

Abstract
Purpose.
Time and spectrally resolved measurements of autofluorescence have the potential to monitor metabolism at the cellular level. Fluorophores that emit with the same fluorescence intensity can be discriminated from each other by decay time of fluorescence intensity after pulsed excitation. We performed time-resolved autofluorescence measurements on fundus samples from a donor with significant extramacular drusen.
Methods.
Tissue sections from two human donors were prepared and imaged with a laser scanning microscope. The sample was excited with a titanium-sapphire laser, which was tuned to 860 nm, and frequency doubled by a BBO crystal to 430 nm. The repetition rate was 76 MHz and the pulse width was 170 femtoseconds (fs). The time-resolved autofluorescence was recorded simultaneously in 16 spectral channels (445–605 nm) and bi-exponentially fitted.
Results.
RPE can be discriminated clearly from Bruch's membrane, drusen, and choroidal connective tissue by fluorescence lifetime. In RPE, bright fluorescence of lipofuscin could be detected with a maximum at 510 nm and extending beyond 600 nm. The lifetime was 385 ps. Different types of drusen were found. Most of them did not contain lipofuscin and exhibited a weak fluorescence, with a maximum at 470 nm. The lifetime was 1785 picoseconds (ps). Also, brightly emitting lesions, presumably representing basal laminar deposits, with fluorescence lifetimes longer than those recorded in RPE could be detected.
Conclusions.
The demonstrated differentiation of fluorescent structures by their fluorescence decay time is important for interpretation of in vivo measurements by the new fluorescence lifetime imaging (FLIM) ophthalmoscopy on healthy subjects as well as on patients.
Fluorophores that emit with the same fluorescence intensity and that exhibit spectral overlap can be discriminated from each other by the decay time of fluorescence intensity after pulse excitation. The in vitro determined elongated decay time of drusen in comparison with that of RPE is also in vivo detectable.
Introduction
Currently, studies of patterns of static autofluorescence intensities from the human retina often are used to diagnose age-related macular degeneration (AMD).1 However, studies of the time-resolved fluorescence of endogenous fluorophores2,3 have greater potential for elucidating the mechanisms of pathogenesis in AMD. One important advantage of time resolved fluorescence measurements is that fluorescence lifetimes are independent of the absorption in neighboring layers, like xanthophyll in the macula. Additionally, weakly emitting fluorophores with long lifetimes can be discriminated from strongly emitting ones with a short lifetime. Moreover, by exploiting the different excitation and emission properties of the fluorophores, most fluorophores can be differentiated completely. To carry out these types of measurements in vivo, a scanning laser ophthalmoscope for 2-dimensional fluorescence lifetime mapping of the human fundus was developed at the Eye Clinic of the Jena University Hospital.4,5 To improve the interpretation of in vivo measurements of time-resolved autofluorescence, comparing measurements of fundus samples are helpful.
We report measurements of time-resolved fluorescence mapping on fixed fundus tissue from two donors, ages 60 and 92.
Materials and Methods
Experimental Setup
To scan the sample, a laser scanning microscope, consisting of an inverted microscope (Axiovert 100M; Carl Zeiss, Jena, Germany) with a 40× water immersion objective (C-Apochromat 40× WI; Carl Zeiss) and the LSM510 scan head, was used. The excitation source is a titanium-sapphire laser (Mira 900D; Coherent, Dieburg, Germany) pumped by a NdYVO4 (Verdi; Coherent, Goettingen, Germany). The repetition rate of the laser was 76 MHz and the pulse width of the pulses (full width at half maximum, FWHM) was measured to be 170 femtoseconds (fs). The emission wavelength of the laser was tuned to 860 nm and frequency doubled by a BBO crystal to 430 nm. This wavelength corresponds to the excitation range of in vivo measurements, which takes into account the transmission of the ocular media. The autofluorescence emitted was descanned and guided to a polychromator (is250, Chromex; Bruker Optik GmbH, Leipzig, Germany), which dispersed the fluorescence light and focused it onto a photomultiplier array (PML 16; Becker & Hickl, Berlin, Germany) consisting of 16 channels. Each spectral channel had a bandwidth of 10 nm. In total, the spectral range from 445–605 nm was covered. Single photon events detected in one of the 16 channels were recorded by a time-correlated single photon counting (TCSPC) board (SPC 150; Becker & Hickl) and saved as data stream. In the data stream the time with respect to the laser pulse and the number of the spectral channel are saved, so that time- and spectrally-resolved histograms can be reconstructed from the data.6
The decay of the normalized fluorescence intensity after excitation with a laser pulse was approximated by a bi-exponential function according equation (1):
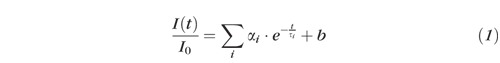 |
with I(t) indicating the detected fluorescence intensity at time t, I0 the detected fluorescence intensity at time t = 0, αi the amplitude, τi the lifetime, and b the background.
