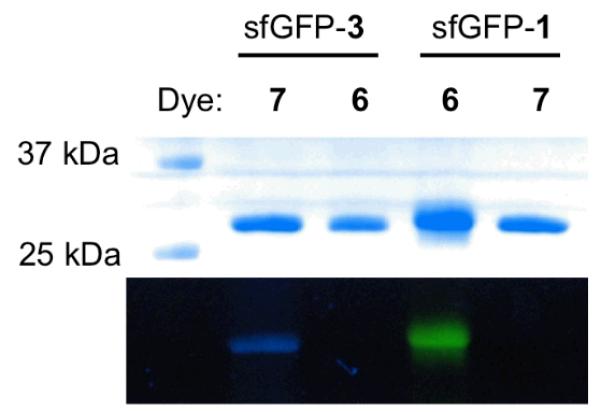
(A) Labeling sfGFP-1 and sfGFP-3 with 6 and 7. Top panel: Coomassie blue stained proteins in a SDS-PAGE gel; bottom panel: fluorescent imaging of the same gel under irradiation of 365 nm UV light. The image shows real colors captured by a regular camera.
Next, we tested whether it is applicable to genetically incorporate both 1 and 3 into a protein in E. coli using QBP as a model protein. To recombinantly synthesize QBP with 1 and 3 incorporated at its 3rd and 141st amino acid positions respectively (QBP(1+3)), E. coli BL21 cells were first transformed with two compatible plasmids that carried genes coding the pair, the pair, and QBP with an amber mutation and an ochre mutation at its 3rd and 141st amino acid positions respectively. Growing the transformed cells in 2YT medium supplemented with 1 mM 1 and 2 mM 3 led to overexpression of QBP(1+3) (Figure 2). The expression yield was 13 mg/L. Providing only one NAA or no NAA in the medium resulted in a negligible QBP expression level.
Figure 2.
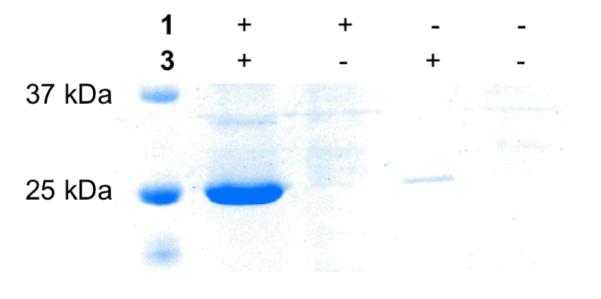
(A) Expression of QBP(1+3) in 2YT medium supplemented with different combinations of NAAs.
With QBP(1+3) in hands, we next performed a one-pot and catalyst-free dual-labeling process of QBP(1+3) using dyes 6 and 7. Incubating QBP(1+3) directly with 6 and 7 at pH 6.4 overnight resulted in close to quantitative conversion of the starting protein to the desired dual-labeled QBP(1+3) (QBP(1+3)-6-7), as proved by the ESI-MS spectrum of the final product (Figure 3E). The clean ESI-MS spectrum of QBP(1+3)-6-7 also indicated that no additional modification took place during the labeling process. Moreover, after affinity purification to remove unreacted dyes, the finally obtained QBP(1+3)-6-7 accounted for 83% of the original QBP(1+3). Two separate reactions to label QBP(1+3) with 6 and 7 individually were also carried out. Each led to close to quantitative conversion (Figures 3C&D). With a much simpler labeling procedure and a much better protein recovery yield, this one-pot catalyst-free dual-labeling method is undoubtedly a much better choice than dual labeling achieved by two sequential CuAAC reactions. One thing that needs to be pointed out though is the pH dependence of an oxime formation reaction with 3. We have also attempted to run the labeling of QBP(1+3) with 7 at pH > 8.0. However, overnight incubation only led to a negligible level of labeling.
Figure 3.

(A) Labeling QBP(1+3) with 6 and 7. Top panel: Coomassie blue stained proteins in a SDS-PAGE gel; bottom panel: fluorescent imaging of the same gel under irradiation of 365 nm UV light. The image shows real colors captured by a regular camera. Deconvoluted ESI-MS spectra of (B) QBP(1+3), (C) QBP(1+3) labeled with 7, (D) QBP(1+3) labeled with 6, and (E) QBP(1+3)-6-7. The theoretic molecular weights of QBP(1+3), QBP(1+3) labeled with 7, QBP(1+3) labeled with 6, and QBP(1+3)-6-7 are 26,028, 26,329, 26,661, and 26962 Da, respectively.
