

Abstract
Summary: The large and growing number of genome-wide datasets highlights the need for high-performance feature analysis and data comparison methods, in addition to efficient data storage and retrieval techniques. We introduce BEDOPS, a software suite for common genomic analysis tasks which offers improved flexibility, scalability and execution time characteristics over previously published packages. The suite includes a utility to compress large inputs into a lossless format that can provide greater space savings and faster data extractions than alternatives.
Availability: http://code.google.com/p/bedops/ includes binaries, source and documentation.
Contact: sjn@u.washington.edu and jstam@u.washington.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Experimental genomic data and subsequent analysis results are frequently stored in variations of the browser extensible data (BED) format (Kent et al., 2002). Many BED analyses require set-like operations using genomic coordinates. Others require nearest-element associations between feature sets and quantitative calculations across related genomic segments. Simple examples include selecting transcription factor binding sites >500 base pairs away from any gene, identifying the closest transcriptional start site for every putative replication origin and computing the average exon expression value per gene. The BEDOPS tool suite addresses these and other typical analysis questions. The suite operates efficiently in memory with BED inputs of any size and number, in sharp contrast to the BEDTools (Quinlan and Hall, 2010) and feature-rich Galaxy (Giardine et al., 2005) packages.
Compressing a large file can save valuable disk space and facilitate data transfers, typically at the expense of data access performance. BEDOPS offers high-quality BED compression into a format that actually reduces access times to the vast majority of data relative to any sequential processing approach.
2 PRINCIPAL BEDOPS UTILITIES
The BEDOPS suite consists of a small number of flexible command-line utilities, most of which we introduce here. Several usage examples are shown in Supplementary Information, while a more extensive set of examples (with data) is available at the BEDOPS website. More advanced cases are also included to show various tools working together in typical pipeline fashion.
To briefly summarize the constituent utility programs, the bedops utility offers set operations, including union, subset and difference; closest-features matches nearest elements between datasets; and bedmap maps source information onto genomically related target regions and calculates one or more statistics or summaries per target region. With standard input and output stream support, these principal utilities can be linked together to create more complex operations while maintaining efficiency and scalability. The starch utility compresses BED files into a concise archive format, while its counterpart, unstarch, streams data from such an archive.
3 PERFORMANCE AND METHODS
Principal BEDOPS utilities require sorted inputs and produce sorted outputs. Each BEDOPS utility minimizes memory consumption by retaining only the information required to compute the next line of output. In contrast, alternative tools load all data from a file into memory and create an index before computing results (Quinlan and Hall, 2010), incurring longer run times and higher memory costs that can lead to failures on large inputs (Fig. 1a and Supplementary Fig. S1). The performance disparity between approaches widens in typical workflows, where tools are chained together to form more complex operations. Connecting two programs with a pipe increases memory consumption in a simple additive manner. For tools that read all data into memory, this poses even greater scalability concerns (Supplementary Fig. S2). In contrast, the memory overhead of principal BEDOPS utilities is typically independent of data input sizes. Thus, BEDOPS pipelines scale to dense datasets over a wide range of hardware devices, from modest personal workstations to high-performance cloud-based servers.
Fig. 1.