The relative contribution Qi of each component to the overall fluorescence decay was calculated according to equation (2):
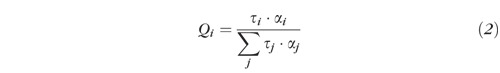 |
For the 2-dimensional fitting procedure, the program SPCImage 2.9.1 (Becker & Hickl) was used. The criterion for optimal fitting was the minimization of χr2:
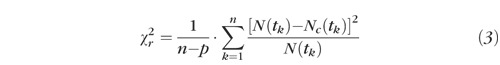 |
with n indicating the number of time channels, p the number of free parameters, N(tk) the detected photons in time channel k, and Nc(tk) the calculated number of photons in time channel k as convolution of model function and instrumental response function.
If the deviation between detected and calculated photons is determined by noise and for n≫p, χr2 is close to 1. The slope of the measured dynamic fluorescence was used as the instrumental response function.
As a result of the fitting process, color coded images of lifetimes (τm, τ1τ2), amplitudes (α1, α2), and relative contributions (Q1, Q2) are obtained.
The mean lifetime, τm, is calculated as:
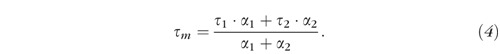 |
In addition, histograms of lifetimes, amplitudes, and relative contributions were calculated. The histograms show which value of lifetime, amplitude or relative contribution is observed most frequently in either the whole image or in selected ranges.
Tissue Samples
All experiments were performed in accordance with the Declaration of Helsinki. Eyes from two donors were studied. A segment spanning from the macula to the pars plicata was collected from a 60-year-old female donor with no known history of ophthalmic disease, but with significant extramacular drusen. In addition, an 8 mm punch was collected from the macula of a 92-year-old female donor with dry AMD. Samples were fixed in 4% paraformaldehyde for 2 hours within 5.5 hours of death, and were processed as described previously.7 Briefly, wedges containing RPE, choroid, and sclera were washed in PBS and cryopreserved with increasing concentrations of sucrose. Following embedment in optimal cutting temperature compound (Ted Pella, Redding, CA), 7 μm sections were collected on a cryostat, coverslipped with aqueous mounting medium, and used for spectral imaging. Slides with mounting medium alone were coverslipped to assess background fluorescence.
Results
Fluorescence Spectra
The fluorescence of different structures from the sample was measured and is presented in Figure 1. In the fluorescence intensity image on the left, the fluorescence intensity is summed over all time and spectral channels, and the fluorescence intensity is encoded by a gray scale. The image on the right displays the mean fluorescence lifetimes determined for each pixel, which are then encoded by color. The color scale used to encode the lifetimes is shown below the figure. Red represents short lifetimes (0 picoseconds [ps]), whereas blue represents longer lifetimes (3500 ps).
Figure 1. .

Human fundus sample. Left: Fluorescence intensity image. Right: Fluorescence lifetime image. Average lifetimes determined for each pixel are encoded by colors ranging from red (0 ps) to blue (3500 ps) as shown by the color scale at the bottom.