To demonstrate the application of QBP(1+3)-6-7 in its unfolding analysis, it was treated with different concentrations of guanidinium chloride (GndCl) and its fluorescent emission spectra under these conditions were measured. When excited by 430 nm light, QBP(1+3)-6-7 displayed improved fluorescent emission from 7 at 470 nm and a diminished fluorescent emission from 6 at 520 nm when the provided GndCl concentration increased (Figure 4A). This suggests the average distance between the two dyes increased when GndCl was used to unfold the protein. The inset of Figure 4A presents a smooth unfolding pattern determined by the change of I470nm/I520nm. Fitting the data to a standard two-state unfolding mechanism resulted in a Cm value of 3.1±0.1 M for GndCl.[13]
Figure 4.

(A) Fluorescent emission spectra of QBP(1+3)-6-7 at different concentrations of GndCl. The excitation wavelength was 430 nm. The inset shows the dependence of I470nm/I520nm on the concentration of GndCl. (B) (A) Fluorescent emission spectra of glutamine-free QBP(1+3)-6-7 and glutamine-bound QBP(1+3)-6-7.
Since QBP consists of two similar globular domains (amino acid residues 3 and 141 are located at two separate domains) linked by two peptide hinges and crystal structure data showed that glutamine binding to QBP causes its significant conformation change from an open conformation to a closed conformation,[14] we then tested whether we could use QBP(1+3)-6-7 to sense this conformation rearrangement process. Providing 1 mM glutamine[15] to saturate QBP(1+3)-6-7 led to its fluorescent emission decrease at 470 nm and increase at 520 nm (Figure 4B), indicating the distance between the two dyes decreased and therefore the protein changed to a more closed conformation. However, this FRET signal change is substantially smaller than what was calculated based on the distance change from the open conformation to the closed conformation. This may be due to two possibilities. First, introducing two dyes to QBP might cause it to take a more closed conformation. Second, the crystal structure of glutamine-free QBP might represent one of many glutamine-free QBP conformations in solution. What we measured might be an average two-chromophore distance change from all these conformations to the close conformation instead of the two-chromophore distance change between the open conformation and the close conformation shown in the crystal structures. Further investigations are necessary to address this observation.
In summary, we have developed an optimal protein dual-labeling method that can be carried out in a one-pot and catalyst-free fashion. The two reactions for the dual labeling process are both biocompatible. No treatment of proteins to avoid non-specific modifications with amino acid side chains such as cysteine thiols is necessary. The two reactions are orthogonal to each other and directed by two genetically incorporated NAAs, assuring the labeling specificity and selectivity and at the same time keeping other residues intact. Moreover, the two labeling reactions are also highly efficient, leading to almost quantitative labeling after an overnight incubation. The recovery yield of the finally labeled protein is also excellent. This simple and straightforward protein dual-labeling method resolves limitations associated with other current dual-labeling strategies and can be easily adopted by other research groups. Its potential applications range from single molecule FRET studies of protein dynamics to biosensor development.
Experimental Section
Except for dye 6 which was purchased from Click Chemistry Tools, all NAAs and dyes were synthesized. The synthesis of these compounds, plasmid constructions, protein expression and purification, protein labeling, and FRET analysis of QBP unfolding are provided in the supporting information.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institute of Health (grant 1R01CA161158), National Science Foundation (grant CHEM-1148684) and the Welch Foundation (grant A-1715). We thank Dr. Peter G. Schultz from Scripps Research Institute for providing us plasmid pEVOL-AzFRS, Dr. Dieter Söll from Yale University for providing us the AcKRS gene, and Dr. Yohannes H. Rezenom from Laboratory for Biological Mass Spectrometry at Texas A&M University for characterizing proteins with electrospray ionization mass spectrometry (ESI-MS).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- [1]a.Stryer L, Haugland RP. Proc Natl Acad Sci U S A. 1967;58:719–726. doi: 10.1073/pnas.58.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Morris SJ, Sudhof TC, Haynes DH. Biochim Biophys Acta. 1982;693:425–436. doi: 10.1016/0005-2736(82)90450-3. [DOI] [PubMed] [Google Scholar]; c Ha T, Enderle T, Ogletree DF, Chemla DS, Selvin PR, Weiss S. Proc Natl Acad Sci U S A. 1996;93:6264–6268. doi: 10.1073/pnas.93.13.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]a.Heim R, Tsien RY. Curr Biol. 1996;6:178–182. doi: 10.1016/s0960-9822(02)00450-5. [DOI] [PubMed] [Google Scholar]; b Suzuki Y, Yasunaga T, Ohkura R, Wakabayashi T, Sutoh K. Nature. 1998;396:380–383. doi: 10.1038/24640. [DOI] [PubMed] [Google Scholar]
