

Abstract
Double strand DNA breaks are a serious threat to cellular viability and yeast systems have proven invaluable in helping to understand how these potentially toxic lesions are sensed and repaired. An important method to study the processing of DNA breaks in the budding yeast Saccharomyces cerevisiae is to introduce a unique double strand break into the genome by regulating the expression of the site-specific HO endonuclease with a galactose inducible promoter. Variations of the HO site-specific DSB assay have been adapted to many organisms, but the methodology has seen only limited use in the fission yeast Schizosaccharomyces pombe because of the lack of a promoter capable of inducing endonuclease expression on a relatively short time scale (∼1 hour). We have overcome this limitation by developing a new assay in which expression of the homing endonuclease I-PpoI is tightly regulated with a tetracycline inducible promoter. We show that induction of the I-PpoI endonuclease produces rapid cutting of a defined cleavage site (>80% after 1 hour), efficient cell cycle arrest, and significant accumulation of the checkpoint protein Crb2 at break-adjacent regions in a manner that is analogous to published findings with DSBs produced by an acute exposure to ionizing irradiation. This assay provides an important new tool for the fission yeast community and, because many aspects of mammalian chromatin organization have been well-conserved in S. pombe but not in S. cerevisiae, also offers an attractive system to decipher the role of chromatin structure in modulating the repair of double stranded DNA breaks.
Keywords: DNA damage, double strand break, chromatin, histone modification, Schizosaccharomyces pombe, tetracycline, endonuclease
Introduction
Every day cells contend with a constant barrage of agents that can damage their genetic material and a failure to effectively respond to DNA damage can lead to debilitating human disorders (Ciccia and Elledge, 2010). Of the many different types of DNA lesions, double strand breaks (DSBs) are probably the most dangerous (Jackson, 2002). DSBs can arise during normal cellular activities, such as DNA replication, or after cells have been exposed to environmental agents, such as ionizing irradiation (IR). DSBs are difficult to repair because both strands of the DNA template are severed and even a single unrepaired DSB can cause cell death (van Gent et al., 2001; Rich et al., 2000). Deciphering how cells contend with potentially toxic DSBs is central to understanding and treating numerous diseases including cancer (Jackson and Bartek, 2009; O'Driscoll and Jeggo, 2006).
A number of different methods have been developed to produce and study cellular DSBs (Polo and Jackson, 2011). IR is probably the most commonly used technique and produces DSBs in a dose dependent manner by a mechanism that is thought to involve the localized attack of each DNA strand by a cluster of •OH radicals (Ward, 1985). An important breakthrough was the discovery that the recruitment of many proteins involved in DSB response can be visualized as microscopic foci that form at regions surrounding sites of IR produced DSBs (Dellaire and Bazett-Jones, 2007). The nature of these microscopic foci is not entirely clear, but probably reflects the binding of hundreds of protein molecules to break adjacent regions as a mechanism to amplify the damage signal. The “IR foci formation” assay has proven to be an invaluable tool, but a limitation of IR experiments is the random nature of break formation and the relative low-resolution microscopy used to follow protein binding.
An alternative approach that overcomes the limitations of IR is to use a site-specific endonuclease to introduce a DSB break at a defined genomic location. The site-specific DSB assay was pioneered in the budding yeast Saccharomyces cerevisiae where expression of the HO endonuclease can be tightly controlled with a galactose-inducible promoter (Sugawara and Haber, 2006; Haber, 2002). Incubation in galactose containing media induces HO expression and constitutive cutting of a unique ∼22 bp cleavage site located in the MAT locus. Because break formation occurs at a defined site, Southern blotting and other assays can be used to follow repair intermediates and chromatin immunoprecipitation (ChIP) can be used to monitor the binding of proteins to the break region with high resolution (Sugawara and Haber, 2006; Haber, 2002). It has also been possible to investigate the influence of the surrounding chromatin landscape on DSB response by relocating the HO cleavage site to different regions of the S. cerevisiae genome (Kim et al., 2007). Site-specific DSB assays have also proven to be an effective tool in higher organisms using a number of different nucleases including I-SceI (Jasin, 1996; Bellaiche et al., 1999), I-PpoI (Berkovich et al., 2007), and AsiSI (Iacovoni et al., 2010; Massip et al., 2010).
A version of the HO system was also initially translated to the fission yeast Schizosaccharomyces pombe by regulating expression of the HO endonuclease with a thiamine repressed nmt promoter (Du et al., 2003; Osman et al., 1996; Prudden et al., 2003). The S. pombe genome lacks a HO recognition site unless it is specifically introduced. DSB formation occurs when cells are transferred from repressive thiamine containing media to media lacking thiamine to induce HO expression. Although the nmt-HO system has provided new insights into DSB biology (Langerak et al., 2011; Moss et al., 2010; Tinline-Purvis et al., 2009; Du et al., 2006; Prudden et al., 2003), the system has not seen widespread use. A major issue is that >20 hours is required for HO induction with the nmt promoter, a time period that is 5-10 times longer than the generation time for a typical S. pombe cell and significantly longer than the ∼1 hour time scale needed for galactose controlled induction in S. cerevisiae. The extended induction period introduces technical complications and limits the ability to perform intensive time course and cell cycle studies. Further, observations indicate that when the nmt promoter is used to drive HO expression and DSB formation over a >20 hour period, the mechanisms used by proteins to bind to the break region can be different from those used when DSB formation occurs in a matter of minutes after an acute exposure to IR (Du et al., 2006).
S. cerevisiae and S. pombe have proven to be invaluable model systems for deciphering the network of DSB response proteins (Ciccia and Elledge, 2010; Harrison and Haber, 2006; Raji and Hartsuiker, 2006; Symington, 2002). A major unresolved question now is to understand how the packaging of DNA into chromatin modulates the sensing and repair of DSBs (Downs et al., 2007; Peterson and Cote, 2004). Cells possess an abundance of enzymatic activities, such as histone post-translational modification, that are used to modify chromatin structure and control access to the DNA template (Bannister and Kouzarides, 2011). Mounting evidence argues that many chromatin modifiers play a key role in protecting genomes from hazardous DSBs and suggest that the location of a break in either an active euchromatin or repressive heterochromatin domain can modulate repair (Xu and Price, 2011; Goodarzi et al., 2009). S. pombe provides an attractive system to address such questions because this yeast shares many key features of mammalian chromatin structure that are notably absent from S. cerevisiae, such as large highly repetitive centromeres, RNAi machinery, di and tri-methylation of histone H4 lysine 20 (H4K20), and heterochromatin domains marked by histone H3K9 methylation and HP1-like proteins, (Grewal, 2000; Grewal and Moazed, 2003; Sanders et al., 2004). However, the lack of a system capable of introducing a site-specific DSB into the S. pombe genome on a relatively short time scale (∼1 hour) has hampered efforts and a fast inducing technology would be beneficial to many researchers.
To overcome the limitations of the nmt-HO system Watson et al very recently described a new site-specific DSB assay for S. pombe in which expression of the HO endonuclease is controlled by the endogenous urg1 promoter that is rapidly turned on adding uracil to growth media ((Watson et al., 2011), see also latter discussion). In parallel work we sought to develop a S. pombe site-specific DSB assay with fast kinetics using a heterologous promoter that does not require the use of a metabolic inducer. We present such a system in which expression of the homing endonuclease I-PpoI is tightly controlled with a tetracycline inducible promoter. We show that induction of the I-PpoI endonuclease produces rapid cutting of a defined cleavage site (>80% after 1 hour), efficient cell cycle arrest, and significant accumulation of the checkpoint protein Crb2 at break-adjacent chromatin. Importantly, Crb2 targeting to the I-PpoI generated break requires the activity of the histone H4K20 methyltransferase Kmt5, an observation that mimics published results with IR generated DSBs (Du et al., 2006; Sanders et al., 2004). These findings illustrate the utility of our site-specific DSB assay and provide a powerful new tool to dissect the complex interplay between chromatin structure and the repair of DNA breaks.
Materials and methods
Plasmids
To produce the tetracycline inducible I-PpoI expression plasmid pSS12 (Figure 1A), the I-PpoI ORF was PCR amplified from plasmid pET-Ppo (Flick et al., 1997) using oligos 470/471 incorporating I-CeuI sites at each end (Table I). The amplified fragment was cloned into the I-CeuI sites of pDUAL-tet-rpsL-neo (Erler et al., 2006) such that the I-PpoI ORF replaced the rpsL-neo fragment. The complete ORF was verified by DNA sequencing. A pDUAL derived tetracycline inducible HO expression plasmid (pSS13) was generated in a similar fashion from plasmid pREP81-X-HO (Prudden et al., 2003) using oligos 468/469 (Table I). The vector control pDUAL-tet plasmid has been described (Erler et al., 2006). A second I-PpoI expression plasmid, pSS16 (Figure 1B), was created such that a linear DNA fragment containing the entire tetracycline-I-PpoI control unit from pSS12 and a clonNAT selectable marker (nat+) could be inserted immediately upstream of the ATG start codon in the ORF of arg3+ (Figure 1D). Plasmids pSS21 and pSS23 (Figure 1C) were created to integrate a linear DNA fragment containing a single I-PpoI cleavage site followed by a hygromycin B selectable marker (hph+) immediately upstream of the ATG start codon in the ORFs of either ura5+ or lys1+, respectively (Figure 1E). Plasmid pSLS466 was also created to integrate a single HO cleavage site at lys1+ in a similar fashion. All plasmids and associated information will be made available through Addgene (http://www.addgene.org/) upon publication.
