

Abstract
Zinc thiolate complexes containing N2S tridentate ligands were prepared to investigate their reactivity toward reactive nitrogen species, chemistry proposed to occur at the zinc tetracysteine thiolate site of nitric oxide synthase (NOS). The complexes are unreactive toward nitric oxide in the absence of dioxygen, strongly indicating that NO cannot be the species directly responsible for S-nitrosothiol formation and loss of Zn2+ at the NOS dimer interface in vivo. S-Nitrosothiol formation does occur upon exposure of zinc thiolate solutions to NO in the presence of air, however, or to NO2 or NOBF4, indicating that these reactive nitrogen/oxygen species are capable of liberating zinc from the enzyme, possibly through generation of the S-nitrosothiol. Interaction between simple Zn2+ salts and pre-formed S-nitrosothiols leads to decomposition of the –SNO moiety, resulting in release of gaseous NO and N2O. The potential biological relevance of this chemistry is discussed.
INTRODUCTION
Nitric oxide (NO) functions in the immune, nervous, and circulatory systems to regulate physiological processes, including macrophage cytotoxicity, neuronal signaling, and vasodilation. Biological NO is synthesized in a five-electron oxidation of L-arginine at the active site of nitric oxide synthases (NOSs), for which there are neuronal, endothelial, and inducible NOS isoforms.1–4 These enzymes are head-to-head homodimers,5 containing a tetrahydrobiopterin binding pocket and a tetracysteine thiolate coordination site for zinc at the dimer interface between oxygenase domains. NOS dimer formation and activity require binding of tetrahydrobiopterin.6,7 The absence of zinc destabilizes the NOS dimer and strongly favors dissociation of the enzyme into its composite monomers.8–11 NOS activity consequently relies on zinc binding to the tetra-cysteine thiolate site, and the enzyme may utilize this structural feature to enable a feedback mechanism for regulating NO production.8–11
Exposure of nitric oxide synthase to NO in aerobic aqueous solutions leads to S-nitrosylation of zinc-binding cysteine residues with concomitant dissociation of zinc from the tetrathiolate site. This chemistry destabilizes the NOS homodimer, which dissociates into monomers and halts NO production. Metallothioneins exposed to either aerobic nitric oxide or biologically relevant S-nitrosothiols also lose Zn2+ from tetracysteine thiolate sites with concomitant formation of S-nitrosothiol.12,13 Comparison of studies conducted aerobically and anaerobically reveals that it is not NO per se, but an oxidation product thereof, that is responsible for thiolate nitrosation in these systems.14–16 The generation, translocation, and action of NO and other reactive nitrogen and oxygen species in biological systems are described in a recent review.17
The reactions of cobalt, nickel, and copper complexes with nitrogen oxides, as well as reactions of zinc thiolates with reactive nitrogen and oxygen species (RNOS), have been the subject of much recent investigation.18–24 Anaerobic nitric oxide is unreactive toward the zinc thiolates, but NO2, or NO in the presence of dioxygen, causes S-nitrosothiol formation. Transnitrosation reactions occur upon exposure of zinc thiolates to external S-nitrosothiols.
In the present investigation we have explored the reactivity of zinc thiolates toward reactive nitrogen species. This work was inspired by the aforementioned studies of NOS and metallothioneins with NO, which expose a need to uncover principles that underlie the chemistry of NO-containing compounds with zinc tetracysteine thiolates. We are also interested in the interaction between zinc thiolates and S-nitrosothiols. Although many of the relevant biological targets of NO contain polythiolate zinc centers, we restrict our studies to zinc complexes containing a single thiolate ligand in order to interrogate a unique site. The interaction of transition metals with S-nitrosothiols has been described for copper,19,25–27 iron,28–30 and iridium,31,32 inter alia,33,34 but much less is known about the reactivity of S-nitrosothiols with zinc. Previous studies of S-nitrosothiol interactions with zinc centers coordinated by monodentate thiolates describe reversible transnitrosation events (eq. 1), whereby transfer of
| (1) |
“NO+” from an exogenous S-nitrosothiol to a coordinated thiolate leads to dissociation of the newly formed S-nitrosothiol, and coordination of the thiolate that is generated by transnitros(yl)ation.22,24 This work provides insight into the transnitrosation chemistry of coordinated zinc thiolates, and it inspires further investigation into the nature of the interaction between Zn2+ and S-nitrosothiols.
We selected for study the previously described ligand PATh,35–37 and an alkyl analog APATh (Chart 1) to generate mononuclear zinc thiolates. Geminal dimethyl groups at the ipso carbon provide steric bulk at the sulfur atom, blocking formation of thiolate-bridged metal dimers or oligomers. In addition, gem-dimethyl groups may stabilize tertiary S-nitrosothiol species that can form upon exposure of the zinc thiolates to RNOS.34 The tridentate coordination mode of the PATh and APATh ligands assures tight binding of zinc,37,38 a feature that favors its retention following exposure to RNOS and facilitates analysis of the products of zinc/S-nitrosothiol reactions. We herein describe the preparation and characterization of the desired zinc thiolates and their reactivity toward biologically relevant nitrogen oxides, specifically NO, NO+, and NO2. Reactions of zinc salts with pre-formed S-nitrosothiols are also presented.
Chart 1.
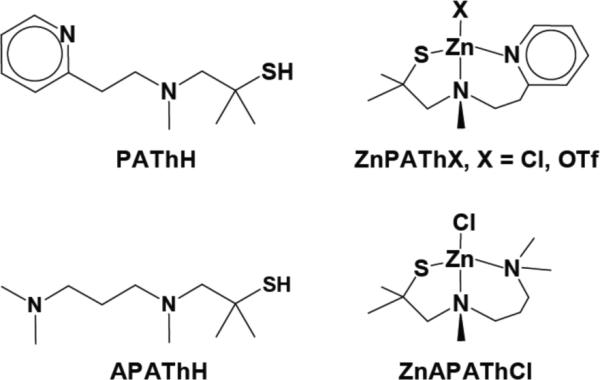
N2S tridentate chelating ligands and the related Zn2+ coordination compounds.
