

SUMMARY
Although many genes predisposing to autism spectrum disorders (ASD) have been identified, the biological mechanism(s) remain unclear. Mouse models based on human disease-causing mutations provide the potential for understanding gene function and novel treatment development. Here we characterize a mouse knockout of the Cntnap2 gene, which is strongly associated with ASD and allied neurodevelopmental disorders. Cntnap2−/− mice show deficits in the three core ASD behavioral domains, as well as hyperactivity and epileptic seizures, as has been reported in humans with CNTNAP2 mutations. Neuropathological and physiological analyses of these mice before the onset of seizures reveal neuronal migration abnormalities, reduced number of interneurons and abnormal neuronal network activity. In addition, treatment with the FDA approved drug risperidone, ameliorates the targeted repetitive behaviors in the mutant mice. These data demonstrate a functional role for CNTNAP2 in brain development and provide a new tool for mechanistic and therapeutic research in ASD.
INTRODUCTION
Autism spectrum disorders (ASD) form a heterogeneous neurodevelopmental syndrome characterized by deficits in language development, social interactions, and repetitive behavior/restricted interests (APA, 2000). Although not necessary for diagnosis, a number of other behavioral abnormalities are frequently associated with ASD, including hyperactivity, epilepsy and sensory processing abnormalities (Geschwind, 2009).
Research into the genetic basis for ASD has identified many genes, including common and rare variants (Sebat et al., 2007; Glessner et al., 2009; Weiss et al., 2009). Association, linkage, gene expression and imaging data support the role of both common and rare variants of contactin associated protein-like 2 (CNTNAP2) in ASD. Originally, a recessive nonsense mutation in CNTNAP2 was shown to cause a syndromic form of ASD, cortical dysplasiafocal epilepsy syndrome (CDFE), a rare disorder resulting in epileptic seizures, language regression, intellectual disability, hyperactivity and, in nearly two-thirds of the patients, autism (Strauss et al., 2006). Several reports have since linked this gene to an increased risk of autism or autism-related endophenotypes (Alarcon et al., 2008; Arking et al., 2008; Bakkaloglu et al., 2008; Vernes et al., 2008). Recently, we have shown that the same CNTNAP2 variant that increases risk for the language endophenotype in autism, leads to abnormal functional brain connectivity in human subjects (Scott-Van Zeeland et al., 2010), consistent with emerging theories of ASD pathophysiology based on altered neuronal synchrony and disconnection (Belmonte et al., 2004).
Cntnap2 encodes a neuronal transmembrane protein member of the neurexin superfamily involved in neuron-glia interactions and clustering of K+ channels in myelinated axons (Poliak et al., 1999; 2003). However, the fact that the gene is expressed embryonically (Poliak et al., 1999; Abrahams et al., 2007; Alarcon et al., 2008) and myelination takes place postnatally, together with the increasing number of reports that link the gene to ASD, suggests an additional role for CNTNAP2 in early brain development. This is supported by the imaging and pathology data in patients with CDFE, in whom nearly half manifest presumed neuronal migration abnormalities on MRI, which was confirmed by histological analysis of brain tissue resected from patients that underwent surgery for epilepsy (Strauss et al., 2006).
The generation of valid animal models is critical for understanding the pathophysiology of ASD and to assess the potential of proposed treatments, as well as developing new effective interventions. Ideally, mouse models should be based on a known genetic cause of the disease (construct validity), reflect key aspects of the human symptoms (face validity) and respond to treatments that are effective in the human disease (predictive validity; Chadman, 2008; Nestler and Hyman, 2010). Here, we demonstrate that the Cntnap2 knockout mouse exhibits striking parallels to the major neuropathological features in CDFE and the core features of ASD. We observe defects in the migration of cortical projection neurons and a reduction in the number of GABAergic interneurons, as well as accompanying neurophysiological alterations. These data show that CNTNAP2 is involved in the development of cortical circuits, and further support alterations in brain synchrony or connectivity in ASD pathophysiology. In addition, treating Cntnap2−/− mice with risperidone rescues the repetitive behavior, but not the social deficits, a dissociation parallel to what is seen in human patients. These data demonstrate the validity of the Cntnap2 KO as a mouse model for ASD and provide initial insight into the underlying mechanisms by which CNTNAP2 affects brain development and function.