Figure 1.

Plasmids and integration strategies. Tetracycline inducible I-PpoI integration plasmids are shown in (A) and (B), I-PpoI cleavage site integration plasmids in (C), and integration strategies in (D) and (E) (not drawn to scale). (A) The I-PpoI ORF was cloned under the control of the CaMV35S promoter (p) and the nmt1 terminator (t) in the pDUAL-tet-rpsL-neo vector (Erler et al., 2006) to create pSS12. The plasmid also contains a tetracycline repressor (TetR) that is constitutively expressed from an ADH1 promoter and terminator. The TETp-I-PpoI NotI fragment from pSS12 (sites shown) is excised and integrated at leu1-32 as detailed (Matsuyama et al., 2004). (B) A second I-PpoI expression plasmid was created by transferring the entire TETp-I-PpoI control unit from pSS12 into a plasmid such that it was flanked by a clonNAT selectable marker (nat+) and sequences homologous to the regions immediately up (5′) and downstream (ORF) of the arg3+ ATG start codon. The TETp-I-PpoI NotI fragment is excised and integrated as shown in (D). PCR primers (355/512 Table I) used to verify correct integration at arg3+ are illustrated as half arrows. (C) To integrate an I-PpoI cleavage site at ura5+, pSS21 was created such that a single cleavage site was flanked by a hygromycin B selectable marker (hph+) marker and sequences homologous to the regions immediately up (5′) and downstream (ORF) of the ura5+ ATG start codon. A NotI fragment with the cleavage site (black triangle) is then excised and integrated at ura5+ as shown in (E). PCR primers used to verify correct integration across each junction (396/611 and 397/612 Tabel I) are illustrated. The pSS23 plasmid was created in a similar fashion for integration of a single I-PpoI cleavage site immediately upstream of the lys1+ ATG start codon (E). Verification primers for lys1+ integration correspond to 396/844 and 397/356 (Table I). For (B) and (C) plasmids, integrations produce an auxotrophic phenotype for the corresponding amino acid that can be used as a second means to verify correct integration. See Material and methods for further details.
Table I.
Oligos.
| Oligo | Sequence |
|---|---|
| 355 arg3 ORF r2 | 5′-TACCAACCTTGGCATTTGCAATC-3′ |
| 356 lys1 ORF r2 | 5′-CAGAAGGAGCGTCTGGAGCATTG-3′ |
| 358 lys1 RT | 5′-GAAGGTTTTAGGCAGTTCGAAC-3′ |
| 361 lys1 RTR | 5′-TTGTTTGACTCTTAAGTCTCTCAG-3′ |
| 396 SV40p rev | 5′-ATGGATATACAAGCTCCCGG-3′ |
| 397 SV40t fwd | 5′-GAGATTTCGATTCCACCGCC-3′ |
| 468 HO ATG I-CeuI | 5′-GATCGATAACTATAACGGTCCTAAGGTAGCGAATGCTTTCTGAAAACACGACTATTC-3′ |
| 469 HO Stop I-CeuI | 5′-GATCGATCGCTACCTTAGGACCGTTATAGTTATTAGCAGATGCGCGCACCTGCGTTG-3′ |
| 470 Ipo ATG I-CeuI | 5′-GATCGATAACTATAACGGTCCTAAGGTAGCGAATGGCGCTCACCAATGCTCAAATC-3′ |
| 471 Ipo Stop I-CeuI | 5′-GATCGATCGCTACCTTAGGACCGTTATAGTTATTATACCACAAAGTGACTGCCCCTTTG-3′ |
| 512 ADHt fwd | 5′-AGTATGAGGTCGCTCTTATTGAC-3′ |
| 516 rRNA.22F | 5′-CGCAATGTGATTTCTGCCCAGTG-3′ |
| 517 rRNA.22R | 5′-GTGGGAATCTCGTTAATCCATTC-3′ |
| 544 ura5 RT fwd | 5′-GCTTTTTTCTTTTGTGAGTATCAACG-3′ |
| 545 ura5 RT rev | 5′-GTAAATACCAACTAACTTCACTCTC-3′ |
| 611 ura5 5′fwd3 | 5′-TGGACTGGAGACTCACATGTCAG-3′ |
| 612 ura5 ORF rev2 | 5′-GCAGTTCCAGCGGTAATAACATC-3′ |
| 618 .5 ura5 up | 5′-GGACAGCCATTGTAACTTATGAAC-3′ |
| 619 .5 ura5 dn | 5′-GTACCATTTATTGACTGGATTAGTC-3′ |
| 620 2.0 ura5 up | 5′-ATTCCATTTCCTCTGGTGAGAATG-3′ |
| 621 2.0 ura5 dn | 5′-CCAATAAATGCTATAAACGAACTCC-3′ |
| 622 10.0 ura5 up | 5′-AGCCTGAAAATGTGGATATTAAAGC-3′ |
| 623 10.0 ura5 dn | 5′-ACAAACTCAGGTCCTAAGAAAAGC-3′ |
| 624 30.0 ura5 up | 5′-AATTCCCAGTTTCCGTGTGCAAG-3′ |
| 625 30.0 ura5 dn | 5′-GTAGGGTCAGATACGTAAATAGTC-3′ |
| 635 lys1 RTR2 | 5′-GAACAAGAGTTTTACGAGGGTCATC-3′ |
| 636 lys1 RT3 | 5′-ACCTTTTAGCCAAAGTGTGCGATC-3′ |
| 637 5.0 ura5 up | 5′-ATCGATATCGCTCGCTGGTAGTAC-3′ |
| 638 5.0 ura5 dn | 5′-ACACTAGCTGGAAATGAGCATAGTG-3′ |
| 724 rRNA.22 fwd2 | 5′-CTGTCTAATTAAAACATAGCATTGC-3′ |
| 725 rRNA.22 rev2 | 5′-CCAGGCTGTGGTTTCGCTAGATAG-3′ |
| 844 lys1 5′ fwd | 5′-GAGTCATCTACAATACTGGC-3′ |
| 914 lys1 5′ I-PpoI | 5′-TCGAACATTTGACACTCTCCG-3′ |
| 916 rDNA 5′ I-PpoI | 5′-CAATGTGATTTCTGCCCAGTG-3′ |
| 917 rDNA 3′ I-PpoI | 5′-GGCTGTGGTTTCGCTAGATAG-3′ |
| 920 his3 RT For | 5′-TTACCAAGCCACTAACACCAG-3′ |
| 921 his3 RT Rev | 5′-GCAGAGACCGTATACATTCCG-3′ |
| 924 I-PpoI hph 3′ | 5′-CCACACCCTAACTGACAAGATC-3′ |
Underlined sequence denotes I-CeuI sites.
Strains, media and manipulations
Standard laboratory methods for fission yeast manipulations were used (Moreno et al., 1991) and all relevant strains are listed in Table II and derived from 1913 (NCYC, Norwich, UK). For routine growth and drug selection after transformation rich media (YES) was used with the following: 60 μg/ml blasticidin S (bsd, InvivoGen), 100 μg/ml clonNAT (nat, Werner BioAgents), 100 μg/ml G418 (kan, Mediatech) and 300 μg/ml hygromycin B (hph, Mediatech). For all anhydrotetracycline (ahTET) experiments, Edinburgh minimal medium with 3.6 g/L sodium glutamate substituted for ammonium chloride and supplemented with the appropriate amino acids was used (EMMG). A 10 mM stock of ahTET (Acros 13803-65-1) was prepared in sterile water and stored at 4°C protected from light. To prepare ahTET-EMMG agar plates, ahTET was added to the media after autoclaving and once plates were poured, they were stored at room temperature protected from light. Integration of pDUAL-derived plasmids at leu1-32 was performed as detailed (Matsuyama et al., 2004). Integration of the HO cleavage site at lys1+, the I-PpoI cleavage site at lys1+ or ura5+ (Figure 1E), and the tetracycline inducible I-PpoI fragment at arg3+ (Figure 1D) were performed by standard transformation using linear NotI fragments from the appropriate plasmids (above). Correct integration at each locus was verified by PCR using oligo pairs spanning each junction site (illustrated Figures 1D and 1E). Oligos used: 355/512 at arg3+ (expected product 628 bp); 396/611 and 397/612 at ura5+ (expected products 773 and 679 bp, respectively); 396/844 and 397/356 at lys1+ (expected products 807 and 810 bp, respectively). Integrations at lys1+, ura5+ and arg3+ induced an auxotrophic phenotype for the corresponding amino acid that provided a second method to verify correct integration. GFP-Crb2 tagging and kmt5 knockouts have been described (Sanders et al., 2004).