EXPERIMENTAL
General Procedures and Starting Materials
Air- and moisture-sensitive materials were handled in an MBraun glovebox under an inert nitrogen atmosphere. 2-(2-Methylaminoethyl)-pyridine and N,N,N'-trimethyl-1,3-propanediamine were purchased from Sigma-Aldrich and used without further purification. Nitrosonium tetrafluoroborate (97%) was obtained from Strem and used as received. Isobutylene sulfide39 and trityl S-nitrosothiol28 were prepared according to published methods. PAThH was synthesized as described35,36 and purified by Al2O3 column chromatography using 2:1 ethyl acetate: pentane to give the desired product as a colorless oil. ZnPAThCl was prepared by a method analogous to the published procedure and characterized by 1H and 13C NMR spectroscopy, as well as X-ray crystallography.40 Nitric oxide was obtained from Airgas and purified according to published methods.28,41 Briefly, NO was passed through an Ascarite column and a 6 ft coil containing silica gel at −78 °C to remove impurities, and NO was then collected and stored under nitrogen in a gas storage bulb. N2O4 was obtained from Sigma-Aldrich and collected in a gas bulb under nitrogen. Purification by successive freeze-pump-thaw steps at −78 °C facilitated the removal of NO2, N2O and N2O3 impurities. Na15NO2 was purchased from Cambridge Isotope Laboratories and used without purification. n-Butanol was obtained from Alfa Aesar and also used as received. n-Butyl nitrite was prepared by modification of a published procedure,42 in which the reaction scale was 1/40th the size of that reported. APATh15NO was prepared in a manner analogous to that reported for APAThNO below, using 15N n-butyl nitrite to nitrosate the thiol. All gas transfers were performed using gas-tight syringes under an inert atmosphere, unless otherwise noted. Acetonitrile (MeCN), tetrahydrofuran (THF), methylene chloride (DCM), pentane, and diethyl ether were saturated with argon, purified by passage through activated alumina, and stored over 4 Å molecular sieves under an inert nitrogen atmosphere prior to use. Deuterated NMR solvents were obtained from Cambridge Isotope Laboratories and used without further purification. Acetonitrile-d3 and methylene chloride-d2 were brought into the glovebox and stored over 4 Å molecular sieves under inert nitrogen atmosphere.
Synthesis of ZnPAThOTf
To a solution of ZnPAThCl (1.0 g, 3.1 mmol, 1 equiv) in ~10 mL of acetonitrile was added thallium triflate (1.10 g, 3.11 mmol, 1 equiv). The resultant suspension was allowed to stir overnight, after which the reaction mixture was filtered. The solvent was removed from the filtrate and the solid was washed with diethyl ether. The material was dried under vacuum, yielding 0.50 g (37%) of the desired product as an off-white powder. 1H NMR (MeCN-d3) δ 1.32 (s, 3H, CH3), 1.52 (s, 3H, CH3), 2.64 (d, J = 13.4 Hz, 1H, CH2), 2.73 (s, 3H, CH3), 2.89 (d, J = 13.4 Hz, 1H, CH2), 2.95 – 3.26 (m, 4H, CH2), 7.52 – 7.56 (m, 2H, py-Hβ + py-Hδ), 8.05 (td, J = 8 Hz, 1.2 Hz, 1H, py-Hγ), 8.50 (d, J = 4.8 Hz, 1H, py-Hε). 13C NMR (MeCN-d3) δ 33.39, 33.89, 34.99, 49.14, 49.92, 60.67, 73.89, 125.31, 127.45, 142.97, 150.06, 161.65. 1F NMR (MeCN-d3) δ −79.41. IR (MeCN, cm−1) 1612, 1571, 1271. Anal. Calc'd. for C13H19F3N2O3S2Zn: C, 35.66; H, 4.37; N, 6.40. Found: C, 35.33; H, 4.23; N, 6.42.
Synthesis of PAThNO
To a solution of PAThH (1.0 g, 4.5 mmol, 1 equiv) in methylene chloride (~10 mL) was added iso-amyl nitrite (0.6 mL, 4.5 mmol, 1 equiv). The reaction mixture was allowed to stir overnight in the dark and the solvent was removed. Silica gel chromatography (1:1 ethyl acetate: pentane) allowed isolation of the pure product as a dark-green oil (0.52 g, 46%). Care was taken to minimize exposure of the reaction mixture and product to light. 1H NMR (MeCN-d3) δ 1.80 (s, 6H, CH3), 2.40 (s, 3H, CH3), 2.87 (s, 4H, CH2), (s, 2H, CH2), 7.10 – 7.14 (m, 1H, py-Hβ), 7.18 (d, J = 8.8 Hz, 1H, py-Hδ), 7.60 (t, J = 5.6 Hz, 1H, py-Hγ), 8.44 (d, J = 4.8 Hz, 1H, py-Hε). 13C NMR (MeCN-d3) δ 27.32, 36.83, 44.83, 60.41, 60.98,69.01, 122.08, 124.27, 137.07, 150.05, 161.43. IR (MeCN-d3, cm−1) 1591, 1569, 1484 (νNO), 1476. Anal. Calc'd. for C12H19N3OS: C, 56.89; H, 7.56; N, 16.58. Found: C, 57.01; H, 7.39; N, 16.48.
Synthesis of PATh2
Sodium hydride (0.214 g, 8.92 mmol, 1 equiv) was added as a solid to a solution of PAThH (2.0 g, 8.9 mmol, 1 equiv) in ~40 mL of tetrahydrofuran and the mixture was allowed to stir overnight. The resulting suspension was left to stand at −30 °C to precipitate unreacted NaH. The solution was decanted by pipette into an aluminum foil-covered flask and iodine (1.36 g, 5.35 mmol, 0.65 equiv) was added to the stirring solution. After 24 h of stirring the solvent was removed and the oily residue was partitioned between diethyl ether and saturated an aqueous sodium thiosulfate solution. The desired product was extracted into the ether layer and the organics were washed with water and dried (MgSO4). The solvent was removed to yield the desired product as a viscous, bright yellow oil (1.4 g, 69%). 1H NMR (CD2Cl2) δ 1.18 (s, 12H, CH3), 2.40 (s, 6H, CH3), 2.47 (s, 4H, CH2), 2.81 – 2.85 (m, 4H, CH2), 2.88 – 2.92 (m, 4H, CH2), 7.08 (dd, J = 7.2 Hz, 5.6 Hz, 1H, py-Hβ), 7.17 (d, J = 8.0 Hz, 1H, py-Hδ), 7.53 – 7.61 (m, 1H, py-Hγ), 8.47 (d, J = 4.8 Hz, 1H, py-Hε). 13C NMR (CD2Cl2) δ 26.52, 36.83, 45.02, 51.53, 60.84, 68.70, 121.44, 123.74, 136.47, 149.64, 161.26. IR (MeCN-d3, cm−1) 1591, 1569, 1475, 1463, 1436. Anal. Calc'd for C24H38N4S2: C, 64.53; H, 8.57; N, 12.54. Found: C, 64.58; H, 8.70; N, 12.58.