RESULTS
Expression of Cntnap2 in mouse brain
Mutant mice lacking the Cntnap2 gene (Caspr2-null mice) were generated by Dr. Elior Peles (Poliak et al., 2003). We backcrossed the original ICR outbred strain onto the C57BL/6J background for 10–12 generations. Cntnap2−/− mice on the C57BL/6J background had a normal appearance; no differences in weight or growth rate were observed when compared with WT littermates. In WT brain, expression of CNTNAP2 was first detected by Western blot around embryonic day 14 (E14). As expected, CNTNAP2 was completely absent in the brain of homozygous mutant animals (Figure S1A). In situ hybridization demonstrated Cntnap2 expression in multiple adult brain regions, primarily cerebral cortex, hippocampus, striatum, olfactory tract and cerebellar cortex (Figure S1B). Embryonic expression was also broad, including the ventricular proliferative zones of the developing cortex and ganglionic eminences (where excitatory projection neurons and inhibitory interneurons arise, respectively) as well as in migrating neurons and most-migratory cells, indicating a possible role in neuron development and/or migration (Figure S1C).
Cntnap2−/− mice exhibit epileptic seizures and abnormal electroencephalogram (EEG) pattern
One of the major phenotypes of CDFE syndrome is the presence of epileptic seizures, which is associated with dense hippocampal astroglyosis (Strauss et al., 2006). In Cntnap2−/− mice spontaneous seizures were commonly observed in animals over 6 months of age. Seizures were consistently induced by mild stressors during routine handling (Movie S1). A behavioral study of the frequency and severity of the seizures (Racine, 1972) is presented in Table S1. Histological analysis of the hippocampal formation in these animals did not show any gross structural abnormalities; although a reduction in parvalbumin positive interneurons was found (see below and Figure S3C). Reactive astrocytosis as indicated by an enhanced expression of glial fibrillary acidic protein (GFAP) was observed throughout hippocampus of mutant mice after the onset of seizures. Reactive astrocytosis was especially dense in the hilus, but was not accompanied by neuronal loss in this structure as indicated by NeuN staining (Figure 1A). EEG recordings from freely moving mutant animals implanted with cortical electrodes at 8 months of age showed generalized interictal spike discharges during slow wave sleep, while no electrical abnormalities were found in the EEG of mutant mice at times before the onset of seizures (Figure 1B). To avoid any confounding effect due to the presence of epileptic seizures, the following neuropathological, physiological and behavioral characterization of Cntnap2 mutants was performed at an age before the onset of seizures.