Table II.
Fission Yeast Strains.
| Strain | Genotype |
|---|---|
| 1913 | h- leu1-32 |
| YSS15 | h- leu1-32 lys1∷HOCS-kanMx6 |
| YSS151 | h- leu1-32∷pDUAL-TETp-I-PpoI |
| YSS154 | h- leu1-32∷pDUAL-TETp |
| YSLS798 | h- leu1-32∷pDUAL-TETp-HO |
| YSLS802 | h- leu1-32∷pDUAL-TETp lys1∷HOCS-kan+ |
| YSLS803 | h- leu1-32∷pDUAL-TETp-I-PpoI lys1∷HOCS-kan+ |
| YSLS804 | h- leu1-32∷pDUAL-TETp-HO lys1∷HOCS-kan+ |
| YSLS790-3 | h- leu1-32∷pDUAL-TETp-I-PpoI rDNAI-PpoImt |
| YSLS792 | h- leu1-32∷pDUAL-TETp-I-PpoI rDNAI-PpoImt lys1∷I-PpoICS-hph+ |
| YSLS793 | h- leu1-32∷pDUAL-TETp-I-PpoI rDNAI-PpoImt lys1∷HOCS-hph+ |
| YSLS702 | h- leu1-32∷pJK148-REP81-GFP-crb2+ crb2Δkan+ |
| YSS197 | h- leu1-32∷pJK148-REP81-GFP-crb2+ crb2Δkan+ arg3∷TETp-I-PpoI-nat+ |
| YSS226 | h- leu1-32∷pJK148-REP81-GFP-crb2+ crb2Δkan+ arg3∷TETp-I-PpoI-nat+ ura5∷I-PpoICS-hph+ |
| YSS271 | h- leu1-32∷pJK148-REP81-GFP-crb2+ crb2Δkan+ arg3∷TETp-I-PpoI-nat+ rDNAI-PpoImt |
| YSS289 | h- leu1-32∷pJK148-REP81-GFP-crb2+ crb2Δkan+ arg3∷TETp-I-PpoI-nat+ rDNAI-PpoImt ura5∷I-PpoICS-hph+ |
| YSLS783 | h- leu1-32∷pJK148-REP81-GFP-crb2+ crb2Δkan+ arg3∷TETp-I-PpoI-nat+ rDNAI-PpoImt ura5∷I-PpoICS-hph+ kmt5Δbsd+ |
ahTET killing assay
Strains were grown to log phase at 30°C in EMMG and 1 × 106 cells were pelleted in a 1.5 ml siliconized tubed by a brief centrifugation, the media was aspirated, cells resuspended in 500 μL of sterile water and 250 μL transferred to a 96 well micro-titer dish. Serial 1:5 dilutions were then prepared in sterile water and 5.0 μL spotted onto EMMG plates containing the appropriate amount of ahTET. Plates were incubated at 30°C for 2-3 days protected from light.
Growth curves
Strains grown to log phase at 30°C in EMMG were diluted back to equivalent densities (0.5 × 106 cells/ml) in fresh media and allowed to recover for 3 hours at 30°C. At the 0 hour time point cultures were split in half, one half was untreated and ahTET (3 μM final) added to the second. Growth at 30°C was then monitored by cell counting and cell aliquots were fixed in methanol for DAPI staining.
I-PpoI cutting assay
To log phase cultures grown at 30°C in EMMG, ahTET (3 μM final) was added to induce I-PpoI expression and 5 × 107 cells harvested at times indicated after induction. Genomic DNA was prepared as described (Bahler et al., 1998), RNase treated and DNA concentrations determined using a NanoDrop Spectrophotometer. For Figure 3, quantitative real-time PCR (qPCR) reactions were performed in triplicate with a portion of each DNA sample using Bio-Rad SYBR Green mix (170-8882) on a Bio-Rad thermal cycler (CFX96) using oligos (Table I) 516/517 spanning the I-PpoI cleavage site in the rDNA repeats and oligos 358/361 that anneal at uncut lys1+. The total amount of product amplified in each reaction was determined versus a standard curve of titrated genomic DNA for each oligo pair and the amount of rDNA product was then normalized against the amount of uncut lys1+ product. The relative amount of rDNA product was then calculated versus the uncut 0 hour control. Cutting at the I-PpoI cleavage site integrated at ura5+ was determined in a similar fashion using oligos 544/545 (Table I).
Figure 3.
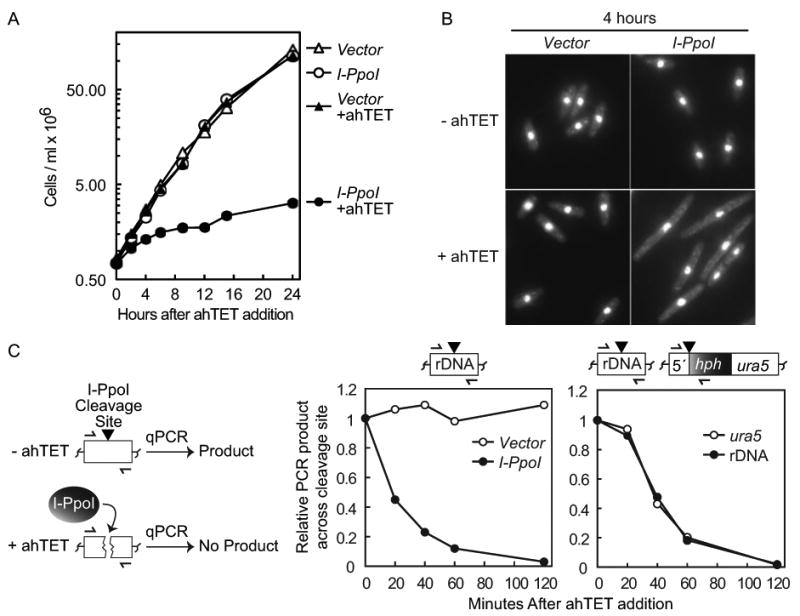
Induction of I-PpoI expression efficiently inhibits cell growth and produces rapid DSB formation. (A) Growth curve after I-PpoI induction. Strains with either an integrated empty vector (YSS154) or a tetracycline inducible I-PpoI allele (YSS151) were grown in EMMG with (+) or without 3 μM ahTET and growth was monitored by cell counting. Time across the x-axis denotes hours after ahTET addition. (B) I-PpoI expression produces cell elongation. Cells at the 4 hour time point in A were fixed in methanol and visualized by DAPI staining. Images have been intentionally overexposed to show the entire cell body. (C) Rapid cutting of endogenous and exogenous I-PpoI cleavage sites. Left, illustration of quantitative real-time PCR (qPCR) I-PpoI cutting assay, half arrows denote oligos that span the I-PpoI cleavage site (black triangle). See text for further details. Middle panel, rapid I-PpoI cleavage at the rDNA repeats. Strains with either integrated empty vector (YSS154) or a tetracycline inducible I-PpoI allele (YSS151) were treated with 3 μM ahTET and cell aliquots taken at the time points indicated after induction. Genomic DNA was prepared and qPCR used to monitor product formation across the I-PpoI cleavage sites located in the rDNA repeats (illustrated top). qPCR product formation at the uncut lys1+ locus was used as a normalization control. See Materials and methods for further details. Right panel, rapid I-PpoI cutting at both a single exogenous I-PpoI cleavage site and at the rDNA repeats. I-PpoI expression was induced in strain YSS226 containing both endogenous rDNA PpoI cleavage sites and a single cleavage site integrated at ura5+ (illustrated top). qPCR was then used to monitor product formation across both cleavage sites as described above.
Figure 5 cutting assays were performed as detailed above except that qPCR reactions were performed with a LC480 II thermal cycler (Roche) using Fast Start Universal SYBER Green Mix (Roche 04913850001). Oligos (Table I) 916/917 and 914/924 were used to monitor product formation across the rDNA and lys1+ I-PpoI cleavage sites, respectively. Oligos 920/921 that anneal in the his3+ ORF were used as a normalization control.
Figure 5.