Synthesis of APAThH
To a stirring solution of N,N,N'-trimethyl-1,3-propanediamine (10 g, 86 mmol, 1 equiv) in approximately 500 mL of MeCN was added dropwise a solution of isobutylene sulfide (10.2 mL, 103 mmol, 1.2 equiv) in 100 mL MeCN at room temperature. The reaction mixture was heated to 45 °C for 16.5 h. The solvent was removed in vacuo and the residue was purified by column chromatography (Al2O3, 2:1 pentane: EtOAc, Rf ~ 0.55) to yield the desired product as a colorless oil (6.3 g, 36%). 1H NMR (MeCN-d3) δ 1.28 (s, 6H, CH3), 1.55 – 1.62 m, 2H, CH2), 2.12 (s, 6H, N-(CH3)2), 2.24 (t, J = 7.2 Hz, 2H, CH2), 2.30 (s, 1H, SH), 2.32 (s, 3H, N-CH3), 2.40 (s, 2H, CH2), 2.50 (t, J = 7.2 Hz, 2H, CH2). 13C NMR (MeCN-d3) δ 26.89, 30.75, 45.10, 45.79, 47.03, 58.20, 59.03, 72.82. IR (MeCN-d3, cm−1) 1463, 1383, 1363. Anal. Calc'd. for C10H24N2S·0.14 EtOAc: C, 58.53; H, 11.68; N, 12.93. Found: C, 58.22; H, 11.41; N, 13.07. The presence of residual ethyl acetate was confirmed by the 1H NMR spectrum and the quantity used for the computed elemental composition was adjusted for the best fit to the results.
Synthesis of ZnAPAThCl
To a solution of APAThH (1.0 g, 4.9 mmol, 1 equiv) in ~5 mL of tetrahydrofuran was added NaH (0.117 g, 4.89 mmol, 1 equiv) as a solid. The suspension was allowed to stir until all solid material had dissolved and gas evolution ceased. Zinc chloride (0.667 g, 4.89 mmol, 1 equiv) was suspended in 10 mL of tetrahydrofuran and the suspension was added to the deprotonated ligand solution. The reaction was allowed to stir overnight. The solvent was removed in vacuo, the residue was taken up in methylene chloride, and the solution was filtered. Solvent was evaporated from the filtrate and the resulting residue was washed with diethyl ether to remove unreacted ligand. The remaining solid was dried in vacuo, and the product was isolated as a white powder (0.86 g, 58%). 1H NMR (MeCN-d3) δ 1.39 (d, J = 7.6 Hz, 6H, CH3), 1.66 – 1.74 (m, 1H, CH2), 1.86 – 1.95 (m, 1H, CH2), 2.28 (d, J = 13.0 Hz, 1H, CH2), 2.45 (bs, 6H, N-(CH3)2), 2.53 (s, 3H, N-CH3), 2.61(d, J = 13.0 Hz, 1H, CH2), 2.66 – 2.80 (m, 3H, CH2), 2.95 – 3.00 (m, 1H, CH2). 13C NMR (MeCN-d3) δ 22.96, 36.70, 37.23, 45.20, 48.72, 48.98, 49.53, 62.34, 62.83, 73.55. IR (CH2Cl2, cm−1) 1469, 1456. Anal. Calc'd. for C10H23ClN2SZn·0.30 CH2Cl2: C, 37.52; H, 7.21; N, 8.50. Found: C, 37.59; H, 7.22; N, 8.27. The presence of residual methylene chloride was confirmed by 1H NMR and the quantity used for the computed elemental composition was adjusted for the best fit to the results.
Synthesis of APAThNO
APAThH (1.0 g, 4.9 mmol, 1 equiv) and iso-amyl nitrite (0.66 mL, 4.9 mmol, 1 equiv) were combined in 10 mL of methylene chloride and the solution was stirred overnight in the dark. The solvent was removed and the green residue was purified by silica gel chromatography (96:4 CH2Cl2:MeOH). The product was isolated as a dark-green oil (0.67 g, 58%). Care was taken to minimize exposure of the reaction mixture and product to light. 1H NMR (CD2Cl2) δ 1.55 – 1.62 (m, 2H, CH2), 1.88 (s, 6H, CH3), 2.20 (s, 6H, N-(CH3)2), 2.27 (t, J = 7.2 Hz, 2H, CH2), 2.32 (s, 3H, N-CH3), 2.50 (t, J = 7.2 Hz, 2H, CH2), 3.01 (s, 2H, CH2). 13C NMR (CD2Cl2) δ 26.06, 27.51, 44.90, 45.46, 57.72, 58.67, 59.81, 69.41; UV-Vis (MeCN) λ (ε) 346 nm (554.1), 556 nm (9.2), 599 nm (16.8). IR (CH3CN, cm−1) 1477 (νNO) {1467 (ν15NO)}, 1388, 1369. Anal. Calc'd. for C10H23N3OS: C, 51.47; H, 9.93; N, 18.01. Found: C, 51.76; H, 9.59; N, 18.23.
Phosphine-Mediated APAThNO Ligation
S-(2-(Diphenylphosphino)phenyl)butanethioate was prepared according to published procedures.43,44 To ZnAPAThCl (50 mg, 0.16 mmol, 1 equiv) in ~15 mL MeCN under inert nitrogen atmosphere was added solid NOBF4 (17 mg, 0.15 mmol, 0.9 equiv). The mixture was allowed to stir for 2 h, at which point the phosphine reagent (120 mg, 0.329 mmol, 2 equiv) was added as a solid. An aliquot of the reaction mixture was immediately analyzed by ESI-MS with anaerobic acetonitrile as the carrier solvent. An [M+H]+ peak at m/z = 582.1 (calc'd 582.27) was observed for the phosphine-ligated APAThNO.
Physical Measurements
1H and 13C NMR spectra were recorded on a 400 MHz Bruker Avance spectrometer. Optical spectra were measured on a Varian Cary 50 Bio UV-visible spectrophotometer in 6SQ Starna cells. The Cary WinUV Scanning Kinetics software was used to record optical time-dependent spectra. FT-IR spectra were recorded on a Thermo Nicolet Avatar 360 spectrometer running the OMNIC software package. A CaF2 cell was used for solution IR spectra. ESI-MS analyses were performed on an Agilent 1100 Series LC/MSD Trap spectrometer. An Agilent Technologies 5975C Mass Selective Detector running in electron impact ionization mode was used for EI-MS studies of the reaction headspace.
UV-Vis Studies
All solutions were prepared under an inert nitrogen atmosphere. Optical spectra were recorded at 25 °C.