Fluorescence spectra were measured at the RPE, Bruch's membrane (BM), druse, and the hyperfluorescent lesion (HL) below the druse, most likely corresponding to basal laminar deposit as indicated by the arrows in Figure 1. These spectra are presented in Figure 2. The fluorescence spectra were calculated by summing up the number of photons recorded in each channel and correcting the values for the relative spectral sensitivity of each channel, including the transmission of the polychromator and the wavelength dependence of the detector.
Figure 2. .

Fluorescence spectra of RPE, BM, hard druse, and HL as indicated in Figure 1 (λexc = 430 nm, 170 fs pulses).
Usually, the highest fluorescence intensity originates from lipofuscin in RPE. However, in this sample, an HL appears on BM and adjacent to the indicated druse. The HL has a fluorescence lifetime that is almost identical to that observed for the druse.
As shown in Figure 2, the shape of the fluorescence spectra, recorded at the druse, BM, and HL are similar. All have a maximum at 468 nm, with a shoulder at 500 nm. The HL appears to have an additional weak emission band with a maximum at 560 nm. The RPE fluorescence spectrum has its maximum at 509 nm, with smaller peaks detectable at 543, 573, and 468 nm. The RPE fluorescence is much more intense for wavelengths longer than 509 nm compared to the intensity of the HL. The relative maximal intensities at the four tissue positions of druse, BM, HL, and RPE are 1, 1.12, 4.08, and 4.38, respectively.
Fluorescence Lifetime
The fluorescence lifetimes were calculated from the sum of photons in all spectral channels. Lifetime images of τm allow for rapid determination of similarities or differences between the various layers of the retina by simple visual inspection. Several types of drusen can be distinguished easily in these images.
Example 1: Soft Druse
A soft druse, covering a large area on BM is visible in Figure 3; the color coding of the average fluorescence lifetimes τm is the same as given in Figure 1. The RPE layer, which is colored orange (corresponding to 385 ps) in the lifetime image, is distinguished easily from the green color (corresponding to 1785 ps) of the soft druse. BM appears as a sharp green line. There also are small patches below BM that are colored orange, indicating that they have nearly the same short lifetime as observed in the RPE. These patches likely are lumens of the choriocapillaris. Attached to BM, there are small orange patches, which may represent an extension of choriocapillaris in the space between BM and the druse.
Figure 3. .

Fluorescence intensity and lifetime τm (0 ≤ τm ≤ 3500 ps) of a fundus sample with a soft druse.
Quantitative analysis of the fluorescence signals from the sample in Figure 3 results in histograms from which lifetimes, amplitudes, and relative contributions can be obtained (Fig. 4).
Figure 4. .

Histograms of lifetimes, amplitudes, and relative contribution of retinal pigment epithelium, Bruch's membrane, and soft druse from the sample in Figure 3. Frequency refers to the number of pixels associated with each parameter.
The fastest component for all three calculated lifetimes (τ1 = 260 ps, τ2 = 1360 ps, and τm = 385 ps) correspond to signal from the RPE, while the longer components (τ1 = 980 ps, τ2 = 4000 ps, and τm = 1785 ps) originate from the soft druse and BM. The broad distribution of τ2 from the soft druse and BM is remarkable because it is such a wide distribution. The major contributing component to the fluorescence decay for RPE and for BM and the soft druse is the lifetime, τ1, which has an amplitude, α1, equal to 90% and 76%, respectively. Whereas the distribution of α1 in RPE is narrow, the distribution for BM and the soft druse is broadened with values down to 20% being observed. The relative contribution, Q1, is maximal at 62% in RPE but in BM and druse, there are two maxima at 39% and 17%.
This global interpretation as presented in Figure 4 was verified by carrying out selective measurements on RPE, BM, and the soft druse. The double peak distribution of Q1 was observed for the druse and BM.