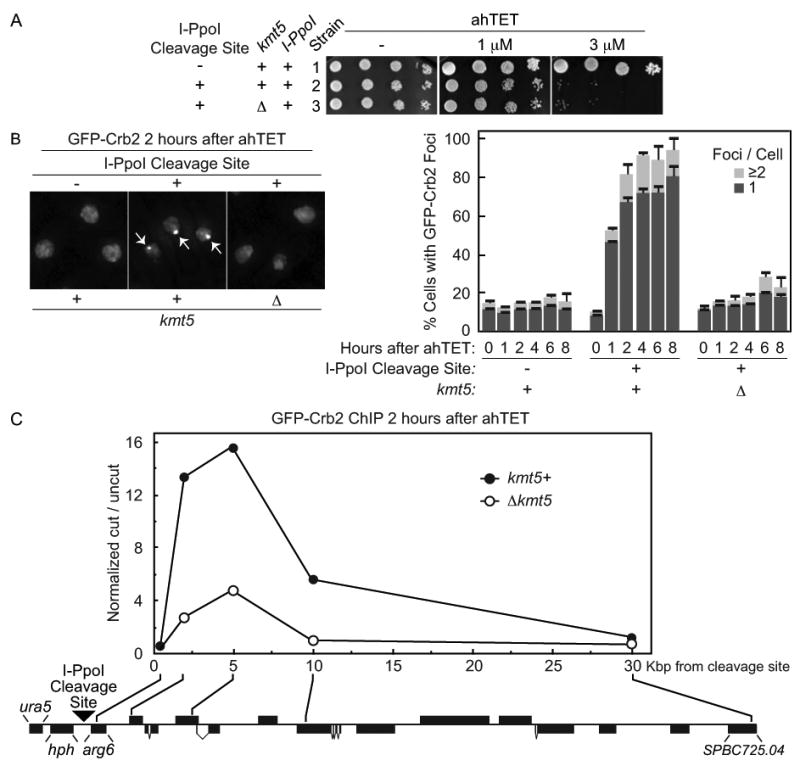
The checkpoint protein Crb2 efficiently accumulates at an I-PpoI induced break in a manner that requires the H4K20 methylase Kmt5. (A) ahTET sensitivity of relevant strains. Strain 1 (YSS271) with an integrated tetracycline inducible I-PpoI allele, I-PpoI resistant rDNA repeats and GFP tagged crb2+ was generated and a single exogenous I-PpoI cleavage site then integrated at ura5+ (illustrated in (C)). to produce strain 2 (YSS289). The Kmt5 ORF was then replaced with a bsdMx6 marker to create Δkmt5 strain 3 (YSLS783). Spot tests were performed as detailed for Figure 2. (B) Live cell microscopy of GFP-Crb2 after induction of I-PpoI expression. The ahTET inducer was added to growing cultures of strains detailed in (A) to induce I-PpoI expression and live cell microscopy was performed at times indicated after ahTET addition. Left, representative images 2 hours after ahTET addition. Note that the nucleus of 3 different cells is denoted in each panel by GFP-Crb2 staining which is entirely nuclear. Arrowheads in middle panel denote GFP-Crb2 foci. Right, quantification of GFP-Crb2 foci, numbers were averaged from at least 3 independent experiments with ≥ 200 cells counted for each point. (C) ChIP of GFP-Crb2 at an I-PpoI induced break. Strains 2 and 3 from A were processed for anti-GFP ChIP 2 hours after induction of I-PpoI expression as described in Materials and methods. The relative enrichment of GFP-Crb2 at the break site was calculated by normalizing the cut/uncut ratio from each oligo pair flanking the cleavage site versus the cut/uncut ratio for an oligo pair at uncut lys1+. The location of the integrated I-PpoI cleavage site (black triangle), the hygromycin B selection mark (hph) and the approximate position of each oligo pair in the ura5-SPBC725.04 region of chromosome II is shown (drawn to scale, black boxes denote annotated ORFs, illustrated in the reverse complement orientation).
Isolation of I-PpoI resistant yeast strains
Typically, 5 × 103 cells were spread onto EMMG plates with 3 μM ahTET and grown at 30°C. Colonies resistant to ahTET were then struck onto fresh EMMG plates with 3 μM ahTET and further validated by performing a spot test killing assay (above). Genomic DNA was prepared from true ahTET resistant clones and rDNA mutation was confirmed by sequencing a PCR product spanning the I-PpoI cleavage site (oligos 724/725 Table I).
Microscopy and ChIP
Live cell microscopy of GFP-Crb2 was performed as described (Sanders et al., 2010). The general procedure for ChIP was adapted from (Pidoux et al., 2004). Cultures were grown to log phase at 30°C in EMMG and then split in half, one half was untreated (uncut) and ahTET (3 μM final) was added to the second (cut). Both cultures were incubated 2 more hours at 30°C, fixed for 30 minutes at 25°C by adding formaldehyde to a final concentration of 1% and glycine was added to a final concentration of 2.5 M to stop fixation. Cells (5 × 108) were harvested, washed with cold PBS, resuspended in 2.5 ml of spheroplasting buffer (0.1 M PIPES pH 7.0, 1.0 mM EGTA, 1 mM magnesium sulfate, 1.2 M sorbitol, 1.2 mg/ml Sigma lysing enzyme, 0.8 mg/ml zymolyase 100T) and digested at 37°C for 20 minutes. Cells were then washed twice in 10 ml cold PEMS (100 mM PIPES pH 7.0, 1.0 mM magnesium chloride, 1.0 mM EDTA, 1.2 M sorbitol). The cell pellet was resuspended in 400 μl of lysis buffer (50 mM HEPES pH 7.5, 140 mM sodium chloride, 1.0 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 2.0 mM PMSF, complete –EDTA protease inhibitor (Roche)) and sonicated for 10 cycles at maximum power (30 seconds on, 60 seconds off) in a Diagenode Bioruptor. The lysate was centrifuged for 5 minutes at 4°C at 13K rpm, the supernatant transferred to a new tube and centrifuged for an additional 15 minutes before collecting the final supernatant. The supernatant was mixed with 50 μl of a 1:1 slurry of Protein A agarose/salmon sperm DNA beads (Millipore 16-157) for 2 hours at 4°C and the beads were pelleted with a brief centrifugation before collecting the cleared lysate. A 40 μl aliquot of the cleared lysate was saved (input) and the remaining 360 μl incubated with 50 μl 1:1 Protein A agarose/salmon sperm DNA beads and 5 μg of anti-GFP (Abcam ab6556) overnight at 4°C. Immunoprecipitates were sequentially washed for 5 minutes with mixing at 25°C with: 1 ml lysis buffer, 1 ml lysis buffer plus 0.5 M NaCl, 1 ml wash buffer (10 mM Tris-HCl pH 8.0, 0.25 M lithium chloride, 0.5% NP-40, 0.5% sodium deoxycholate, 1.0 mM EDTA) and 1 ml TE pH 8.0. Beads were resuspended in 300 μl of TES (50 mM Tris-HCl pH 8, 10 mM EDTA, 1% SDS) plus 100 mg/ml Proteinase K and 460 μl of TES-Proteinase K was added to the 40 μL input sample. Both input and precipitate samples were then incubated at 68°C for 2 hours in a Thermomixer (Eppendorf) at 1300 rpm. The supernatant (300 μl) from the precipitate sample was removed and a fresh 200 μl of TES added before an additional 20 minute incubation. Another 200 μl aliquot was removed from the precipitate sample and combined with the previous 300 μl aliquot. The total 500 μl precipitate pool and the 500 μl input sample were then purified using QIAquick spin column kit (Qiagen) and eluted into 100 μl TE with 20 μg RNase.
qPCR reactions were performed with a portion of each input and precipitate sample from both cut and uncut cells as described above for Figure 3 using oligo pairs at 0.5 (618/619), 2 (620/621), 5 (637/638), 10 (622/623), or 30 (624/625) kb upstream of the I-PpoI cleavage site at ura5+ (Table I). PCR product formation at uncut lys1+ was also monitored with oligos 635/636 (Table I). Data analysis was performed using the ΔΔCt method outlined in the QIAGEN EpiTect ChIP qPCR Assay Handbook. Briefly, averaged Ct values were calculated from triplicate reactions and used to determine the cut/uncut ratio for each oligo pair. The relative enrichment was then calculated by normalizing the cut/uncut ratio for each oligo pair flanking the I-PpoI cleavage site at ura5+ against the cut/uncut ratio determined for the oligo pair at uncut lys1+. Note that control experiments with non-specific IgG routinely showed background levels with a relative enrichment of ∼1 (data not shown).
Results and discussion
A tetracycline inducible I-PpoI expression system
To devise a system to introduce a site-specific DSB in the S. pombe genome on a relatively short time course, we first scanned the literature for a promoter capable of producing a rapid induction of target gene expression when integrated as a single copy. We identified a strong candidate in a heterologous tetracycline inducible system in which an ADH1 promoter drives constitutive expression of a Tetracycline repressor (TetR) that normally binds to and represses the plant viral cauliflower mosaic virus 35S (CaMV35S) promoter (Figure 2A and (Erler et al., 2006; Faryar and Gatz, 1992)). High-level promoter activity is achieved by addition of tetracycline to the growth media to inactivate the Tet repressor. Compared to the nmt promoter, the tetracycline-controlled system offered a significantly shorter induction time, the distinct advantage that laborious media changes would not be required and the ability to avoid the potential pleiotropic effects of metabolic inducers. Several key improvements made by Erler and colleagues (Erler et al., 2006) to the originally system (Faryar and Gatz, 1992) also made the tetracycline inducible promoter particularly attractive. First, Erler et al demonstrated anhydrotetracycline (ahTET) to be a superior induction ligand; second, they also demonstrated faster induction rates using defined (EMM) versus rich media (YES); and thirdly, inserted the system into a previously constructed pDUAL vector (Matsuyama et al., 2004) for simple chromosomal integration.