Zinc Thiolate Reactions with NO(g)
Stock solutions of zinc thiolates were prepared at 10 times the desired reaction concentration. A 2.7 mL volume of acetonitrile was transferred to a cuvette and 300 μL of the zinc thiolate solution was injected into the cell. The cell was sealed with an air-tight Teflon cap containing a septum, and 1 mL of headspace was removed from the sealed cuvette by a gas-tight syringe. Nitric oxide (1 mL) was withdrawn from the storage bulb by a gas-tight syringe and the syringe needle was covered with a septum. The syringe and cuvette were removed from the glovebox and the cuvette was transferred to the UV-Vis spectrophotometer. After one optical scan of the zinc thiolate solution, nitric oxide was quickly injected into the cuvette. The cuvette was shaken to mix the reagents and returned to the spectrophotometer. Optical spectra were recorded at regular intervals. For studies with NO + air, a 1 mL volume of air was injected into the reaction cuvette following completion of the NO study, the cuvette was shaken, and the cell was returned to the spectrophotometer. Optical spectra were again recorded at regular intervals.
Zinc Thiolate Reactions with NOBF4
Stock solutions of NOBF4 in acetonitrile were prepared at 1.1 times the desired reaction concentration. Solutions of zinc thiolates were prepared at 10 times the desired reaction concentration. A 2.7-mL portion of the NOBF4 solution was transferred to a cuvette and the cell was sealed with an air-tight Teflon cap containing a septum. A 300-μL portion of zinc thiolate solution was withdrawn by a gas-tight syringe and the needle was covered by a septum. The cuvette was transferred to the UV-Vis spectrophotometer and, after one optical scan of the nitrosonium tetrafluoroborate solution, the zinc thiolate solution was quickly injected. The cuvette was shaken to mix the reagents and returned to the spectrophotometer. Optical spectra were recorded at regular intervals.
Zinc Thiolate Reactions with NO2(g)
The procedure and quantities used identical as those described for the reactions with nitric oxide, except that NO2 (g) was added instead of NO (g).
S-Nitrosothiol Reactions with Zn(OTf)2
All solutions were prepared under an inert nitrogen atmosphere.
PAThNO
A 2.52 mM PAThNO solution in acetonitrile was prepared and 2.5 mL of the stock solution were sealed in a cuvette. A 1-mL solution of Zn(OTf)2 (2.3 mg, 0.0063 mmol) dissolved in acetonitrile was withdrawn into a gas-tight syringe and the needle was covered by a septum. The cuvette and syringe were removed from the glovebox and the cuvette was placed into the spectrophotometer. After one optical scan of the PAThNO solution, the zinc solution was injected into the cuvette. The cell was shaken to mix the reagents and returned to the spectrophotometer. The final solution was 1.8 mM in both reagents. Optical spectra were recorded at regular intervals.
APAThNO
The procedure was identical to that described for PAThNO, with the following exceptions. The prepared solution of S-nitrosothiol was 2.1 mM in APAThNO and the acetonitrile solution of zinc(II) salt was 5.25 mM in Zn(OTf)2. The final solution was 1.5 mM in both reagents.
FT-IR Time Dependent Studies of S-Nitrosothiol Reactions with Zinc Salts
All reactions were performed under an inert nitrogen atmosphere. To a stirring solution of 100 mg (0.429 mmol) of APAThNO in ~3 mL acetonitrile-d3 was added 1 equiv of solid ZnX2 (X = Cl, OTf). Aliquots of the reaction mixtures were obtained over the duration of the study and injected into a CaF2 solution IR cell under an inert nitrogen atmosphere. The cell was sealed, removed from the glovebox, and placed into the FT-IR spectrometer for analysis. For the study with APATh15NO, the conditions were analogous except that 50 mg (0.21 mmol) of the S-nitrosothiol were used.
1H NMR Studies
All samples were prepared under an inert nitrogen atmosphere.
Comparison of the reactivity of ZnPAThOTf with NO+ with that of PAThNO with Zn(OTf)2. Reactivity of ZnPAThOTf with NO+
A 1-mL portion of a 7.5 mM stock solution of ZnPAThOTf (0.008 mmol) in acetonitrile-d3 was added to 0.9 mg (0.008 mmol) of NOBF4 . The solution was allowed to stir for 1 h, at which point an aliquot was analyzed by 1H NMR spectroscopy.
Reactivity of PAThNO with Zn(OTf)2
PAThNO (0.100 g, 0.395 mmol) in ~2 mL of acetonitrile-d3 was added to Zn(OTf)2 (0.144 g, 0.395 mmol). The solution was allowed to stir for 1 h, at which point an aliquot was analyzed by 1H NMR spectroscopy.
Disulfide titrations with Zn(OTf)2
All samples were prepared under an inert nitrogen atmosphere. Stock solutions of the disulfide PATh2 (15 mM) and Zn(OTf)2 (75 mM) were prepared in acetonitrile-d3. Samples for 1H NMR spectroscopic analysis were prepared by adding 250 μL of the PATh2 stock solution and the appropriate volume of MeCN-d3 to NMR tubes containing 0.5, 1, 2, or 5 equivalents of Zn(OTf)2 in a total reaction volume of 500 μL. The final concentration of PATh2 in each NMR tube was 7.5 mM. Samples were analyzed after 1 h of mixing.
EI-MS Studies of Zinc Thiolate with NO+ and of S-Nitrosothiol with Zinc Salts
All solutions were prepared under an inert nitrogen atmosphere. NOBF4 (0.154 g, 1.32 mmol) was placed into a custom-made gas-tight cell in ~3 mL of methylene chloride in a glovebox under an inert nitrogen atmosphere. The cell was removed from the glovebox and exposed to an inert argon purge. This procedure ensured that high concentrations of nitrogen would not mask peaks in the m/z = 30 region. ZnAPAThCl (0.40 g, 1.3 mmol) was dissolved in ~1 mL of methylene chloride under an inert nitrogen atmosphere and was transferred by gas-tight syringe to the reaction cell under an inert argon flow. The cell was sealed and the reaction mixture was allowed to stir for 1.5 h prior to analysis. The cell was then connected to an inert He gas flow and to the mass spectrometer, and the connecting cables were purged thoroughly prior to analysis of the reaction headspace. Headspace analysis was undertaken with the mass spectrometer operating in selective ion mode. Analogous reaction conditions were employed for the reactions of APAThNO with ZnCl2 and with Zn(OTf)2. ZnCl2 (58.4 mg, 0.428 mmol, 1 equiv) and acetonitrile were added to the reaction cell in the glovebox and a solution of APAThNO (100 mg, 0.214 mmol, 1 equiv) was injected into the cell after an argon purge. A sample of Zn(OTf)2 (77.9 mg, 0.214 mmol, 1 equiv) was exposed to APAThNO (50 mg, 0.21 mmol, 1 equiv) in acetonitrile in a parallel experiment. For the control experiment with APAThNO, a solution containing 50 mg of the S-nitrosothiol was added to the reaction cell in the glovebox. The cell was then sealed, removed from the glovebox, and purged with argon. The headspace of the stirring solution was analyzed by EI-MS after 2 h. All reactions were carried out in the dark to prevent photodecomposition of S-nitrosothiol.