Example 2: Two Hard Drusen
Figure 5 presents the total fluorescence intensity image on the left and the fluorescence lifetime map on the right for another region of the retina containing hard drusen. The total fluorescence image shows two hard drusen on the surface of BM that have weak fluorescence intensity. High fluorescence intensity is observed for the RPE, and a feature resembling a very small hard druse that appears to be on BM and between the two larger drusen. From the fluorescence lifetime map, it is clear that τm is much shorter in the RPE than in the HL, drusen, and BM. Therefore, the bright fluorescence in the HL does not originate from lipofuscin in the RPE but from the same material as in the drusen and BM.
Figure 5. .

Images of fluorescence intensity (left) and lifetime τm (right) of a human fundus sample with hard drusen.
Data of the fluorescence decay signals were collected while focusing separately on either the hard drusen or HL, as well as on RPE and Bruch's membrane, and binning a 7 × 7 region of the detector. The most probable values of the fitting parameters are given in the Table. The intervals in the table mean that no clear maximum exists in the histogram for the corresponding parameter.
Table. .
Most Frequent Lifetimes and Amplitudes of Selected Structures in Figure 5
|
τm (ps) |
α1 (%) |
τ1 (ps) |
τ2 (ps) |
|
| Left druse | 1531–2142 | 74 | 937 | 2460–5118 |
| Right druse | 1889 | 75 | 974 | 2424–5103 |
| HL | 1532–2050 | 46–75 | 498 | 2231–4853 |
| BM | 1531–2142 | 76 | 957 | 2557–5190 |
| RPE | 440 | 88 | 330 | 1455 |
The data in the Table demonstrate clearly that the observed lifetimes in RPE are different from those observed in drusen, HL, and BM. The distribution of τm is nearly identical for the drusen, HL, and BM. This most likely indicates that the HL, drusen, and BM consist of similar substances.
The following images in Figure 6 are the color coded maps for the values of lifetimes, amplitudes and relative contributions for the components of the fluorescence decay observed for the tissue sample with two hard drusen (example 2). Note that the color coding is the same as used previously, where more red corresponds to the shorter lifetime/smaller amplitude and more blue corresponds to the longer lifetime/larger amplitude. The lifetime images (set A) show that τ1 and τ2 are shorter in the RPE than in other regions. In contrast, the amplitude, α1, is much higher in the RPE than the amplitude, α2. In other regions, both amplitudes are approximately the same (set B).
Figure 6. .

Images of lifetime, amplitude, and relative contribution components for the fluorescence decay measured on the sample in Figure 5.
Example 3: Drusen with Differing Fluorescence Intensities
Figure 7 shows the fluorescence intensity image and the lifetime map for a sample of tissue that has two hard drusen of different fluorescence intensities, so that one appears dark and the other appears brighter. The lifetime map shows that τm for both drusen are nearly identical and, accordingly, they appear in the same color in the lifetime images. In the dark druse, there is a small spot that exhibits a lifetime corresponding to that observed in the RPE.
Figure 7. .

Total fluorescence intensity image (left) and lifetime map with 0 < τm < 3500 ps (right) for the tissue sample containing two hard drusen with different fluorescence intensities.
The fluorescence spectra for both drusen are nearly identical as shown in Figure 8, but the intensity is much higher in the bright druse, as is expected. The shape of the emission spectra indicates that the drusen are detectable as bright spots if the detector collects the fluorescence between 470 and 500 nm.
Figure 8. .

Fluorescence spectra of dark and bright drusen in Figure 7.
Because of the overall low fluorescence intensity from the drusen in this sample, there are a low number of pixels registered in this signal. This results in the signal-to-noise ratio being smaller and introduces greater error into the fits of these data. However, the histogram of τ1 does appear to have a single, dominant value, τ1 = 958 ps, in the dark druse, whereas the lifetime τ1 is distributed between 240 and 1160 ps in the bright druse (data not shown).
In addition, although the plots are noisy, a difference was found in the histogram of the amplitude, α1. A clear maximum of α1 was found at 78% for the dark druse whereas two maxima of α1 were found at 53% and 73% in the bright druse (data not shown).