Figure 2.
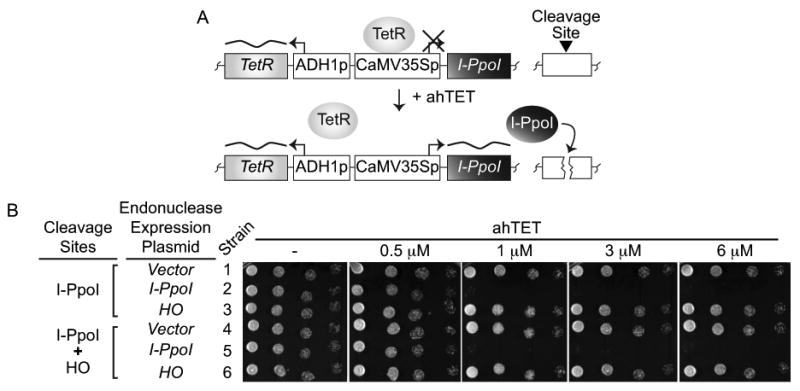
A tetracycline inducible I-PpoI expression system. (A) Assay schematic. The homing endonuclease I-PpoI is under the control of a tetracycline inducible CaMV35S promoter (p) and an ADH1 promoter drives constitutive expression of the tetracycline repressor (TetR) that normally represses I-PpoI expression. Addition of anhydrotetracycline (ahTET) to growth media triggers induction of I-PpoI expression and cutting at endogenous I-PpoI cleavage sites (black triangle) located in the rDNA repeats or at a single exogenous cleavage site integrated elsewhere. (B) Induction of I-PpoI expression with ahTET produces efficient killing of S. pombe cells. Either empty vector or plasmids containing tetracycline inducible I-PpoI or HO alleles were integrated into strains containing endogenous rDNA I-PpoI cleavage sites with or without a single exogenous HO cleavage site integrated at lys1+. Serial dilutions of cells (1:5) were spotted onto EMMG agar plates containing the indicated amount of ahTET and grown at 30°C. See materials and methods for further details. Strains: 1-YSS154; 2-YSS151; 3-YSLS798; 4-YSLS802; 5-YSLS803; 6-YSLS804.
To evaluate the tetracycline inducible promoter, we first compared the ability of the system to regulate expression of two different site-specific endonucleases: the S. cerevisiae HO nuclease and the homing endonuclease I-PpoI from the slime mold Physarum polycephalum. HO was chosen because of its established use as a site-specific endonuclease in S. pombe (Du et al., 2003; Osman et al., 1996; Prudden et al., 2003). I-PpoI was selected because it is an effective site-specific nuclease in budding yeast (Muscarella and Vogt, 1993) and mammalian cells (Berkovich et al., 2007). I-PpoI also has the convenient property of recognizing an endogenous ∼15 bp cleavage site that is present in the S. pombe ribosomal DNA (rDNA) repeats and 100% conserved in all eukaryotes (Ellison and Vogt, 1993). Each endonuclease ORF was cloned into the pDUAL-tet-rpsL-neo vector (Erler et al., 2006) and constructs integrated into the leu1-32 locus of strains with or without an exogenous HO cleavage site also integrated at lys1+ (see Materials and methods). Control strains with an integrated empty vector were also generated.
As an initial means to monitor endonuclease expression, we assessed the ability of cells to grow on agar plates containing increasing amounts of the ahTET inducer (Figure 2B). We reasoned that strains with a nuclease expression plasmid would be hypersensitive to the inducer because growth on ahTET media should kill cells by producing constitutive endonuclease expression and DSB formation. Figure 2B shows that colony formation of cells with the integrated I-PpoI expression plasmid was severely inhibited on ahTET media relative to vector control cells that grew equally well irrespective of the inducer (compare strains 1 and 4 versus 2 and 5). Surprisingly, cells containing the HO expression plasmid were equally resistant to ahTET regardless of the presence or absence of an HO cleavage site integrated at lys1+ (compare stains 3 and 6 Figure 2B). We also observed no cell killing when the HO cleavage site was integrated at arg3+(data not shown). These findings indicate that the tetracycline inducible promoter can drive I-PpoI expression, but not HO expression, to levels sufficient to produce efficient target site cleavage. It is currently unclear why the tetracycline promoter-HO combination is not effective, but we note that expressing HO from the nmt41 promoter can cause cell death in contrast to the tetracycline inducible promoter (data not shown). From this finding we suspect that the lack of cell death with tet-HO construct is because the HO endonuclease does not accumulate to high levels when the tetracycline inducible promoter is used to drive expression.
I-PpoI induction produces rapid cell cycle arrest and cleavage site cutting
The observations above argue that the I-PpoI endonuclease is uniquely suited for the tetracycline inducible promoter. We further investigated the capabilities of the tetracycline-I-PpoI combination by quantitatively measuring cell growth after induction of I-PpoI expression. Cultures of vector control and I-PpoI expression strains were either untreated or treated with the ahTET inducer (+ahTET Figures 3A and 3B). Growth was then monitored at time points after induction by cell counting and by imaging DAPI stained cells. As expected, Figure 3A shows that vector control cells grew equally well irrespective of ahTET. Importantly, without ahTET the growth rate of vector control and I-PpoI expression plasmid cells was essentially identical (Figure 3A and compare strains 1 and 2 Figure 4C). These data indicate that I-PpoI expression is sufficiently repressed to have no observable effect on cell growth in the absence of the inducer. In contrast, in the presence of ahTET growth of the I-PpoI expression strain was severely limited (Figure 3A). Growth inhibition was very efficient as ahTET treated I-PpoI cells displayed only ∼1 population doubling over the first 12 hour course of the experiment compared to ∼5 doublings for untreated cells. DAPI staining revealed that after only 4 hours of growth in ahTET media cells with the I-PpoI plasmid were very elongated compared to untreated and vector control cells (Figure 3B). The elongated phenotype is indicative of G2/M arrested cells and argues that I-PpoI induced cutting triggered efficient activation of the G2/M DNA damage checkpoint.
Figure 4.
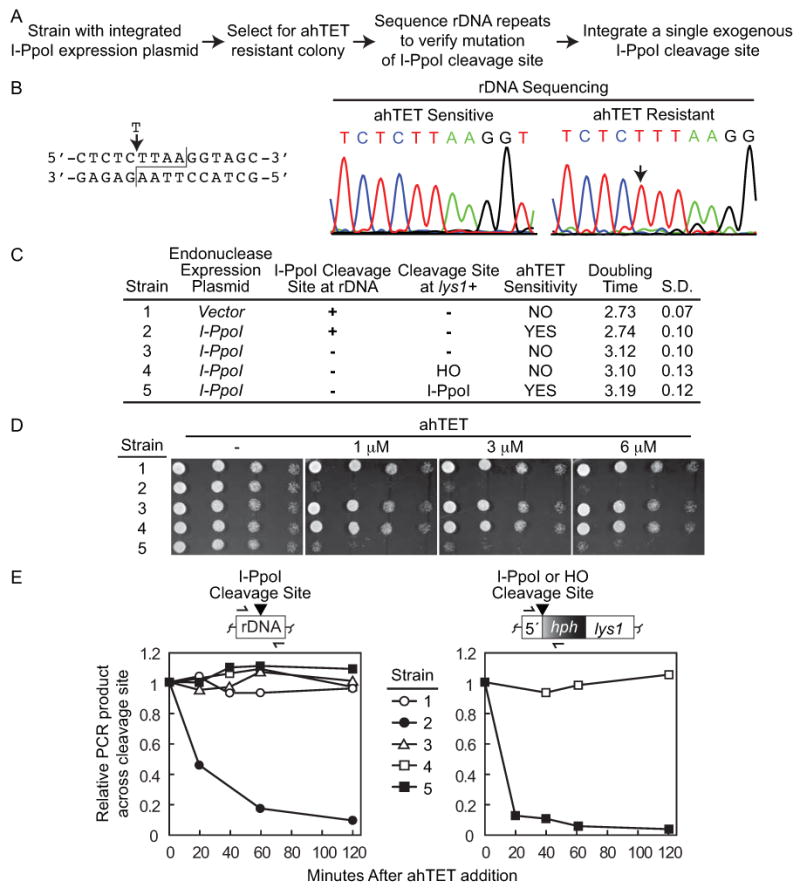
Isolation of rDNA mutations resistant to I-PpoI cleavage. (A) Strategy used to produce strains containing I-PpoI resistant rDNA repeats and a single exogenous I-PpoI cleavage site. (B) Sequence of I-PpoI resistant rDNA repeats. Left, I-PpoI rDNA cleavage site with T insertion of resistant strains indicated. Right, typical DNA sequencing chromatograms of rDNA repeats from ahTET sensitive and resistant strains (strains 2 and 3 in C, respectively) with the T insertion site indicated by the arrow. (C) Characteristics of relevant strains. The procedure detailed in A was used to generate strain 3 (YSLS790-3) containing mutated rDNA repeats resistant to I-PpoI cutting from the parental I-PpoI expressing strain 2 (YSS151). Either a single HO (strain 4, YSLS793) or I-PpoI (strain 5, YSLS792) cleavage site was then integrated back into the genome at lys1+(illustrated in right panel of (E)). Strain 1 (YSS154) with an integrated empty vector was assayed as a control. Strains 3, 4, and 5 are all derived from strain 2. Doubling times were determined at 30°C in EMMG in the absence of ahTET and averaged from at least 3 independent experiments with the standard deviation (S.D.) shown. (D) Integration of a single exogenous I-PpoI cleavage site back into the genome of a strain with I-PpoI resistant rDNA repeats restores ahTET sensitivity. Spot tests were performed as described for Figure 2 with strains detailed in (C). (E) I-PpoI rapidly cleaves a single exogenous cleavage site but not mutated rDNA repeats. I-PpoI cutting assays were performed as detailed for Figure 3C using strains detailed in (C) and oligos that span either the I-PpoI cleavage sites in the rDNA repeats (left) or a single exogenous HO or I-PpoI cleavage site integrated at lys1+ (right). Cleavage sites are denoted by the black triangle. Note that data is from a single experiment where all five strains were processed simultaneously so that the rate of I-PpoI cutting observed at the rDNA and lys1+ cleavage sites is directly comparable.
