

Abstract
Motivation: Comparative analyses of gene expression data from different species have become an important component of the study of molecular evolution. Thus methods are needed to estimate evolutionary distances between expression profiles, as well as a neutral reference to estimate selective pressure. Divergence between expression profiles of homologous genes is often calculated with Pearson's or Euclidean distance. Neutral divergence is usually inferred from randomized data. Despite being widely used, neither of these two steps has been well studied. Here, we analyze these methods formally and on real data, highlight their limitations and propose improvements.
Results: It has been demonstrated that Pearson's distance, in contrast to Euclidean distance, leads to underestimation of the expression similarity between homologous genes with a conserved uniform pattern of expression. Here, we first extend this study to genes with conserved, but specific pattern of expression. Surprisingly, we find that both Pearson's and Euclidean distances used as a measure of expression similarity between genes depend on the expression specificity of those genes. We also show that the Euclidean distance depends strongly on data normalization. Next, we show that the randomization procedure that is widely used to estimate the rate of neutral evolution is biased when broadly expressed genes are abundant in the data. To overcome this problem, we propose a novel randomization procedure that is unbiased with respect to expression profiles present in the datasets. Applying our method to the mouse and human gene expression data suggests significant gene expression conservation between these species.
Contact: marc.robinson-rechavi@unil.ch; sven.bergmann@unil.ch
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Changes in gene expression have been suggested to underlie many differences in gene function or in phenotype. More generally, expression is an important component of gene function, and studying the evolution of gene expression is a key step in evolutionary genomics. While there has been a great deal of research concerning the primary treatment of expression data in general (see Garber et al. (2011), and Quackenbush (2002) for reviews), there has been little investigation into the methods used more specifically to quantify expression evolution (Pereira et al., 2009). This can make it difficult to critically assess contradictory results, such as the reports that broadly expressed genes are more conserved (Khaitovich et al., 2005) or less conserved (Liao et al., 2010; Liao and Zhang, 2006b) than specifically expressed genes.
To assess whether and how much expression has been conserved between two orthologous genes by selection, we need an expectation for expression similarity under neutral evolution. Thus, the estimation of gene expression conservation requires two components: (i) a measure of gene expression similarity; and (ii) the expected value of the divergence level under neutrality.
The two most common measures of similarity between expression profiles of orthologous genes are Pearson's correlation coefficient (Chan et al., 2009; Liao and Zhang, 2006a, b; Xing et al., 2007; Yanai et al., 2004; Yang et al., 2005; Zheng-Bradley et al., 2010) and Euclidean distance (Jordan et al., 2005; Liao and Zhang, 2006a; Yanai et al., 2004). The results obtained with Pearson's and Euclidean distances have been reported to be poorly correlated (Liao and Zhang, 2006a; Pereira et al., 2009). This poses the question which of these measures provides a better description of expression similarity. It has been demonstrated that Pearson's correlation coefficient, in contrast to Euclidean distance, underestimates the expression similarity between orthologous genes with a conserved uniform pattern of expression. In consequence, use of the Euclidean distance has been encouraged (Pereira et al., 2009).
For neutral evolution, one expects that similarity between expression profiles of orthologous genes gradually decreases with time. For species that have diverged for sufficiently long time no detectable similarity in expression is expected to remain; this has been postulated to be the case between mouse and human (100 million years; Jordan et al., 2005). It has been suggested that such large neutral divergence could be approximated by calculating the distance between expression profiles of randomly chosen pairs of genes from the species compared. The standard approach used to generate random pairs of genes is to permute the orthology relationship between them (Chan et al., 2009; Liao and Zhang, 2006a, b; Xing et al., 2007; Zheng-Bradley et al., 2010).