RESULTS AND DISCUSSION
Preparation of Compounds
To prepare for studies of the reactivity of the zinc thiolate moiety we first synthesized the PAThH ligand from isobutylene sulfide and 2-(2-methylaminoethyl)-pyridine according to previously published procedures.35,36 Deprotonation of the ligand followed by ZnCl2 addition afforded the chloride complex ZnPAThCl in decent yield (Scheme 1a). Structural parameters for ZnPAThCl match previously reported values.40 Reaction of thallium triflate with an acetonitrile solution of ZnPAThCl gave ZnPAThOTf, which was obtained in 37% yield (Scheme 1b). The S-nitrosothiol PAThNO was prepared as a green oil for spectral comparison with zinc thiolate/nitrogen oxide reaction products by treating PAThH with iso-amyl nitrite (Scheme 1c).
Scheme 1.

Synthesis of PATh species.
We also synthesized the new ligand APAThH, an alkyl propylaminothiolate, to eliminate C=C and C=N stretches that appear in the characteristic infrared N–O spectral stretching region and thereby facilitate the interpretation of IR spectroscopic studies (Figure S1 in the Supporting Information). APAThH retains the coordinating atoms afforded by PATh but replaces the spectrally interfering pyridyl moiety of the latter with a tertiary amine. Condensation of N,N,N'-trimethyl-1,3-diaminopropane with isobutylene sulfide readily yielded APAThH (Scheme 2a). The zinc complex ZnAPAThCl was prepared by analogy to the synthesis of ZnPAThCl (Scheme 2b). We were unable to obtain X-ray quality crystals of ZnAPAThCl. Given the similarity between the coordination environments of PAThH and APAThH, however, we expect the structural characteristics of ZnAPAThCl to be similar to those of ZnPAThCl.40 The S-nitrosothiol APAThNO was obtained by transnitrosation of APAThH with iso-amyl nitrite (Scheme 2c), and the disulfide PATh2 (Scheme 1d) was prepared by iodine oxidation of [PATh]− thiolate. These compounds were synthesized for spectral comparison with the products of reactions between the zinc complexes and RNOS.
Scheme 2.

Synthesis of APATh species.
Exposure of Zinc PATh and APATh Complexes to Nitric Oxide under Various Conditions
No reaction was observed by UV-Vis spectroscopy upon addition of excess nitric oxide to anaerobic acetonitrile solutions of ZnPAThCl, ZnPAThOTf, and ZnAPAThCl. Injection of air into the cuvette led to the immediate appearance of peaks in the UV-Vis spectrum that are characteristic of tertiary S-nitrosothiols (eqs. 2, 3, Fig. 1, and Figures S2 and S3 in the Supporting Information).34 The “0 min” trace reflects immediate appearance of a spectral band arising from the S-nitrosothiol, which then decays over time.
Figure 1.

UV-Vis reactivity profiles for anaerobic 1.5 mM ZnPAThCl + excess NO (g), left, and 1.5 mM ZnPAThCl + excess NO (g) + air, right. Recorded in acetonitrile.
| (2) |
| (3) |
The lack of reactivity under anaerobic conditions is consistent with previous findings,45 which reveal Zn2+-based model systems that utilize β-diketiminate24 and tris(pyrazolyl)borate22 scaffolds to be inert to anaerobic NO (g). This finding has important implications for nitric oxide in biology. Specifically, this result requires that it is not NO itself, but an oxidation product thereof, that is responsible for S-nitrosothiol formation at biological zinc thiolate sites, such as those in NOS and metallothioneins. NO has been previously described as a poor nitrosating agent for thiols.46 The nitric oxide mediated thiolate nitros(yl)ation reaction is an electron-releasing process (eq. 4), and, in the absence of a viable electron acceptor, no reaction is observed. In biological milieu, however, dioxygen is present to oxidize NO to NO2, for example, thereby enabling S-nitrosothiol formation (eq. 5, 6). In accord with the redox nature of NO, the reported reaction between the model complex iPr2TpZn–StBu and NO/O2 mixtures, or NO2 (g), led to S-nitrosothiol formation and isolation of iPr2TpZn–(O2NO).22 Our results are consistent with this S-nitros(yl)ation chemistry, although we did not explore the fate of the Zn2+ ion in the NO/O2 chemistry.
| (4) |
| (5) |
| (6) |
Reactions of Zinc PATh and APATh Complexes with NOBF4
We next examined the reactivity of nitrosonium tetrafluoroborate with zinc thiolates. Exposure of ZnPAThCl, ZnPAThOTf, and ZnAPAThCl to 1 equiv of NOBF4 in acetonitrile led to the appearance of peaks in the optical spectra corresponding to tertiary S-nitrosothiols (Figure S4 in the Supporting Information). S-Nitrosothiol formation was demonstrated for the reaction of ZnAPAThCl with NO+ by trapping APAThNO and studying the products by mass spectrometry (Scheme 3).43,44 Utilization of phosphine-mediated N=O bond scission and ESI-MS analysis of products confirmed that APAThNO formed upon exposure of ZnAPAThCl to NO+ (Figure S5 in the Supporting Information). We therefore conclude that optical peaks attributed to tertiary S-nitrosothiol formation in reactions of zinc thiolates and RNOS are correctly assigned.
Scheme 3.