To measure the rate and efficiency of I-PpoI cutting, we used quantitative real-time PCR (qPCR) to monitor the amplification of a product across the rDNA cleavage sites after ahTET induction (illustrated Figure 3C, see also Materials and methods). The left graph in Figure 3C shows that product formation across the rDNA cleavage site remained constant in vector control cells. In contrast, I-PpoI expressing cells displayed a rapid decrease in PCR product formation (>80% after 1 hour), indicating efficient DSB formation after I-PpoI induction. We further investigated whether an exogenous I-PpoI cleavage site integrated at ura5+ could be efficiently cut. The right graph in Figure 3C shows that loss of the PCR product across both the endogenous and exogenous cleavage sites occurred at nearly identical rates when monitored simultaneously in a strain containing both ura5+ and rDNA cleavage sites. Note that there is some small variability between experiments in the rate of cleavage at early time points after ahTET induction but that >80% cutting is reproducible observed at later points (compare 20-40 minute points versus 60-120 minutes Figure 3C). The observations in Figure 3 indicate that tetracycline induced I-PpoI expression produces rapid (∼1 hour) formation of a site-specific DSB.
Generation of I-PpoI resistant rDNA repeats
The presence of the endogenous I-PpoI cleavage sites in the rDNA repeats provided a convenient means to initially assess cutting, but we reasoned that an ideal site-specific DSB system would use a unique exogenous cleavage site that could be easily integrated into different genomic locations. We next sought to enhance the applicability of the I-PpoI based assay by first eliminating the I-PpoI cleavage sites in the rDNA repeats, and then integrating back a single exogenous cleavage site. The strategy (Figure 4A and Material and methods) was based upon the previous observation that non-cleavable rDNA point mutations could be easily isolated after expression of the I-PpoI endonuclease in S. cerevisiae cells (Muscarella and Vogt, 1993). The mechanism that mediates mutation of the highly repeated rDNA loci is unclear, but is thought to be a two-step process that involves the formation of a mutation in a single rRNA gene that rapidly spreads to the entire population through gene-conversion (Muscarella and Vogt, 1993). Clones resistant to ahTET were isolated and DNA sequencing of a PCR product spanning the rDNA cleavage site used to confirm mutation. I-PpoI resistant cells arose at a rate of ∼1:1,000 and 23 of 30 ahTET resistant clones sequenced from three independent screens contained mutated rDNA repeats, with all but one mutant harboring a single T insertion in the I-PpoI cleavage site (Figure 4B). The only other mutation recovered was an insertion of two T's at the same position (data not show). Strikingly, the single T insertion was also observed in the rDNA repeats of I-PpoI resistant S. cerevisiae cells (Muscarella and Vogt, 1993). Figure 4D shows that cells with the I-PpoI expression plasmid and mutated rDNA repeats were resistant to ahTET similar to vector control cells that contain the cleavage sites but lack the endonuclease (compare strains 1 and 3). To directly investigate I-PpoI cutting at the mutated rDNA repeats, the qPCR assay from Figure 3C was used and revealed that the mutated rDNA repeats are not cleaved by I-PpoI in contrast to the very efficient cleavage that is observed for wt rDNA repeats (Figure 4E left, compare strains 3-5 versus strain 2). Importantly, the rDNA mutation produced only a modest increase in strain generation time in the absence of ahTET (compare strains 2 and 3 Figure 4C), indicating that overall cell fitness was not severely impaired.
We next asked if integrating a single exogenous I-PpoI cleavage site back into the genome of a strain with mutated rDNA repeats would recover ahTET sensitivity. As a control, we also integrated a single HO cleavage site that should not be cut by the I-PpoI endonuclease. Figure 4D shows that when the I-PpoI cleavage site was integrated at lys1+, but not the HO cleavage site, cells displayed ahTET sensitivity similar to the parental strain containing normal rDNA cleavage sites (compare strains 2, 4, and 5). Using the qPCR cutting assay we also observed very rapid DSB formation at the exogenous I-PpoI cleavage site (>90% after 1 hour) but not at the HO cleavage site after inducing I-PpoI expression (right panel Figure 4E). Interestingly, the rate of cutting at the single exogenous I-PpoI cleavage site is notably faster than that observed for the highly repeated rDNA cleavage sites (Figure 4E, compare strain 5 right versus strain 2 left). In total, Figure 4 demonstrates the ability to rapidly and efficiently produce a unique DSB in the S. pombe genome using the tetracycline inducible I-PpoI system.
Binding of Crb2 to the I-PpoI lesion site mimics IR induced DSBs
We next investigated how closely an I-PpoI produced break reflected DSBs generated by acute IR exposure. To examine this question, we asked if the accumulation of the checkpoint protein Crb2 could be visualized at sites of I-PpoI induced DSBs as seen with IR produced DSBs (Du et al., 2003). Strains were generated that harbored GFP tagged crb2+, a tetracycline inducible I-PpoI expression construct, and mutated rDNA repeats resistant to I-PpoI cleavage with or without a single exogenous I-PpoI cleavage site integrated at ura5+ (illustrated Figure 5C, see also Materials and methods). Figure 5A shows that only cells with the integrated I-PpoI cleavage site were sensitive to ahTET (compare strains 1 and 2), indicating that DSB formation required the presence of the exogenous cleavage site. Live cell microscopy was then used to monitor GFP-Crb2 localization before and after I-PpoI expression was induced with ahTET. Figure 5B shows that Crb2 localized throughout the nucleus and that after ahTET treatment sites of very efficient focal accumulation could be observed in a manner that required the exogenous I-PpoI cleavage site. Consistent with the presence of a single integrated I-PpoI cleavage site, the majority of cells contained only one ahTET induced Crb2 focus. Importantly, the number of detectable Crb2 foci in the absence of ahTET is not significantly different regardless of the presence or absence of the I-PpoI cleavage site (compare 0 time points Figure 5B right). This observation argues that without the inducer there is minimal background I-PpoI cutting due to promoter leakiness.
We next used ChIP to monitor Crb2 binding to break adjacent chromatin with higher resolution (see Materials and methods). I-PpoI cutting at the single exogenous cleavage site was induced in GFP-Crb2 cells and anti-GFP ChIP used to precipitate Crb2 bound DNA. Parallel ChIP experiments were also performed with uninduced cells. Oligo pairs that annealed at 0.5, 2, 5, 10, or 30 kb upstream of the I-PpoI cleavage site (illustrated Figure 5C), or at an uncut locus, were then used in real-time PCR reactions to monitor Crb2 chromatin binding before (uncut) and after (cut) I-PpoI induction. Figure 5C shows that Crb2 was significantly enriched at break adjacent regions after I-PpoI cutting with peak accumulation occurring 5 Kbp away from the cleavage site (see kmt5+). These observations indicate that I-PpoI induced cutting triggers the rapid binding of the Crb2 protein to break adjacent chromatin similar to treating cells with IR.
Crb2 binding to the I-PpoI induced break was further probed by determining the requirement for histone modification. Others and we have shown that focal accumulation of Crb2 at DSBs produced by acute IR exposure strictly requires binding to two different modified histones (Du et al., 2006; Greeson et al., 2008; Sanders et al., 2010; Sofueva et al., 2010). In contrast, Crb2 binding to break adjacent chromatin can occur independently of histone modification when >20 hours is needed to produce a DSB using the thiamine controlled nmt promoter to drive HO expression (Du et al., 2006). To examine the requirement for histone modification in Crb2 targeting to the I-PpoI produced DSB, we deleted the kmt5+ gene and assessed GFP-Crb2 binding to the break site using microscopy and ChIP (Δkmt5 Figures 5B and 5C). The Kmt5 methylase catalyzes the dimethyl histone H4K20 modification that is a direct binding target of Crb2 (Botuyan et al., 2006; Greeson et al., 2008; Sanders et al., 2004). Loss of kmt5+ severely compromises focal accumulation of Crb2 at IR induced DSBs, but not at breaks produced by HO after >20 hours of nmt driven expression (Du et al., 2006; Sanders et al., 2004).