Here, we show formally and empirically that, in contrast to previous reports (Liao and Zhang, 2006a; Pereira et al., 2009), there exists a relationship between the Pearson's correlation coefficient and the Euclidean distance, which depends on the data normalization. We also extend the previous study of Pereira et al. (2009) by considering more than just the uniform pattern of expression. We demonstrate that in fact both distance measures depend on the expression specificity of analyzed genes. Next, we discuss these observations in the context of the assessment of gene expression conservation. We show that the comparison of expression profiles for randomly permuted gene pairs is biased when broadly expressed genes are abundant in the data, a distribution characteristic of many datasets. To overcome this problem, we propose a novel procedure to generate random gene pairs. This procedure is not biased by the over- or underrepresentation of any expression profile in the datasets. Finally, we use our approach to provide clear evidence for constrained evolution of gene expression between mouse and human.
2 METHODS
2.1 Gene expression data
We used the human and mouse gene expression data from the GNF Gene Expression Atlas of Su et al. (2004) as a case study. This study was performed on the Affymetrix HG-U133A array as well as on the custom array GNF1H for human, and on the custom array GNF1M for mouse. In total, expression profiles for 79 human and 61 mouse organs were measured, with 44 928 probe sets for human and 36 182 probe sets for mouse. We only took into account organs belonging to the homologous organ groups (HOGs) defined in the Bgee database (Bastian et al., 2008). Using the mapping available in the Bgee database we could connect 36 human organs and 30 mouse organs to 27 HOGs. See Supplementary Table S1 for the list of HOGs and their corresponding organs. Microarray data were normalized with the gcrma R package (Wu et al., 2004).
To assign the probe sets to their corresponding human or mouse genes we used the mapping available in Bgee. We kept only probe sets which matched to a unique Ensembl gene. A total of 15 121 probe sets corresponding to 13 853 mouse genes, and 23 920 probe sets corresponding to 15 338 human genes were found.
To estimate the expected values of distances for gene pairs with conserved expression patterns, we used data from replicated experiments, performed in each species. Thus, for each probe set we had two vectors of values representing its expression over the organs. The datasets contained 36 organs and 23 920 probe set pairs for human, and 30 organs and 15 121 probe set pairs for mouse. The results of the study on mouse gene expression data are presented in the Supplementary Materials.
To study gene expression evolution between mouse and human we merged human and mouse organs into 27 HOGs. For every probe set in each HOG the arithmetic mean of the gcRMA normalized expression values was calculated (each HOG was represented by at least two microarrays). We used a subset of 8942 one-to-one orthologous gene pairs (see Human–Mouse Orthologous Genes). If the gene was matched by more than one probe set on the microarray, we randomly picked one probe set to represent that gene.
2.2 Human–mouse orthologous genes
Homology information of human and mouse genes was retrieved from Ensembl release 55 (Hubbard et al., 2009), using BioMart (Smedley et al., 2009). A total of 8942 pairs of human–mouse one-to-one orthologous genes had expression information in the datasets we used.
2.3 Normalization procedures
For a given gene we consider a vector x of expression intensities xi across n different organs indexed by i=1,… n. The Manhattan normalization of x is calculated by dividing it by its L1 norm:
In some studies (Liao and Zhang, 2006a; Pereira et al., 2009) this normalization is called relative abundance. The Euclidean normalization of vector x is calculated by dividing the vector by its L2 norm:
Finally, we introduce a so-called z-like normalization of x which corresponds to the Euclidean normalization of x minus its mean value:
2.4 Pearson's and Euclidean distances
The Pearson's distance (dP) between two expression profiles is defined as 1−r, where
| (1) |
is the Pearson's correlation coefficient between vectors x and y. Here the vector elements xi and yi are the expression signal intensities of two genes in the condition i,  and
and 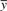 are the sample means, sx and sy are the sample SDs. zx and zy are the z-scores of vectors x and y.
are the sample means, sx and sy are the sample SDs. zx and zy are the z-scores of vectors x and y.
The Euclidean distance (dE) between two expression profiles is defined as
| (2) |
with notations as for Equation (1).