Traceless ligation of APAThNO by phosphine-mediated N=O bond scission.43,44
Reactions of S-Nitrosothiols with Zinc(II) Salts
PAThNO and APAThNO were prepared to examine the interaction between these pre-formed tertiary S-nitrosothiols and Zn2+. Optical spectroscopic studies of reaction mixtures containing 1 equiv of Zn(OTf)2 and PAThNO or APAThNO revealed a decrease in the intensity of the peaks at ~340 nm compared to optical spectra of S-nitrosothiol solutions lacking Zn2+ (Fig. 2, eq. 7). This spectroscopic change corresponds to decay of the nO → π* transitions for each S-nitrosothiol34 in the presence of Zn(OTf)2. Peaks at 602 nm in the spectra of PAThNO and APAThNO disappear completely upon addition of Zn(OTf)2. A new peak, corresponding to an
| (7) |
as-of-yet unidentified species, grows in at 330 nm after 1 h in the APAThNO/Zn(OTf)2 reaction spectrum (Fig. 2). IR spectroscopic analysis of the APAThNO reaction with ZnCl2 revealed the appearance of three bands in the 1600 cm−1 region of the spectrum, and two bands were similarly observed for the reaction of APAThNO with Zn(OTf)2 (Figure S6 in the Supporting Information). Concomitant disappearance of the υNO feature of APAThNO at 1477 cm−1 was observed for reactions with both zinc(II) salts (Figure S6 in the Supporting Information). Additionally, inner-sphere coordination of a single triflate anion was deduced for the reaction with Zn(OTf)2, judging by the appearance of a band at 1311 cm−1, a signature for covalent binding of the sulfonate moiety.47 There is a corresponding decrease in the intensity of the asymmetric sulfonate ion stretching frequency at 1287 cm−1 from the outer-sphere triflate anion.47 Use of 15N-isotopically labeled APAThNO revealed that the bands in the 1600 cm−1 infrared spectral region for reactions with ZnCl2 and Zn(OTf)2 showed negligible changes compared to the unlabeled nitrosothiol, indicating that these peaks are not caused by stretches of N-containing moieties in the product(s) (Figure S7 in the Supporting Information).
Figure 2.

Disappearance of characteristic S-nitrosothiol peaks occurs upon exposure of 1.8 mM PAThNO (left) and 1.5 mM APAThNO (right) to 1 equiv of Zn(OTf)2 in acetonitrile. Reactions were monitored for 1 h.
There is a dependence on the anion in the reactions of zinc salts with the S-nitrosothiol, as inferred from the presence of an additional band in the IR spectra of the ZnAPAThCl/NOBF4 and APAThNO/ZnCl2 reaction mixtures that is absent in solutions containing a triflate counterion (Figures S6 and S7 in the Supporting Information). The better coordinating ability of chloride compared to triflate may control the availability of binding sites for the –SNO moiety on zinc, or Cl− may participate directly in the reaction.
To further explore the chemistry between Zn2+ and the –SNO moiety of S-nitrosothiols we recorded 1H NMR spectra for reactions of ZnPAThOTf with NOBF4 and of PAThNO with Zn(OTf)2 (Figure S8 in the Supporting Information). All of the peaks observed in the PAThNO/Zn(OTf)2 reaction mixture are present in the ZnPAThOTf/NOBF4 1H NMR spectrum. Spectroscopic comparison of mixtures suggests that PAThNO binds zinc(II) and, based on infrared spectroscopic evidence, induces dissociation of one triflate counteranion. In the related ZnPAThOTf reaction with NOBF4, NO+ attacks the thiolate, such that the reactive species in both instances is [Zn(PAThNO)(OTf)]+. In addition, the spectrum contains a new singlet at 2.05 ppm. This peak may arise from the gem-dimethyl groups of a metastable intermediate, possibly coordinated S-nitrosothiol. We were unable to trap and identify this species.
The complexity of the NMR spectra of these reaction mixtures precluded definitive identification of the products. However, being aware of the propensity of S-nitrosothiols to decompose to the corresponding disulfides over time,34 we prepared the disulfide PATh2 as a control to assess whether exposure of zinc thiolates to nitrosonium ion, or of S-nitrosothiol to Zn2+, could result in disulfide formation. 1H NMR spectral titration studies of PATh2 with Zn(OTf)2 revealed formation of various species, depending on the ratio of zinc to disulfide in solution (Figure S9 in the Supporting Information). The 1H NMR spectra of neither the free disulfide nor of disulfide solutions containing Zn2+ matched the 1H NMR spectra of the zinc thiolate/NOBF4 or S-nitrosothiol/Zn(OTf)2 reaction mixtures (Figure S10 in the Supporting Information). From these results we conclude that disulfide is not a product of S-nitrosothiol exposure to Zn2+. It therefore appears likely that zinc promotes heterolytic, not homolytic, cleavage of the S–N bond of S-nitrosothiols. These results complement previous studies of the transnitrosation reactions between monodentate zinc(II)-bound thiolates and exogenous S-nitrosothiols (eq. 1, vide supra).22,24
Finally, we analyzed the reaction headspace of ZnAPAThCl/NOBF4, APAThNO/ZnCl2, and APAThNO/Zn(OTf)2 reaction mixtures by EI-MS to determine whether gaseous species are generated by S-nitrosothiols in the presence of Zn2+. NO and N2O were detected in all instances (Figures S11 – S13 in the Supporting Information). NO2 was not observed. The ZnCl2/APAThNO reaction was also monitored for the possible formation of Cl2 or NOCl (Figure S12 in the Supporting Information). Neither was observed, indicating that the chloride anion is unlikely to participate in any redox chemistry. The headspace of a control solution of APAThNO was examined by EI-MS and neither NO nor N2O were present (Figure S14 in the Supporting Information). This control experiment confirms that NO and N2O are true reaction products and not the result of S-nitrosothiol autodecomposition. A possible reaction pathway that accounts for Zn2+-mediated decomposition of S-nitrosothiols with concomitant generation of N2O, and not disulfide, involves thiolate oxidation to generate transient RS[O]+ species, which may undergo further decomposition. The existence of reactive RS[O]+ species is described in the literature.48 Two equiv of S-nitrosothiol may react to form N2O, RS[O]+, and RS−. The resultant thiolate may coordinate Zn2+ to regenerate the starting complex. Equation 8 describes the proposed reaction. We also suggest that Zn2+, by forming a stable complex with the resulting thiolate, may activate S-nitrosothiols to release NO+, which may, in turn, lead to catalytic S-nitrosothiol decomposition and generation of NO.49
| (8) |
Reactions of Zinc PATh and APATh Complexes with NO2
Exposure of ZnPAThCl, ZnPAThOTf, and ZnAPAThCl to excess NO2 led to immediate appearance and subsequent decay of optical features attributed to S-nitrosothiols (Figure S15 in the Supporting Information). The decomposition of S-nitrosothiols in the presence of excess nitrogen dioxide has been reported.50,51 N2O4 equilibrates with NO2 and oxidizes S-nitrosothiols to thionitrates (eq. 9), which lack the optical features of S-nitrosothiols.