- [3]a.Ting AY, Kain KH, Klemke RL, Tsien RY. Proc Natl Acad Sci U S A. 2001;98:15003–15008. doi: 10.1073/pnas.211564598. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Harpur AG, Wouters FS, Bastiaens PIH. Nature Biotechnology. 2001;19:167–169. doi: 10.1038/84443. [DOI] [PubMed] [Google Scholar]
- [4]a.Roy R, Hohng S, Ha T. Nat Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Margittai M, Widengren J, Schweinberger E, Schroder GF, Felekyan S, Haustein E, Konig M, Fasshauer D, Grubmuller H, Jahn R, Seidel CA. Proc Natl Acad Sci U S A. 2003;100:15516–15521. doi: 10.1073/pnas.2331232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5]a.Schuler B, Pannell LK. Bioconjug Chem. 2002;13:1039–1043. doi: 10.1021/bc025509t. [DOI] [PubMed] [Google Scholar]; b Talaga DS, Lau WL, Roder H, Tang J, Jia Y, DeGrado WF, Hochstrasser RM. Proc Natl Acad Sci U S A. 2000;97:13021–13026. doi: 10.1073/pnas.97.24.13021. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jager M, Nir E, Weiss S. Protein Sci. 2006;15:640–646. doi: 10.1110/ps.051851506. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jager M, Michalet X, Weiss S. Protein Sci. 2005;14:2059–2068. doi: 10.1110/ps.051384705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Brustad EM, Lemke EA, Schultz PG, Deniz AA. J Am Chem Soc. 2008;130:17664–17665. doi: 10.1021/ja807430h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7]a.Neumann H, Wang K, Davis L, Garcia-Alai M, Chin JW. Nature. 2010;464:441–444. doi: 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]; b Wan W, Huang Y, Wang Z, Russell WK, Pai PJ, Russell DH, Liu WR. Angew Chem Int Ed Engl. 2010;49:3211–3214. doi: 10.1002/anie.201000465. [DOI] [PubMed] [Google Scholar]
- [8]a.Wang L, Brock A, Herberich B, Schultz PG. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]; b Wang L, Schultz PG. Angew Chem Int Ed Engl. 2004;44:34–66. doi: 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]; c Xie J, Schultz PG. Methods. 2005;36:227–238. doi: 10.1016/j.ymeth.2005.04.010. [DOI] [PubMed] [Google Scholar]; d Xie J, Schultz PG. Nat Rev Mol Cell Biol. 2006;7:775–782. doi: 10.1038/nrm2005. [DOI] [PubMed] [Google Scholar]
- [9]a.Srinivasan G, James CM, Krzycki JA. Science. 2002;296:1459–1462. doi: 10.1126/science.1069588. [DOI] [PubMed] [Google Scholar]; b Kavran JM, Gundllapalli S, O’Donoghue P, Englert M, Soll D, Steitz TA. Proc Natl Acad Sci U S A. 2007;104:11268–11273. doi: 10.1073/pnas.0704769104. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Neumann H, Peak-Chew SY, Chin JW. Nat Chem Biol. 2008;4:232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]; d Wang YS, Wu B, Wang Z, Huang Y, Wan W, Russell WK, Pai PJ, Moe YN, Russell DH, Liu WR. Mol Biosyst. 2010;6:1557–1560. doi: 10.1039/c002155e. [DOI] [PubMed] [Google Scholar]; e Wang YS, Russell WK, Wang Z, Wan W, Dodd LE, Pai PJ, Russell DH, Liu WR. Mol Biosyst. 2011;7:714–717. doi: 10.1039/c0mb00217h. [DOI] [PubMed] [Google Scholar]
- [10].Huang Y, Wan W, Russell WK, Pai PJ, Wang Z, Russell DH, Liu W. Bioorg Med Chem Lett. 2010;20:878–880. doi: 10.1016/j.bmcl.2009.12.077. [DOI] [PubMed] [Google Scholar]
- [11].Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc Natl Acad Sci U S A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Umehara T, Kim J, Lee S, Guo LT, Soll D, Park HS. FEBS Lett. 2012;586:729–733. doi: 10.1016/j.febslet.2012.01.029. [DOI] [PubMed] [Google Scholar]
- [13].van Mierlo CP, van Dongen WM, Vergeldt F, van Berkel WJ, Steensma E. Protein Sci. 1998;7:2331–2344. doi: 10.1002/pro.5560071110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14]a.Hsiao CD, Sun YJ, Rose J, Wang BC. J Mol Biol. 1996;262:225–242. doi: 10.1006/jmbi.1996.0509. [DOI] [PubMed] [Google Scholar]; b Sun YJ, Rose J, Wang BC, Hsiao CD. J Mol Biol. 1998;278:219–229. doi: 10.1006/jmbi.1998.1675. [DOI] [PubMed] [Google Scholar]
- [15].Weiner JH, Furlong CE, Heppel LA. Arch Biochem Biophys. 1971;142:715–717. doi: 10.1016/0003-9861(71)90538-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.