2.5 Organ specificity of gene expression
To measure the expression specificity of human genes we used the organ specificity index τ (Yanai et al., 2005). The τ of a given gene with an expression vector x is defined as follows:
| (3) |
where
The value of τ varies between 0 and 1, with higher values indicating higher organ specificity.
2.6 τ-group composition
To study the relation between dE and τ we used replicated expression data for human genes (36 organs, 23 920 probe sets). We sorted the probe set pairs according to the organ specificity index τ [Equation (3)] of the first replicate, and we divided the probe set pairs into three τ-groups of equal size (e.g. the first group contained 1/3 of the probe set pairs with the first replicate having lowest τ). For each group we recorded the minimum and maximum τ value of the first replicate, and used these values to filter out probe sets with the two replicates having τ values from different groups. The resulting τ-groups were of similar, but not equal, size (Table 1). An alternative τ-group composition, with a more balanced distributions of τ values (first group containing genes with τ∈[0, 0.2); second group with τ∈[0.2, 0.6); and third group with τ∈[0.6, 1]) leads to unbalanced sizes of three groups. Nevertheless, for both approaches the results are qualitatively the same (Supplementary Figs. S6 and S7).
Table 1.
Composition of three τ-groups of human probe set (ps) pairs
| Organ specificity (τ) | Number of ps pairs | |
|---|---|---|
| τ-group 1 | 0.003≤τ≤0.117 | 6348 |
| τ-group 2 | 0.117<τ≤0.295 | 5280 |
| τ-group 3 | 0.295<τ≤0.879 | 6692 |
2.7 Randomization procedures
Changes in gene expression patterns between randomly chosen genes from two species have been suggested as an approximation for the result of neutral expression evolution (Jordan et al., 2005). We used two different randomization procedures to create such sets of random gene pairs. First, we permuted the gene order within replicates (or within species). We refer to these as randomly permuted pairs. Second, we performed what we refer to as ‘τ-uniform sampling’. We first randomly chose an organ specificity index (τ), uniformly from the interval of (τmin, τmax), where τmin and τmax are the lowest and the highest values of the observed τ, respectively. Next, we picked the gene with the value of τ closest to the randomly chosen τ within one dataset (i.e. within one replicate, or one species). Then, independently, we repeated the procedure for the second dataset. Thus, we obtained two randomly chosen genes which form a new random pair. Repeating the procedure provides the ‘τ-uniform’ random gene pairs.
3 RESULTS AND DISCUSSION
3.1 Correlation between Pearson's and Euclidean distances depends on data normalization
To compare gene expression between species, over many different conditions, it is important to normalize the expression levels between the conditions to obtain a common scale between species. This is distinct from the preprocessing normalization (within condition), which is typically done using methods such as LOESS (Yang et al., 2002) or gcRMA (Wu et al., 2004), and is not specific to inter-species evolutionary studies. In the following, we only consider the impact of the between conditions normalization on the evolutionary comparisons. We discuss three normalization procedures commonly used for evolutionary studies: Manhattan normalization [also referred to as ‘relative abundance’ (Liao and Zhang, 2006a)], Euclidean normalization and z-like normalization (see Section 2.3 for mathematical definition of all three normalizations).
One can use any of these normalizations before calculating the Pearson's or Euclidean distance between two gene expression profiles. However, the choice of normalization can affect the results. Pearson's distance (dP) between two expression profiles remains the same, regardless of whether and how the data are normalized, and it ranges between 0 and 2. The reason is that r is defined on the z-scores [see Equation (1) in Section 2.4], which are invariant with respect to linear transformation. In contrast, the Euclidean distance between two expression profiles (dE) changes its value depending on the normalization used, even though the interval of possible dE values is always between 0 and 2.