| (9) |
Potential Biological Relevance
Consistent with prior results, we find that an isolated zinc thiolate bond is unreactive toward nitric oxide in the absence of an oxidizing agent. We propose, on the basis of spectral changes of S-nitrosothiols in the presence of Zn2+, that zinc, in its role as a Lewis acid, activates the RSNO moiety, possibly stabilizing RS− with release of NO+. Biological consequences for nitric oxide synthase may be the need for zinc to dissociate from nitrosated thiolates in order to maintain physiological reversibility of the nitrosation reaction. Without such dissociation, interaction of Zn2+ with S-nitrosothiols could result in thiolate oxidation that would block conversion back to the biologically useful form of the enzyme.52,53 Irreversible modification of zinc thiolates in transcription factors following oxidative stress provides a precedent for the biological irreversibility that we propose here.54,55
SUMMARY AND CONCLUSIONS
Nitric oxide under anaerobic conditions is unreactive toward the zinc thiolates examined in this work, which implies that it is not NO itself, but an oxidized form thereof, that is the active species responsible for S-nitrosothiol formation and demetallation in NOS and metallothioneins in biology. Zinc thiolate reactivity with NO in the presence of dioxygen supports this hypothesis, and studies with NO2 indicate that nitrogen dioxide is a viable species for inducing zinc thiolate nitrosation in vivo. Exposure of ZnPAThCl, ZnPAThOTf, and ZnAPAThCl to NO+ in the form of nitrosonium tetrafluoroborate also leads to thiolate nitrosation. Addition of NOBF4 to ZnPAThOTf, or of Zn(OTf)2 to PAThNO, most likely generates a coordinated S-nitrosothiol as a transient species, but for both PAThNO and APAThNO, interaction of the –SNO moiety with Zn2+ promotes its decomposition. NO and N2O were detected in the headspace of solutions containing Zn2+ and S-nitrosothiol, irrespective of whether the latter was prepared independently or generated in situ. The absence of disulfide products from reaction mixtures is characteristic of heterolytic, not homolytic, cleavage of the S–N bond in the presence of Zn2+, the role of which may be to activate the –SNO moiety toward heterolysis, possibly by stabilizing the thiolate formed in the reaction. Oxidized thiolates, RS[O], may be the ultimate reaction products, as proposed in equation 8.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Science Foundation grant CHE0907905 (SJL) and by National Institute of Health grant 1S10RR013886-01 awarded to the MIT Department of Chemistry Instrumentation Facility. We thank Dr. Yogesh Surendranath for technical assistance with the EI-MS studies, as well as Drs. Zachary Tonzetich, Joel Rosenthal, Michael Pluth, and Andrew Tennyson for valuable advice and discussion.
Footnotes
SUPPORTING INFORMATION AVAILABLE The infrared spectrum of PAThNO and related species; optical spectra of reactions of NO ± air, NO2, and NOBF4 with zinc thiolates; the ESI-MS spectrum of the reaction between ZnAPAThCl, NOBF4, and phosphine; solution IR comparisons of the reactions of APAThNO and APATh15NO with ZnCl2 and Zn(OTf)2; 1H NMR spectra of ZnPAThOTf/NOBF4, PAThNO/Zn(OTf)2, and PATh2/Zn(OTf)2 reactions; and EI-MS analyses of the headspaces of reaction mixtures containing ZnAPAThCl/NOBF4, APAThNO/ZnCl2, APAThNO/Zn(OTf)2, and APAThNO are available in the supporting information. This material is available free of charge at http://pubs.acs.org.
REFERENCES
- (1).Bredt DS, Snyder SH. Annu. Rev. Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]
- (2).Lehninger AL, Nelson DL, Cox MM. Lehninger Principles of Biochemistry. 4 ed W. H. Freeman & Co.; 2004. [Google Scholar]
- (3).Moncada S, Palmer RMJ, Higgs EA. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- (4).Nathan C, Xie Q.-w. Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- (5).Ghosh DK, Stuehr DJ. Biochemistry. 1995;34:801–807. doi: 10.1021/bi00003a013. [DOI] [PubMed] [Google Scholar]
- (6).Ghosh DK, Wu C, Pitters E, Moloney M, Werner ER, Mayer B, Stuehr DJ. Biochemistry. 1997;36:10609–10619. doi: 10.1021/bi9702290. [DOI] [PubMed] [Google Scholar]
- (7).Crane BR, Rosenfeld RJ, Arvai AS, Ghosh DK, Ghosh S, Tainer JA, Stuehr DJ, Getzoff ED. EMBO J. 1999;18:6271–6281. doi: 10.1093/emboj/18.22.6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hemmens B, Goessler W, Schmidt K, Mayer B. J. Biol. Chem. 2000;275:35786–35791. doi: 10.1074/jbc.M005976200. [DOI] [PubMed] [Google Scholar]
- (9).Mitchell DA, Erwin PA, Michel T, Marletta MA. Biochemistry. 2005;44:4636–4647. doi: 10.1021/bi0474463. [DOI] [PubMed] [Google Scholar]
- (10).Ravi K, Brennan LA, Levic S, Ross PA, Black SM. Proc. Natl. Acad. Sci. USA. 2004;101:2619–2624. doi: 10.1073/pnas.0300464101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tummala M, Ryzhov V, Ravi K, Black SM. DNA Cell Biology. 2008;27:25–33. doi: 10.1089/dna.2007.0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chen Y, Irie Y, Keung WM, Maret W. Biochemistry. 2002;41:8360–8367. doi: 10.1021/bi020030+. [DOI] [PubMed] [Google Scholar]
- (13).Khatai L, Goessler W, Lorencova H, Zangger K. Eur. J. Biochem. 2004;271:2408–2416. doi: 10.1111/j.1432-1033.2004.04160.x. [DOI] [PubMed] [Google Scholar]