Figure 5B shows that the ability of Crb2 to form discrete microscopic foci after induction of I-PpoI expression was significantly impaired by the Δkmt5 mutation. The requirement for kmt5+ in Crb2 binding to the I-PpoI produced break was mirrored in higher resolution ChIP experiments (Figure 5C). Importantly, the reduction in Crb2 targeting observed in Δkmt5 cells is not due to a lack of I-PpoI cutting because kmt5+and Δkmt5 cells were equally sensitive to ahTET (Figure 5A). These findings indicate that efficient targeting of Crb2 to the I-PpoI produced DSB requires the activity of the H4K20 methyltransferase Kmt5 and contrasts with the dispensability of kmt5+ when the slower inducing nmt-HO system is used to produce a site-specific DSB (Du et al., 2006). In total, the observations from Figure 5 argue that the tetracycline inducible I-PpoI site-specific DSB assay effectively mimics IR generated DSBs.
Summary
Here we present a new assay to efficiently introduce a single site-specific DSB into the S. pombe genome using the homing endonuclease I-PpoI under the control of a tetracycline inducible promoter. With this assay DSB formation at a unique I-PpoI cleavage site occurs with fast kinetics after inducing endonuclease expression (>90% after 1 hour, Figure 4E). Importantly, the tetracycline inducible promoter is tightly repressed because there are no observable defects in cell growth (Figure 4C) or increase in focal accumulation of the Crb2 checkpoint protein in the absence of the ahTET inducer (Figure 5B and data not shown). Two major advantages of this new assay compared to the thiamine controlled nmt-HO system (Du et al., 2003; Osman et al., 1996; Prudden et al., 2003) are the relatively rapid rate of DSB formation (∼1 hour versus >20 hours) and the use of a non-metabolic inducer that avoids potential pleiotropic affects and that does not require inconvenient media changes. The advantage of rapid DSB formation is reflected in our demonstration that efficient binding of the Crb2 protein to regions flanking the I-PpoI induced DSB requires histone modification activity similar to observations made with IR generated DSBs (Figure 5). The ease at which a site-specific DSB can be introduced with the tet-I-PpoI system makes it possible to perform intensive time course studies and to investigate DSB response at different stages of the S. pombe cell cycle, an experiment that is not feasible with the slower inducing nmt-HO system. Because the single I-PpoI cleavage site can be integrated into essentially any genomic region, it will also be possible to harness S. pombe genetics to investigate how the sensing and repair of DSBs differs between active euchromatin and repressive heterochromatic regions of the genome.
One consideration with the I-PpoI system is that once ahTET has been added to growing cultures, removing the inducer does not efficiently turn off I-PpoI activity (data not shown). This limits the ability to use the I-PpoI system as describe here to monitor the completion of DSB repair events. Introducing an ectopic homologous template into cells should alleviate this issue by promoting DSB repair, a strategy that has been successfully used in S. cerevisiae (Ira et al., 2003) and that we are actively pursuing. Another important consideration is that the use of a strain with I-PpoI resistant rDNA repeats appears to be essential for success. We initially attempted to monitor Crb2 focal accumulation in a strain in which I-PpoI cutting occurred at both the rDNA repeats and at a single exogenous cleavage site. Surprisingly, even though DSB formation clearly occurred at both sites (see Figure 3C right panel), no significant Crb2 focal accumulation could be observed and only a very weak (∼2 fold) enrichment of Crb2 could be measured by ChIP at the exogenous cleavage site (data not shown). These results contrast to the very efficient Crb2 accumulation that is observed with both microscopy and ChIP when I-PpoI cleavage occurs only at a unique exogenous site (Figure 5). It seems likely that when I-PpoI cutting occurs in the rDNA repeats, the high number of breaks may compete away the available pool of Crb2 from the single exogenous site and/or dilute out the Crb2 signal such that binding to break regions can no longer be efficiently visualized. Others have also observed difficulty in visualizing the binding of DSB response proteins to sites within the rDNA repeats (Irmisch et al., 2009; Ampatzidou et al., 2006), suggesting that our observations reflect a general property of the highly repeated rDNA loci. Thus, prior elimination of the I-PpoI cleavage sites in the rDNA repeats greatly increases the applicability of the system by enhancing factor binding to the exogenous break site and by providing a framework to easily manipulate the location of a single site-specific DSB. Through three independent screens, we have found isolating rDNA mutations to be a straightforward and efficient process that is easily adaptable to almost any starting strain. Strikingly, the major rDNA mutation we have observed was also isolated in a screen for I-PpoI resistant mutants in S. cerevisiae (Muscarella and Vogt, 1993), suggesting a similar mutational mechanism. Importantly, the effect of the rDNA mutation on overall cell physiology is small and mutations appear to be stable as we have not observed any reversion events and continuous selection is not required. However, the meiotic stability of rDNA mutations is unknown because we have not yet attempted to mate rDNA mutation strains. Rather, we have focused on making de novo rDNA mutations directly in the appropriate strain because of the relative ease of the procedure.
During the preparation of this manuscript Watson et al reported a new S. pombe site-specific DSB assay using the HO endonuclease under the control of the uracil inducible urg1 promoter (Watson et al., 2011). Similar to the tet-I-PpoI system described here, the urg1-HO assay can produce a unique DSB with relatively fast kinetics (∼1 hour). A major advantage of the urg1-HO system compared to the I-PpoI assay is that the S. pombe genome lacks endogenous HO cleavage sites, potentially facilitating strain construction and eliminating the need to introduce mutations into the rDNA repeats as detailed here for the I-PpoI endonuclease. However, the HO endonuclease must be introduced into the native urg1 locus using a multi-step process whereas the tet-I-PpoI construct can be integrated at many different genomic locations in a single transformation. A limitation of the urg1-HO system is that minimal media lacking uracil is required for strain growth, making it incompatible with commonly used uracil auxotrophic markers such as ura4-D18. Because the tet-I-PpoI system does not require a metabolic inducer cell growth during strain construction can be performed with either rich or minimal media. We have found that the ability to use rich media significantly speeds up strain construction and all routine procedures compared to using only minimal media. Induction experiments can also be performed in rich media, though with slightly slower kinetics compared to minimal media ((Erler et al., 2006) and our unpublished observations). These elements make the tet-I-PpoI system potentially applicable to many more existing strains and experimental conditions compared to the urg1-HO system. Another consideration is that the urg1 promoter is induced during meiosis and meiotic HO expression is detrimental even when an HO cleavage site is not present in the genome (Watson et al., 2011). The meiotic compatibility of the tet-I-PpoI expression construct is unknown (see also above). Thus, both the urg1-HO and the tet-I-PpoI systems have unique benefits and we believe that the availability of two distinct fast inducing site-specific DSB assays will prove advantageous to many in the S. pombe community.
In summary, we have engineered a new heterologous system to rapidly introduce a unique DSB into the S. pombe genome that is tightly regulated and has minimal issues related to promoter leakiness. Our development of the tetracycline controlled I-PpoI system avoids the potential complications associated with metabolic inducers and is compatible with a wide variety of strain genotypes, adding an important new instrument to the fission yeast toolbox that is applicable to a broad range of questions.
Acknowledgments
We would like to give a special thank you to Dr. Rodney Rothstein for the suggestion of generating I-PpoI resistant strains and to Dr. Tim Humphrey, Dr. Barry Stoddard, and Dr. Francis Stewart for gifts of plasmids. We are also grateful to all of our colleagues in the Department of Biochemistry at Case Western Reserve University and the Department of Molecular Genetics at the Cleveland Clinic Foundation for their helpful advice and encouragement during the course of this study. This work was supported by Career Development Award [0017/2006-C] from the International Human Frontier Science Program Organization (S.L.S.), the Case Comprehensive Cancer Center and Radiation Resource Core Facility (NIH Grant No. P30 CA43703), and the National Institutes of Health (Grant Nos GM50752 and AG19960 to K.W.R.). Imaging facilities were provided by the Center For Aids Research at Case Western Reserve University.