Example 4: Drusen with Fluorescence on the Border Only
Figure 9 presents the total fluorescence intensity image (left) and lifetime map (right) for a tissue sample containing drusen that exhibit fluorescence on the border only. There is no spatially confined area that exhibits fluorescence throughout as observed for the hard drusen (Figs. 5 and 6). The border between the drusen and the RPE fluoresces along a narrow line and has the same lifetime τm, as BM. There appears to be a substantial degree of non-fluorescent material inside of the large druse and a few, very small irregularities on the apical surface of the druse. A small amount of fluorescent material is observed inside of the small druse. This material exhibits a short lifetime τm, that is identical within experimental error of τm in RPE. Thus, the material is most likely to be lipofuscin.
Figure 9. .

Fluorescence intensity image (left) and lifetime map with 0 < τm < 3500 ps (right) for the tissue sample containing drusen with fluorescence on the border only.
Correspondence between Fluorescent Regions and Anatomical Structures
The correspondence between fluorescing regions and anatomical fundus structures of a human donor eye is demonstrated in Figure 10. Figure 10A is the fluorescence intensity image and Figure 10B is the image of mean fluorescence lifetime, τM. The fluorescence decay was bi-exponentially approximated. The HE stained image is presented in Figure 10C. It is visible clearly that the bright fluorescence with a short lifetime originates from the retinal pigment epithelium. The identification of the fluorescent layers is in agreement with those reported by Marmorstein et al.8
Figure 10. .

Correspondence between fluorescence intensity (A), fluorescence decay time τM (B), and HE-stained human fundus sample (C). The fluorescence was detected between 500 and 560 nm. Left druse, hyalinized feature; right druse, calcification.
In Vivo Human Fluorescence Lifetime Imaging (FLIM) Measurements
To demonstrate that the method works, we added in vivo FLIM measurements of an AMD patient having macular drusen and of a healthy subject for comparison. The lifetime of the drusen is longer than the surroundings, as expected. The measurements were performed using the fluorescence lifetime laser scanning ophthalmoscope, described previously.5 The decay of fundus fluorescence was triple-exponentially fitted.
As demonstrated in Figure 11, drusen are detectable with longer mean lifetime τM also in en face measurements.
Figure 11. .

In vivo detection of drusen in a lifetime image of an AMD patient (left). The lifetime image of a healthy fundus is shown in the right image for comparison. Both subjects are wearing artificial intraocular lenses.
Discussion
FLIM is a new retinal imaging method that uses the fluorescence lifetime as an additional parameter for discriminating endogenous or exogenous fluorophores in addition to the fluorophore-specific excitation and emission.9 Most applications of FLIM are performed in microscopy,10 but there have been several recent developments of fluorescence lifetime imaging of endogenous fluorophores for the in vivo investigation of the human eye.2,3,5,11–13 Thus, FLIM offers an improvement to standard fluorescence intensity detection used in early diagnostics of retinal disorders, such as AMD1,3 or occlusion of retinal branch arteries.13
In our study, fluorescence spectra and fluorescence lifetimes are determined for drusen, RPE, and Bruch's membrane in histological sections of a human donor eye. The measurements are performed on a 2-photon microscope using the second harmonic wavelength at 430 nm and pulses of 170 fs full width at half maximum as the excitation. This wavelength spectrally is near the wavelength of 448 nm, which is used in the fluorescence lifetime laser scanner ophthalmoscope.4 Fluorescence spectra and lifetimes are measured simultaneously using a polychromator, adapted with 16 photomultipliers measuring the fluorescence decay in TCSPC. Because TCSPC is an extremely sensitive technique, fluorescence spectra and lifetimes can be determined from weakly emitting structures. Thus, the fluorescence spectra and lifetimes of lipofuscin in RPE and drusen can be detected, and the differences between these measured quantities can be used to distinguish between drusen and RPE in in vivo studies. For this reason, different types of drusen were studied, including soft drusen, hard drusen, dark and bright drusen, and drusen emitting on the border only.