The correlation between dP and dE is poor for Manhattan (Supplementary Fig. S1A; see also Liao and Zhang, 2006a; Pereira et al., 2009) and Euclidean normalizations (Supplementary Fig. S1B). In contrast, z-like normalization leads to an interdependent relationship between dP and dE, defined by
| (4) |
(see Theoretical Analysis in Supplementary Material, and Supplementary Fig. S1C). As dP gives the same results for all three normalizations, and for z-like normalization it is equal to dE2/2, we focused on the Euclidean distance. If not stated otherwise, the Euclidean distance was calculated for all three normalizations: Manhattan, Euclidean and z-like, referred to as dEM, dEE and dEZ, respectively.
3.2 Commonly used measures of gene expression similarity depend on the organ specificity of the genes
Intuitively, one might assume that the distance between two orthologous genes which have conserved the expression profile of their last common ancestor should be close to zero, and that this should hold regardless of the gene expression pattern. To assess if this is indeed the case, we performed an empirical study. We used human microarray data with the expression information from 36 different organs in two replicates (Su et al., 2004). The replicates were used to ‘simulate’ pairs of genes with conserved expression profiles. We calculated the organ specificity index τ [Equation (3)] for each pair of replicates, and then divided them into three τ-groups of similar size (see Section 2.6 for details). The first two groups contained broadly expressed genes (τ≤0.295), and only the third group consisted of genes with more specific expression patterns (τ>0.295; Table 1).
We measured the Euclidean distances (dEM, dEE and dEZ) for probe set pairs within each τ-group. The resulting levels of expression similarity between replicates strongly depended on the organ specificity level. Values of dEM and dEE were significantly lower for broadly expressed genes than for organ-specific genes (p<10−16, Mann–Whitney U test, Fig. 1A and B; Supplementary Fig. S5A and B). In contrast, values of dEZ were significantly higher for broadly expressed genes than for organ-specific genes (p<10−16, Mann–Whitney U test; Fig. 1C; Supplementary Fig. S5C). See Supplementary Figure S3 for the correlation analysis between the Euclidean distances and organ specificity index.
Fig. 1.

The distribution of expression similarity between human replicates depends on their organ specificity. (A) dEM and (B) dEE are significantly lower for broadly expressed genes (group 1) than for organ-specific genes (group 3). For randomly permuted gene pairs dEM and dEE also differ between the three τ-groups. They are significantly lower for random pairs in group 1 than in group 3. (C) dEZ is significantly higher for broadly expressed genes (group 1) than for organ-specific genes (group 3). dEZ for randomly permuted pairs is high in all three groups, even in the first τ-group, where random pairs consist of two broadly expressed genes (this is a consequence of low r for uniformly expressed genes). Note that the scale of the x-axis differs strongly between graphs.
3.3 The rate of neutral expression evolution estimated with randomly permuted gene pairs depends on the organ specificity of the genes
The rate of neutral expression evolution is typically approximated by calculating the distance between expression profiles of randomly paired genes. The random choice of the genes is assumed to remove any similarity between them (Jordan et al., 2005). The standard approach to generate random gene pairs is to permute the ortholog relationship between the genes in the datasets. We created random probe set pairs by permuting the probe set order within each of the three τ-groups separately, and we then calculated the Euclidean distances (dEM, dEE and dEZ) between their expression profiles. We found that dEM and dEE were significantly lower for random pairs from the first τ-group, than for random pairs from the third τ-group (p<10−16, Mann-Whitney U test; Fig. 1A and B; Supplementary Fig. S5A and B). This is because the first τ-group consisted of broadly expressed genes. Consequently, even the randomly matched probe set pairs tended to have similar expression patterns and thus low distances. In contrast, the third τ-group consisted of genes with more specific expression patterns, and so the random pairs were truly different.
dEZ between random pairs was not affected by organ specificity, in the sense that in all three τ-groups the median dEZ was around 1.4 (Fig. 1C; Supplementary Fig. S5C). Values of dEZ were high even in the first τ-group, although it consisted of random pairs with similar, broad patterns of expression. The reason is that 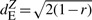 is a decreasing function of r, which for broadly expressed gene pairs reflects mainly the noise of the measurement and is close to 0 (for details see Pereira et al., 2009 and Supplementary Fig. S2). Thus, random gene pairs from the first τ-group tend to have high dEZ values (around
is a decreasing function of r, which for broadly expressed gene pairs reflects mainly the noise of the measurement and is close to 0 (for details see Pereira et al., 2009 and Supplementary Fig. S2). Thus, random gene pairs from the first τ-group tend to have high dEZ values (around 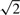 ).