- (14).Aravindakumar CT, Ceulemans J, Ley M. d. Biochem. J. 1999;344 doi: 10.1042/0264-6021:3440253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Williams DLH. Chem. Soc. Rev. 1985;14:171–196. [Google Scholar]
- (16).Zhu J, Meeusen J, Krezoski S, Petering DH. Chem. Res. Toxicol. 2010;23:422–431. doi: 10.1021/tx900387k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tennyson Andrew G., Lippard Stephen J. Chem. Biol. 2011;18:1211–1220. doi: 10.1016/j.chembiol.2011.09.009. [DOI] [PubMed] [Google Scholar]
- (18).Melzer MM, Jarchow-Choy S, Kogut E, Warren TH. Inorg. Chem. 2008;47:10187–10189. doi: 10.1021/ic8014332. [DOI] [PubMed] [Google Scholar]
- (19).Melzer MM, Li E, Warren TH. Chem. Commun. 2009:5847–5849. doi: 10.1039/b911643e. [DOI] [PubMed] [Google Scholar]
- (20).Puiu SC, Warren TH. Organometallics. 2003;22:3974–3976. [Google Scholar]
- (21).Tennyson AG, Dhar S, Lippard SJ. J. Am. Chem. Soc. 2008;130:15087–15098. doi: 10.1021/ja803992y. [DOI] [PubMed] [Google Scholar]
- (22).Varonka MS, Warren TH. Inorg. Chem. 2009;48:5605–5607. doi: 10.1021/ic900664r. [DOI] [PubMed] [Google Scholar]
- (23).Wiese S, Kapoor P, Williams KD, Warren TH. J. Am. Chem. Soc. 2009;131:18105–18111. doi: 10.1021/ja903550n. [DOI] [PubMed] [Google Scholar]
- (24).Varonka MS, Warren TH. Inorg. Chim. Acta. 2007;360:317–328. [Google Scholar]
- (25).Stubauer G, Giuffrè A, Sarti P. J. Biol. Chem. 1999;274:28128–28133. doi: 10.1074/jbc.274.40.28128. [DOI] [PubMed] [Google Scholar]
- (26).Baciu C, Cho K-B, Gauld JW. J. Phys. Chem. B. 2005;109:1334–1336. doi: 10.1021/jp0443759. [DOI] [PubMed] [Google Scholar]
- (27).Williams DLH. Chem. Commun. 1996:1085–1091. [Google Scholar]
- (28).Harrop TC, Tonzetich ZJ, Reisner E, Lippard SJ. J. Am. Chem. Soc. 2008;130:15602–15610. doi: 10.1021/ja8054996. [DOI] [PubMed] [Google Scholar]
- (29).Vanin AF, Papina AA, Serezhenkov VA, Koppenol WH. Nitric Oxide. 2004;10:60–73. doi: 10.1016/j.niox.2004.02.005. [DOI] [PubMed] [Google Scholar]
- (30).Szaciłowski K, Chmura A, Stasicka Z. Coord. Chem. Rev. 2005;249:2408–2436. [Google Scholar]
- (31).Perissinotti LL, Estrin DA, Leitus G, Doctorovich F. J. Am. Chem. Soc. 2006;128:2512–2513. doi: 10.1021/ja0565976. [DOI] [PubMed] [Google Scholar]
- (32).Perissinotti LL, Leitus G, Shimon L, Estrin D, Doctorovich F. Inorg. Chem. 2008;47:4723–4733. doi: 10.1021/ic7024999. [DOI] [PubMed] [Google Scholar]
- (33).Lee J, Chen L, West AH, Richter-Addo GB. Chem. Rev. 2002;102:1019–1066. doi: 10.1021/cr0000731. [DOI] [PubMed] [Google Scholar]
- (34).Szaciłowski K, Stasicka Z. Prog. React. Kinet. Mech. 2001;26:1–58. [Google Scholar]
- (35).Chang S, Karambelkar VV, diTargiani RC, Goldberg DP. Inorg. Chem. 2001;40:194–195. doi: 10.1021/ic001079b. [DOI] [PubMed] [Google Scholar]
- (36).Chang S, Karambelkar VV, Sommer RD, Rheingold AL, Goldberg DP. Inorg. Chem. 2002;41:239–248. doi: 10.1021/ic010321r. [DOI] [PubMed] [Google Scholar]
- (37).diTargiani RC, Chang S, Salter MH, Hancock RD, Goldberg DP. Inorg. Chem. 2003;42:5825–5836. doi: 10.1021/ic034337o. [DOI] [PubMed] [Google Scholar]
- (38).Andersen RJ, diTargiani RC, Hancock RD, Stern CL, Goldberg DP, Godwin HA. Inorg. Chem. 2006;45:6574–6576. doi: 10.1021/ic060497z. [DOI] [PubMed] [Google Scholar]
- (39).Mills DK, Font I, Farmer PJ, Hsiao Y-M, Tuntulani T, Buonomo RM, Goodman DC, Musie G, Grapperhaus CA, Maguire MJ, Lai C-H, Hatley ML, Smee JJ, Bellefeuille JA, Darensbourg MY, Hancock RD, Eng S, Martell AE. In: Inorg. Synth. Marcetta YD, editor. 2007. pp. 89–98. [Google Scholar]
- (40).Ji M, Vahrenkamp H. Eur. J. Inorg. Chem. 2005;2005:1398–1405. [Google Scholar]
- (41).Lorković IM, Ford PC. Inorg. Chem. 2000;39:632–633. doi: 10.1021/ic9910413. [DOI] [PubMed] [Google Scholar]
- (42).Noyes WA. Org. Synth. 1943;2:103. [Google Scholar]
- (43).Zhang J, Wang H, Xian M. Org. Lett. 2009;11:477–480. doi: 10.1021/ol802663q. [DOI] [PubMed] [Google Scholar]
- (44).Zhang J, Wang H, Xian M. J. Am. Chem. Soc. 2009;131:3854–3855. doi: 10.1021/ja900370y. [DOI] [PubMed] [Google Scholar]
- (45).Tennyson AG, Lippard SJ. Unpublished work.
- (46).Hogg N. Annu. Rev. Pharmacol. Toxicol. 2002;42:585–600. doi: 10.1146/annurev.pharmtox.42.092501.104328. [DOI] [PubMed] [Google Scholar]
- (47).Lawrance GA. Chem. Rev. 1986;86:17–33. [Google Scholar]
- (48).Derbesy G, Harpp DN. J. Org. Chem. 1995;60:1044–1052. [Google Scholar]
- (49).Zhao Y-L, McCarren PR, Houk KN, Choi BY, Toone EJ. J. Am. Chem. Soc. 2005;127:10917–10924. doi: 10.1021/ja050018f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Oae S, Shinhama K. Org. Prep. Proced. Int. 1983;15:165–198. [Google Scholar]
- (51).Oae S, Shinhama K, Fujimori K, Kim YH. Bull. Chem. Soc. Jpn. 1980;53:775–784. [Google Scholar]
- (52).Sengupta R, Ryter SW, Zuckerbraun BS, Tzeng E, Billiar TR, Stoyanovsky DA. Biochemistry. 2007;46:8472–8483. doi: 10.1021/bi700449x. [DOI] [PubMed] [Google Scholar]
- (53).Benhar M, Forrester MT, Hess DT, Stamler JS. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Kröncke KD. Arch. Biochem. Biophys. 2007;463:183–187. doi: 10.1016/j.abb.2007.03.008. [DOI] [PubMed] [Google Scholar]
- (55).Kröncke K-D, Klotz L-O, Suschek CV, Sies H. J. Biol. Chem. 2002;277:13294–13301. doi: 10.1074/jbc.M111216200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.