It was found that the fluorescence of drusen and Bruch's membrane is maximal for excitation at 430 nm with the emission monitored between 470 and 510 nm. The signal intensity decreases dramatically as the excitation is shifted to longer wavelengths. The most intense maximum of the fluorescence emission in the druse is at 468 nm, with a second band near 500 nm and a weak shoulder at 560 nm. In Bruch's membrane, the most intense maximum also is observed at 468 nm, but with a broad shoulder around 570 nm.
RPE has a broad fluorescence spectrum with a maximum at 509 nm. It has been reported that the all-trans retinal dimer exhibits fluorescence maximum at 510 nm after excitation at 430 nm.14 In addition, several overlapping bands are observed at 468, 541, and 573 nm, which indicate the existence of several different fluorophores contributing to RPE fluorescence. The maximum at 573 nm may contain contributions in part from A2E, which was determined to have a fluorescence maximum at 600 nm after excitation at 448 nm,4,12 or by in vivo measurements with an excitation wavelength of 488 nm.15
RPE, drusen, and Bruch's membrane can be distinguished by determining the lifetimes and amplitudes using a double exponential fit of the fluorescence decay after excitation by femtosecond laser pulses. When the mean lifetime, τm, is calculated, it is found that different values exist between RPE and the other structures. For example, constructing histograms that present how often each value of lifetime is determined in a selected region versus lifetime value, the most frequent lifetime, τm = 440 ps, was determined for RPE. The lifetimes of the other structures are longer than 1000 ps. For one druse (left druse in Fig. 5), a small distribution of τm was found with the most frequent value at 1548 ps. The distribution of τm for the right druse in Figure 5 and for Bruch's membrane is broader, and most frequent at approximately 1880 ps. This indicates that drusen consist of different fluorophores or of the same fluorophores in different microenvironments.
It was demonstrated that bright and a dark drusen have nearly the same fluorescence spectrum but the lifetime distributions are different. Applying FLIM, it could be demonstrated further that the fluorescence from regions with the same brightness as RPE do not contain contributions from lipofuscin because they have lifetimes like drusen. The material in the HL most likely corresponds to basal laminar deposit. As the lifetime in RPE is determined by lipofuscin, in most cases no lipofuscin is detectable in drusen because they have longer lifetimes than RPE. In some cases, however, structures within drusen appeared to contain lipofuscin, which may shed light on the origin(s) of drusen deposits.
The lifetime images in Figures 1 and 3 show orange patches that are vascular lumens of the choriocapillaris. These small orange patches are attached to BM (Fig. 3) and indicate an extension of choriocapillaris in the space between BM and the druse. The existence of choriocapillaris in the space between the RPE cells and the elastic layer of BM was described previously in the far periphery.16
The visibility of different kinds of drusen by multimodal imaging has been described previously.17
Since the fluorescence lifetime is sensitive to the viscosity, the effect of fixation was tested on single layers of fundus samples of porcine eyes. Even though the lifetimes of all layers of the fixed samples always was significantly longer than in unfixed samples, the relationship of lifetimes τRPE < τoutersegment < τinnersegment was the same in fixed and unfixed samples. It can be concluded that the lifetime of drusen also always is longer than from RPE, independent of the exact value of lifetimes.
No change was found for the fluorescence spectra of RPE in fixed and unfixed samples.
To demonstrate that the method works, we added in vivo FLIM measurements of an AMD patient having macular drusen and of a healthy subject for comparison. The lifetime of the drusen was longer than the surroundings, as expected. The measurements were performed using the fluorescence lifetime laser scanning ophthalmoscope, described previously.5
In summary, we report on FLIM of retinal tissue from a donor with significant extramacular drusen, and demonstrated that this technique is able to discriminate between different fluorophores from different spatial regions of the tissue in a straightforward manner. We suggest that this is a promising new technique that allows much greater information to be obtained than in traditional fluorescence intensity measurements.
Footnotes
Supported by National Institutes of Health Grant EY017451.