References
- Ampatzidou E, Irmisch A, O'Connell MJ, Murray JM. Smc5/6 is required for repair at collapsed replication forks. Mol Cell Biol. 2006;26:9387–9401. doi: 10.1128/MCB.01335-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahler J, Wu JQ, Longtine MS, et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellaiche Y, Mogila V, Perrimon N. I-SceI endonuclease, a new tool for studying DNA double-strand break repair mechanisms in Drosophila. Genetics. 1999;152:1037–1044. doi: 10.1093/genetics/152.3.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovich E, Monnat RJJ, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9:683–690. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- Botuyan MV, Lee J, Ward IM, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellaire G, Bazett-Jones DP. Beyond repair foci: subnuclear domains and the cellular response to DNA damage. Cell Cycle. 2007;6:1864–1872. doi: 10.4161/cc.6.15.4560. [DOI] [PubMed] [Google Scholar]
- Downs JA, Nussenzweig MC, Nussenzweig A. Chromatin dynamics and the preservation of genetic information. Nature. 2007;447:951–958. doi: 10.1038/nature05980. [DOI] [PubMed] [Google Scholar]
- Du LL, Nakamura TM, Moser BA, Russell P. Retention but not recruitment of Crb2 at double-strand breaks requires Rad1 and Rad3 complexes. Mol Cell Biol. 2003;23:6150–6158. doi: 10.1128/MCB.23.17.6150-6158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du LL, Nakamura TM, Russell P. Histone modification-dependent and -independent pathways for recruitment of checkpoint protein Crb2 to double-strand breaks. Genes Dev. 2006;20:1583–1596. doi: 10.1101/gad.1422606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison EL, Vogt VM. Interaction of the intron-encoded mobility endonuclease I-PpoI with its target site. Mol Cell Biol. 1993;13:7531–7539. doi: 10.1128/mcb.13.12.7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erler A, Maresca M, Fu J, Stewart AF. Recombineering reagents for improved inducible expression and selection marker re-use in Schizosaccharomyces pombe. Yeast. 2006;23:813–823. doi: 10.1002/yea.1396. [DOI] [PubMed] [Google Scholar]
- Faryar K, Gatz C. Construction of a tetracycline-inducible promoter in Schizosaccharomyces pombe. Curr Genet. 1992;21:345–349. doi: 10.1007/BF00351693. [DOI] [PubMed] [Google Scholar]
- Flick KE, McHugh D, Heath JD, et al. Crystallization and preliminary X-ray studies of I-PpoI: a nuclear, intron-encoded homing endonuclease from Physarum polycephalum. Protein Sci. 1997;6:2677–2680. doi: 10.1002/pro.5560061226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi AA, Noon AT, Jeggo PA. The impact of heterochromatin on DSB repair. Biochem Soc Trans. 2009;37:569–576. doi: 10.1042/BST0370569. [DOI] [PubMed] [Google Scholar]
- Greeson NT, Sengupta R, Arida AR, et al. Di-methyl H4 lysine 20 targets the checkpoint protein CRB2 to sites of DNA damage. J Biol Chem. 2008;283:33168–33174. doi: 10.1074/jbc.M806857200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SI. Transcriptional silencing in fission yeast. J Cell Physiol. 2000;184:311–318. doi: 10.1002/1097-4652(200009)184:3<311::AID-JCP4>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Grewal SI, Moazed D. Heterochromatin and epigenetic control of gene expression. Science. 2003;301:798–802. doi: 10.1126/science.1086887. [DOI] [PubMed] [Google Scholar]
- Haber JE. Uses and abuses of HO endonuclease. Methods Enzymol. 2002;350:141–164. doi: 10.1016/s0076-6879(02)50961-7. [DOI] [PubMed] [Google Scholar]
- Harrison JC, Haber JE. Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet. 2006;40:209–235. doi: 10.1146/annurev.genet.40.051206.105231. [DOI] [PubMed] [Google Scholar]
- Iacovoni JS, Caron P, Lassadi I, et al. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010;29:1446–1457. doi: 10.1038/emboj.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, et al. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell. 2003;115:401–411. doi: 10.1016/s0092-8674(03)00886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmisch A, Ampatzidou E, Mizuno K, et al. Smc5/6 maintains stalled replication forks in a recombination-competent conformation. EMBO J. 2009;28:144–155. doi: 10.1038/emboj.2008.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP. Sensing and repairing DNA double-strand breaks. Carcinogenesis. 2002;23:687–696. doi: 10.1093/carcin/23.5.687. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet. 1996;12:224–228. doi: 10.1016/0168-9525(96)10019-6. [DOI] [PubMed] [Google Scholar]
- Kim JA, Kruhlak M, Dotiwala F, et al. Heterochromatin is refractory to gamma-H2AX modification in yeast and mammals. J Cell Biol. 2007;178:209–218. doi: 10.1083/jcb.200612031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langerak P, Mejia-Ramirez E, Limbo O, Russell P. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 2011;7:e1002271. doi: 10.1371/journal.pgen.1002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massip L, Caron P, Iacovoni JS, et al. Deciphering the chromatin landscape induced around DNA double strand breaks. Cell Cycle. 2010;9:2963–2972. doi: 10.4161/cc.9.15.12412. [DOI] [PubMed] [Google Scholar]
- Matsuyama A, Shirai A, Yashiroda Y, et al. pDUAL, a multipurpose, multicopy vector capable of chromosomal integration in fission yeast. Yeast. 2004;21:1289–1305. doi: 10.1002/yea.1181. [DOI] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- Moss J, Tinline-Purvis H, Walker CA, et al. Break-induced ATR and Ddb1-Cul4(Cdt)(2) ubiquitin ligase-dependent nucleotide synthesis promotes homologous recombination repair in fission yeast. Genes Dev. 2010;24:2705–2716. doi: 10.1101/gad.1970810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscarella DE, Vogt VM. A mobile group I intron from Physarum polycephalum can insert itself and induce point mutations in the nuclear ribosomal DNA of saccharomyces cerevisiae. Mol Cell Biol. 1993;13:1023–1033. doi: 10.1128/mcb.13.2.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Driscoll M, Jeggo PA. The role of double-strand break repair - insights from human genetics. Nat Rev Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- Osman F, Fortunato EA, Subramani S. Double-strand break-induced mitotic intrachromosomal recombination in the fission yeast Schizosaccharomyces pombe. Genetics. 1996;142:341–357. doi: 10.1093/genetics/142.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CL, Cote J. Cellular machineries for chromosomal DNA repair. Genes Dev. 2004;18:602–616. doi: 10.1101/gad.1182704. [DOI] [PubMed] [Google Scholar]
- Pidoux A, Mellone B, Allshire R. Analysis of chromatin in fission yeast. Methods. 2004;33:252–259. doi: 10.1016/j.ymeth.2003.11.021. [DOI] [PubMed] [Google Scholar]
- Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudden J, Evans JS, Hussey SP, et al. Pathway utilization in response to a site-specific DNA double-strand break in fission yeast. EMBO J. 2003;22:1419–1430. doi: 10.1093/emboj/cdg119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raji H, Hartsuiker E. Double-strand break repair and homologous recombination in Schizosaccharomyces pombe. Yeast. 2006;23:963–976. doi: 10.1002/yea.1414. [DOI] [PubMed] [Google Scholar]
- Rich T, Allen RL, Wyllie AH. Defying death after DNA damage. Nature. 2000;407:777–783. doi: 10.1038/35037717. [DOI] [PubMed] [Google Scholar]
- Sanders SL, Arida AR, Phan FP. Requirement for the phospho-H2AX binding module of Crb2 in double-strand break targeting and checkpoint activation. Mol Cell Biol. 2010;30:4722–4731. doi: 10.1128/MCB.00404-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SL, Portoso M, Mata J, et al. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–614. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Sofueva S, Du LL, Limbo O, et al. BRCT Domain Interactions with Phospho-Histone H2A Target Crb2 to Chromatin at Double-Strand Breaks and Maintain the DNA Damage Checkpoint. Mol Cell Biol. 2010;30:4732–4743. doi: 10.1128/MCB.00413-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Haber JE. Repair of DNA double strand breaks: in vivo biochemistry. Methods Enzymol. 2006;408:416–429. doi: 10.1016/S0076-6879(06)08026-8. [DOI] [PubMed] [Google Scholar]
- Symington LS. Role of RAD52 Epistasis Group Genes in Homologous Recombination and Double-Strand Break Repair. Microbiology and Molecular Biology Reviews. 2002;66:630–670. doi: 10.1128/MMBR.66.4.630-670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinline-Purvis H, Savory AP, Cullen JK, et al. Failed gene conversion leads to extensive end processing and chromosomal rearrangements in fission yeast. EMBO J. 2009;28:3400–3412. doi: 10.1038/emboj.2009.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gent DC, Hoeijmakers JH, Kanaar R. Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet. 2001;2:196–206. doi: 10.1038/35056049. [DOI] [PubMed] [Google Scholar]
- Ward JF. Biochemistry of DNA lesions. Radiat Res Suppl. 1985;8:S103–11. [PubMed] [Google Scholar]
- Watson AT, Werler P, Carr AM. Regulation of gene expression at the fission yeast Schizosaccharomyces pombe urg1 locus. Gene. 2011;484:75–85. doi: 10.1016/j.gene.2011.05.028. [DOI] [PubMed] [Google Scholar]
- Xu Y, Price BD. Chromatin dynamics and the repair of DNA double strand breaks. cc. 2011;10:261–267. doi: 10.4161/cc.10.2.14543. [DOI] [PMC free article] [PubMed] [Google Scholar]