).
3.4 A large fraction of broadly expressed genes leads to an underestimation of expression conservation
Our analysis shows that if the fraction of broadly expressed genes is large, the level of gene expression conservation is likely to be underestimated. This is especially important if we consider the fact that housekeeping genes (broadly expressed) are more frequent than organ-specific genes (Ramsköld et al., 2009). We found such skewed distributions not only in the human data considered here (Fig. 3A), but also in several other datasets, e.g. most mouse genes are broadly expressed over different organs, most Arabidopsis genes are broadly expressed over different light conditions, and most zebrafish genes are broadly expressed over different developmental stages (Supplementary Fig. S4).
Fig. 3.

Random gene pairs have their τ values differently distributed depending on the randomization procedure used. (A) τ distribution for human replicates. The τ pairs are distributed along the diagonal, which is expected for replicates. (B) τ distribution for randomly permuted gene pairs. The τ pairs are biased towards low values, which are the most frequent values in human datasets. (C) τ distribution for τ-uniform random pairs. The τ pairs are uniformly distributed, and not biased towards the low values.
To illustrate the extent to which the abundance of broadly expressed genes affects measures of gene expression conservation, we re-analyzed all the human probe set pairs, without dividing them into τ-groups. We created random probe set pairs by permuting the probe set order within both replicates, and we calculated the Euclidean distances (dEM, dEE and dEZ) both for the pairs of replicates and for the random pairs. Ideally, one would expect to detect very high similarity between replicates, and very low similarity between random pairs.
For Manhattan and Euclidean normalizations, distances for most human random pairs were very small, indistinguishable from the distances between replicates (Fig. 2A and B; Supplementary Fig. S8A and B). This contradicts the assumption that differences between randomly paired genes are to approximate well the rate of neutral divergence, with very low similarity (i.e. high distance) expected (Jordan et al., 2005). For the z-like normalization, distances between random pairs were high, which is consistent with the assumption of pseudo-neutrality (Jordan et al., 2005). However the dEZ values for the replicates were similarly high (Fig. 2C; Supplementary Fig. S8C), whereas they are expected to be low. Thus, the presence of numerous broadly expressed genes causes systematically low values of dEM and dEE between randomly paired genes, and systematically high values of dEZ between conserved gene pairs. The first is a consequence of the fact that it is easier to randomly choose two broadly expressed genes, and thus to get a low value of dEM or dEE. The second is a consequence of low values of r for uniformly expressed genes, leading to the high values of dEZ (as discussed in the Section 3.3). In all cases, the level of gene expression conservation is underestimated.
Fig. 2.

Overrepresentation of broadly expressed human genes causes underestimation of the conservation of expression when randomly permuted pairs are used to approximate the neutral evolution rate. (A, B) For most randomly permuted pairs (grey) the distance (dEM and dEE) is small, indistinguishable from the distances between replicates (green). For τ-uniform random pairs (blue) dEE and dEM are higher, which is more consistent with the assumption about neutral evolution (Jordan et al., 2005). (C) dEZ is high both for randomly permuted gene pairs and for the group of replicates. The distribution of dEZ does not change with the new random pairs set.
Although we show this effect using a specific set of human microarray data, our conclusions are very general and hold for any study in which a significant fraction of the genes is uniformly expressed over conditions (see Fig. S2 and its caption for a mathematical explanation).