Disclosure: D. Schweitzer, None; E.R. Gaillard, None; J. Dillon, None; R.F. Mullins, None; S. Russell, None; B. Hoffmann, None; S. Peters, None; M. Hammer, None; C. Biskup, None
References
- 1. Bindewald A, Bird AC, Dandekar SS, et al. Classification of fundus autofluorescence patterns in early age-related macular disease. Invest Ophthalmol Vis Sci. 2005;46:3309–3314 [DOI] [PubMed] [Google Scholar]
- 2. Schweitzer D, Hammer M, Schweitzer F, et al. In vivo measurement of time-resolved autofluorescence at the human fundus. J Biomed Opt. 2004;9:1214–1222 [DOI] [PubMed] [Google Scholar]
- 3. Schweitzer D, Quick S, Schenke S, et al. Comparison of parameters of time-resolved autofluorescence between healthy subjects and patients suffering from early AMD. Ophthalmologe. 2009;106:714–722 [DOI] [PubMed] [Google Scholar]
- 4. Schweitzer D, Schenke S, Hammer M, et al. Towards metabolic mapping of the human retina. Microsc Res Tech. 2007;70:410–419 [DOI] [PubMed] [Google Scholar]
- 5. Schweitzer D. Metabolic mapping Holz FG, Spaide RF. Medical Retina-Focus on Retinal Imaging. Berlin, Germany: Springer; 2010:106–123 [Google Scholar]
- 6. Biskup C, Kelbauskas L, Zimmer T, et al. Interaction of PSD-95 with potassium channels visualized by fluorescence lifetime-based resonance energy transfer imaging. J Biomed Opt. 2004;9:753–759 [DOI] [PubMed] [Google Scholar]
- 7. Mullins RF, Skeie JM, Malone EA, Kuehn MH. Macular and peripheral distribution of ICAM-1 in the human choriocapillaris and retina. Molecular Vision. 2006;12:224–235 [PubMed] [Google Scholar]
- 8. Marmorstein AD, Marmorstein LY, Sakaguchi H, Hollyfield JG. Spectral profiling of autofluorescence associated with lipofuscin, Bruch's membrane, and sub-RPE deposits in normal and AMD eyes. Invest Ophthalmol Vis Sci. 2002;43:2435–2441 [PubMed] [Google Scholar]
- 9. Lakowicz JR. Principles of Fluorescence Spectroscopy. 3rd ed New York, NY: Kluwer Academic Press,2007. [Google Scholar]
- 10. Becker W. Advanced Time-Correlated Single Photon Counting Techniques. Springer Series in Chemical Physics 81. Berlin, Germany: Springer;,2005. [Google Scholar]
- 11. Schweitzer D, Jentsch S, Schenke S, Hammer M, Biscup C, Gaillard ER. Spectral and time-resolved studies on ocular structures. SPIE/OSA. 2007;6628:662807–1 –662807-12 [Google Scholar]
- 12. Schweitzer D. Quantifying fundus autofluorescence Lois N, Forrester FF. Fundus Autofluorescence. Philadelphia: Williams & Wilkins; 2009:78–95 [Google Scholar]
- 13. Schweitzer D, Quick S, Klemm M, Hammer M, Jentsch S, Dawczynski J. Time-resolved autofluorescence in retinal vessel occlusion. Ophthalmologe. 2010;107:1145–1152 [DOI] [PubMed] [Google Scholar]
- 14. Kim SR, Jang YP, Sparrow JR. Photooxidation of RPE lipofuscin bisretinoids enhances fluorescence intensity. Vision Research. 2010;50:729–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Delori FC, Dorey CK, Staurenghi G, Arend O, Goger DG, Weiter JJ. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Invest Ophthalmol Vis Sci. 1995;36:718–729 [PubMed] [Google Scholar]
- 16. Schraermeyer U, Addicks K, Kociok N, Esser P, Heimann K. Capillaris are present in Bruch's membrane at the ora serrata in the human eye. Invest Ophthalmol Vis Sci. 1998;39:1076–1084 [PubMed] [Google Scholar]
- 17. Spaide RF, Curcio CA. Drusen characterization with multimodal imaging. Retina. 2010;30:1441–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]