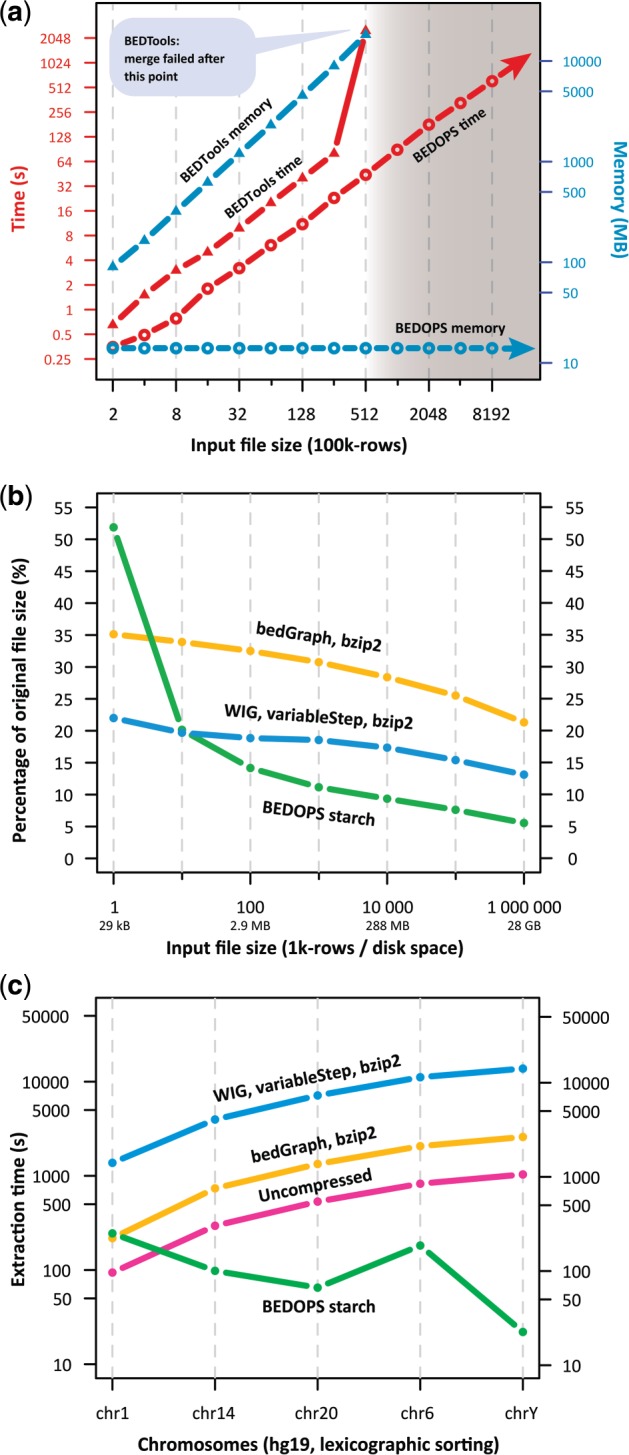
Performance results using subsets of 46-way phyloP (Pollard et al., 2010) human conservation data as inputs. (a) The time and memory resources used to merge overlapping and adjoining genomic segments as a function of input size. (b) Compressed file sizes on disk as a percentage of original uncompressed sizes. (c) Single-chromosome data extraction times from files containing >2.8×109 sorted records. BEDOPS accessed target chromosome data only, whereas conventional approaches processed inputs sequentially
For unsorted data, we provide a speed-optimized sorting utility for data that fit into memory, as well as a method to sort datasets of any size (Supplementary Fig. S3 and Supplementary Methods).
BEDOPS offers a data compression format, starch, that achieves a smaller footprint on disk than popular alternatives, such as compressed bedGraph or WIG format (Fig. 1b). The starch utility transforms a BED file into a more compressible form before applying a standard compression technique (Supplementary Methods). Under the assumption that chromosomes provide a very natural data partition for many genomics analyses, the concise starch format is organized around a chromosome-specific indexing scheme, enabling fast single-chromosome data extractions with improved performance even over a sequentially processed BED file with no compression (Fig. 1c). BEDOPS also includes bedextract, a utility that quickly retrieves information by chromosome directly from a sorted BED file.
4 FILE FORMATS
BEDOPS supports a relaxed variation of the BED specification, to which several popular formats, including WIG, SAM/BAM (Li et al., 2009), VCF (Danacek et al., 2011) and GFF (http://www.sanger.ac.uk/Software/formats/GFF), readily convert (Supplementary Methods). Thus, data currently stored in any of these formats can be transformed and analyzed using features offered by BEDOPS.
5 CONCLUSION
Computing overlap and proximity relationships between datasets is fundamental to many genomic analyses. Working with large datasets complicates computations and necessitates scalable solutions. The BEDOPS suite facilitates common analysis tasks and functions efficiently with inputs of arbitrary size. The suite also includes efficient data storage and retrieval techniques to help manage and query sizable datasets.
Funding: National Institutes of Health Grants (1U54HG004592 and 5U01ES017156).
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Danacek P., et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardine B., et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J., et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard K.S., et al. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.