3.5 An alternative construction of random gene pairs improves the estimation of expression conservation
To overcome the limitation of using randomly permuted gene pairs to estimate the expression divergence under neutrality, we propose a new procedure to create random gene pairs. This procedure is unbiased regardless of over- or underrepresentation of any expression profiles in the datasets. Consequently, it provides a better approximation of the expression divergence under neutral evolution between distant species. To generate a single random pair of genes, one randomly chooses two expression specificity values, τ1 and τ2, uniformly from the interval of (τmin, τmax), where τmin and τmax are the lowest and the highest values of the observed τ, respectively. Next, one picks the two genes from the two datasets that have the closest τ values to τ1 and τ2, respectively. The resulting pairs of genes have the two τ values uniformly distributed, and not biased as for randomly permuted gene pairs (Fig. 3B and C).
We applied our new procedure 23 920 times to create as many random probe set pairs for human datasets. Then, we calculated the Euclidean distances (dEM, dEE, and dEZ) both for replicates and random probe set pairs. We found that, relative to classical randomly permuted pairs, the distribution of dEE and dEM for τ-uniform random pairs differs strongly from that for replicates (Fig. 2A and B), with a high frequency of large distance values, as expected for very divergent pairs. Of note, dEM and dEE give the same shape of distribution (Figs. 1A and B, and 2A and B). While both of these measures could be combined with τ-uniform sampling to estimate gene expression conservation, for mathematical consistency we prefer the use of dEE.
The estimation of gene expression conservation with dEZ cannot be corrected by creating the set of random gene pairs differently, because dEZ varies significantly with organ specificity for replicates, i.e. for conserved genes, and not for random gene pairs. Thus, we do not recommend using dEZ, and consequently the Pearson's correlation coefficient, in any study which aims to detect similarity between genes expressed uniformly over all conditions.
Of note, neither the standard procedure used to generate random pairs, nor our new proposed approach takes into consideration the time passed since the divergence of two organisms. Therefore, the estimated ‘neutral’ divergence will be the same for closely related species (e.g. human and chimp) and more distant species (e.g. human and mouse).
3.6 Results of the comparative study of human and mouse gene expression differ strongly according to the choice of randomization method
To demonstrate the importance of our novel approach, we investigated how much evidence of selectively constrained gene expression evolution we can detect between human and mouse. We selected 8942 one-to-one orthologous gene pairs from the human and mouse datasets (Su et al., 2004). We created two sets of random gene pairs, using both random permutation and the procedure of τ-uniform sampling, and we calculated the Euclidean distance (dEE) for orthologous gene pairs and for both sets of random pairs (see Fig. S9 for analogous analysis with dEM). If the dEE value for a human–mouse orthologous gene pair is smaller than the fifth percentile of dEE for randomly paired genes, there is some evidence that the expression evolution of this pair has been constrained (Liao and Zhang, 2006a). Using randomly permuted gene pairs did not provide clear evidence for constrained evolution (Fig. 4). Only 8% of orthologous pairs were identified to have a conserved expression pattern, which was close to the random expectation of 5%. In contrast,with τ-uniform random pairs, 29% of orthologous genes were identified to have conserved expression (Fig. 4).
Fig. 4.

The choice of the randomization method changes the conclusions about gene expression evolution between mouse and human. There is no clear evidence for constrained evolution if we compare the distribution of dEE for orthologous (green) and randomly permuted gene pairs (grey). Whereas, comparison of dEE distribution for orthologous (green) and τ-uniform random pairs (blue) suggest that expression evolution is far from neutral.
The number of detected genes with conserved expression pattern may seem surprisingly low in comparison to Liao and Zhang (2006a), who reported that as much as 84% of genes showed conserved expression between human and mouse. However, we note that Liao and Zhang (2006a) used two different metrics to calculate the distance between orthologous genes and between randomly paired genes — the so called net distance and the Euclidean distance, respectively. We show that this inconsistency caused an overestimation of the expression conservation between human and mouse (see Supplementary Materials and Supplementary Fig. S10). Consequently, we believe that correcting for the randomization process yields more accurate results than a one-sided correction of the distance.
We are aware that the alternative way of creating random gene pairs proposed in this article has some weaknesses, such as visible artificial peaks in the dEE distribution (Fig. 4), which are the consequence of the non-uniform distribution of τ between 0 and 1. This is because with the τ-uniform sampling one chooses the genes with less frequent τ values more often than genes with more frequent τ values. For example here, the number of narrowly expressed genes was increased at the expense of decreasing the number of broadly expressed genes. Consequently, when only a few genes have a τ value in some non-negligible range, these few genes might repeat many times in the randomized set, and discrete effects may manifest themselves causing artificial peaks. Note that the peaks would disappear if τ values were uniformly distributed between 0 and 1, but then there would be no need for τ-uniform sampling of gene pairs at all. Note also that the peaks do not affect the analysis, as they do not change the overall shape of the distribution of distance values between the randomized gene pairs (Fig. 4).
Finally, one may argue that the τ-uniform sampling contradicts the very purpose of randomization because it makes a probability of choosing a gene higher, if its τ value is underrepresented in the dataset. But the aim of the set of randomized gene pairs is not to be ‘just random’, but to display maximal divergence between gene pairs, i.e. to simulate the neutral evolution defined in Jordan et al. (2005). In contrast to the standard approach, the τ-uniform sampling makes the distribution of distance values between gene pairs actually independent of the τ distribution observed in the analyzed dataset. Thus, we believe that the distance between τ-uniform random gene pairs approximates better a large neutral divergence.
4 CONCLUSIONS
The Euclidean distance should be used with caution as an estimator of gene expression conservation because it varies as a function of expression specificity. Our results strongly suggest that to assess whether gene expression evolves neutrally, one should use dEE (Euclidean distance preceded by Euclidean normalization) and compare its distribution for orthologous and τ-uniform random pairs. Importantly, we validated this approach on real data, and recovered clear evidence for gene expression conservation between mouse and human. Previous small differences reported between real and random gene pairs were likely caused by the way the random pairs were constructed (Liao and Zhang, 2006a, b). Although in this study we applied our approach to microarray data analysis, the issues highlighted here are also relevant to data acquired with RNA-seq technology (Mortazavi et al., 2008).
We would like to emphasize that while it is possible to verify whether the expression of a given set of genes was under selective pressure, there is no straightforward way to compare the strength of selection acting on two groups of genes with different expression patterns. Indeed, if we compare a group of broadly expressed genes with a group of narrowly expressed genes, with similar high conservation of expression, the latter will always have higher dEE values (and lower dEZ values). This methodological problem suggests a need to re-interpret results from previous evolutionary studies comparing the evolution of broadly and narrowly expressed genes. In particular, studies which have reported higher conservation of organ-specific genes (Liao et al., 2010; Liao and Zhang, 2006b; Movahedi et al., 2011) could have been biased by the fact of using the Pearson's correlation coefficient (equivalent to dEZ) as a measure of conservation.
In this article, we thoroughly analyzed, formally and experi-mentally, the common measures of expression conservation, and we showed the superiority of the Euclidean distance paired with the Euclidean normalization. We also highlighted the limitation of using randomly permuted pairs to approximate neutrally evolving genes, and proposed a new methodology to better estimate the rate of neutral evolution. With the increase of expression data for many species, our work is likely to become very useful for evolutionary studies of gene expression.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank P. Lichocki for fruitful discussion. The authors also thank F. Bastian, N. Galtier and O. Riba-Grognuz for critical comments on the manuscript.
Funding: Etat de Vaud; Swiss National Science Foundation [ProDoc grant 1206624/1]; Swiss Institute of Bioinformatics [to S.B.]
Conflict of Interest: none declared.
Footnotes
† The authors wish it to be known that, in their opinion, the last two authors should be regarded as joint Last Authors.
REFERENCES
- Bastian F., et al. Data Integration in the Life Sciences. Vol. 5109. Springer; 2008. Bgee: integrating and comparing heterogeneous transcriptome data among species; pp. 124–131. of Lecture Notes in Computer Science. [Google Scholar]
- Chan E.T., et al. Conservation of core gene expression in vertebrate tissues. J. Biol. 2009;8:33. doi: 10.1186/jbiol130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M., et al. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods. 2011;8:469–77. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- Hubbard T.J.P., et al. Ensembl 2009. Nucleic Acids Res. 2009;37:D690–D697. doi: 10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan I.K., et al. Evolutionary significance of gene expression divergence. Gene. 2005;345:119–126. doi: 10.1016/j.gene.2004.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P., et al. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science. 2005;309:1850–1854. doi: 10.1126/science.1108296. [DOI] [PubMed] [Google Scholar]
- Liao B.-Y., et al. Contrasting genetic paths to morphological and physiological evolution. Proc. Natl. Acad. Sci. USA. 2010;107:7353–7358. doi: 10.1073/pnas.0910339107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao B.-Y., Zhang J. Evolutionary conservation of expression profiles between human and mouse orthologous genes. Mol. Biol. Evol. 2006a;23:530–540. doi: 10.1093/molbev/msj054. [DOI] [PubMed] [Google Scholar]
- Liao B.-Y., Zhang J. Low rates of expression profile divergence in highly expressed genes and tissue-specific genes during mammalian evolution. Mol. Biol. Evol. 2006b;23:1119–1128. doi: 10.1093/molbev/msj119. [DOI] [PubMed] [Google Scholar]
- Mortazavi A., et al. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Movahedi S., et al. Comparative network analysis reveals that tissue specificity and gene function are important factors influencing the mode of expression evolution in arabidopsis and rice. Plant Physiol. 2011;156:1316–30. doi: 10.1104/pp.111.177865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira V., et al. A problem with the correlation coefficient as a measure of gene expression divergence. Genetics. 2009;183:1597–1600. doi: 10.1534/genetics.109.110247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush J. Microarray data normalization and transformation. Nat. Genet. 2002;32(Suppl):496–501. doi: 10.1038/ng1032. [DOI] [PubMed] [Google Scholar]
- Ramsköld D., et al. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput. Biol. 2009;5:e1000598. doi: 10.1371/journal.pcbi.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D., et al. Biomart–biological queries made easy. BMC Genomics. 2009;10:22. doi: 10.1186/1471-2164-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su A.I., et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., et al. A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 2004;99:909–917. [Google Scholar]
- Xing Y., et al. Assessing the conservation of mammalian gene expression using high-density exon arrays. Mol. Biol. Evol. 2007;24:1283–1285. doi: 10.1093/molbev/msm061. [DOI] [PubMed] [Google Scholar]
- Yanai I., et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21:650–659. doi: 10.1093/bioinformatics/bti042. [DOI] [PubMed] [Google Scholar]
- Yanai I., et al. Incongruent expression profiles between human and mouse orthologous genes suggest widespread neutral evolution of transcription control. OMICS. 2004;8:15–24. doi: 10.1089/153623104773547462. [DOI] [PubMed] [Google Scholar]
- Yang Y. H., et al. Normalization for CDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., et al. Gene expression evolves faster in narrowly than in broadly expressed mammalian genes. Mol. Biol. Evol. 2005;22:2113–2118. doi: 10.1093/molbev/msi206. [DOI] [PubMed] [Google Scholar]
- Zheng-Bradley X., et al. Large scale comparison of global gene expression patterns in human and mouse. Genome Biol. 2010;11:R124. doi: 10.1186/gb-2010-11-12-r124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.