Figure 1. Cntnap2−/− mice show epileptic seizures and abnormal EEG pattern.
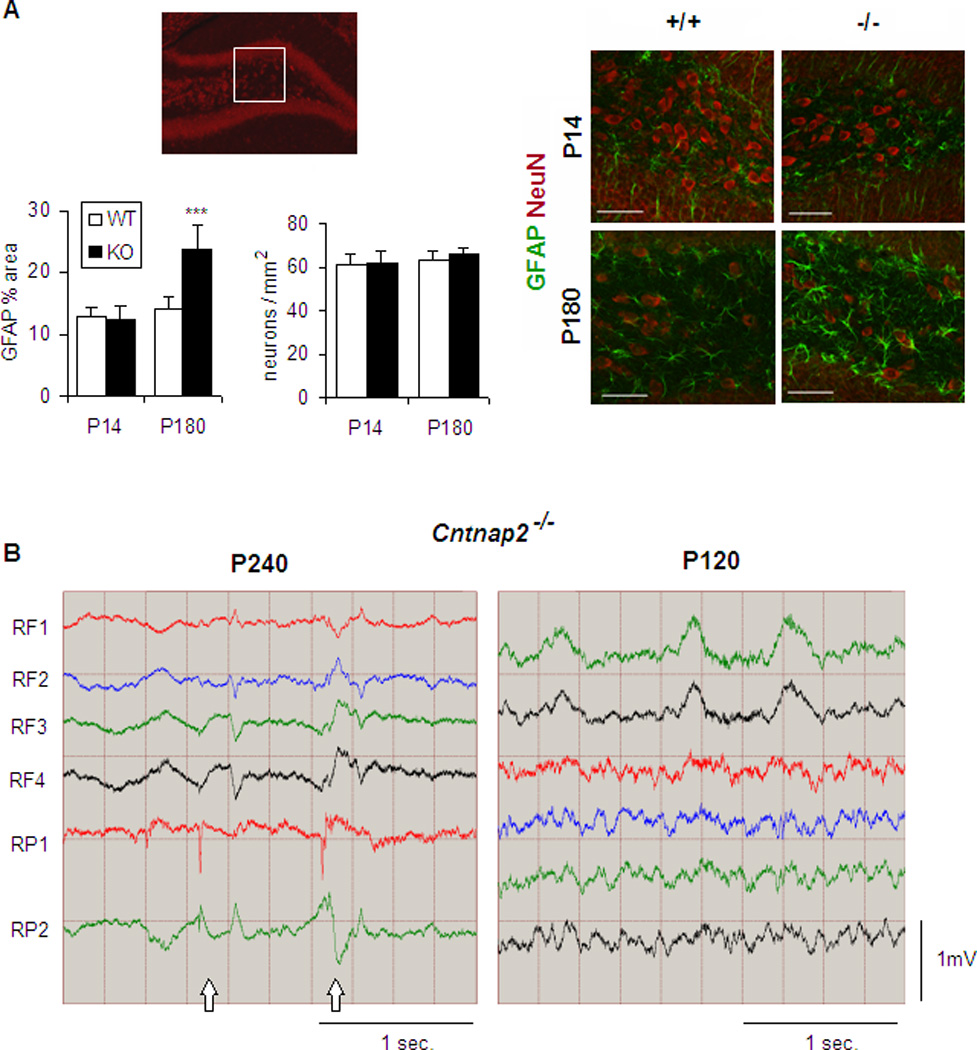
(A) Presence of reactive astrocytes in the hipocampal hilus (inset) of P180 but not P14 mutant mice without significant changes in neuronal density. GFAP, glial fibrillary acidic protein; NeuN, neuronal nuclei. Scale bar: 50 µm. GFAP quantification is shown as % of area occupied by reactive astrocytes. n=4 mice/genotype for each age. Data are presented as mean ± S.E.M. ***p<0.001 (B) EEG recording from mutant mice show abnormal spike discharges (arrows) after seizure onset. n=3 mice for each age. RF, right frontal; RP, right parietal. See also Table S1, Movie S1 and Figure S3C.
Cntnap2−/− mice show neuronal migration abnormalities
We performed detailed histological analyses of Cntnap2 KO brain and found no gross morphological changes in the brain structure of mutant animals by conventional staining techniques (cresyl violet staining), consistent with previous reports (Poliak et al. 2003). Neuronal nuclei (NeuN) immunohistochemistry (IHC) revealed the presence of ectopic neurons in the corpus callosum of mutant mice at postnatal day 14 (P14), after neuronal migration is completed, which persisted through adulthood (Figure 2A). Interestingly, ectopic neurons of unknown origin in white matter were also reported in CDFE syndrome patients (Strauss et al., 2006).
Figure 2. Cntnap2−/− mice show neuronal migration abnormalities.

(A) Presence of ectopic neurons in the corpus callosum of Cntnap2−/− mice. NeuN, neuronal nuclei; CTX, cortex; STR, striatum. Scale bar: 20µm. (B) Expression of Cux1, a marker for upper layer projection neurons, in somatosensory cortex of WT and Cntnap2−/− mice. Note the abnormal distribution of CUX1 positive cells in groups (arrowheads) and rows (arrow) in deep cortical layers of mutant mice. Scale bar: 50µm (C) Neuronal birth-dating analysis. BrdU injected at E16.5 was immunostained at P7. Note abnormal distribution of neurons in groups (arrowheads) and rows (arrow) in deep cortical layers of mutant animals. Scale bar: 50µm. Data are presented as mean ± S.E.M. *p<0.05, **p<0.01, ***p<0.001. n=3 mice/genotype for each age. See also Figure S2.
Patients with CDFE syndrome also show neuronal migration abnormalities, such as abnormal arrangements of neurons in clusters or migratory rows in the deep layers of cortex (Strauss et al., 2006). We assessed laminar positioning of cortical projection neurons in WT and Cntnap2 KO mice with antibodies against CUX1, a marker for upper cortical layers, and FOXP2, a marker for deep cortical layers (Molyneaux et al., 2007). Cntnap2−/− mice show significantly higher numbers of CUX1+ cells in deep cortical layers (V-VI; Figure 2B). In contrast, deeper layer FOXP2+ cells show the same pattern in both genotypes (Figure S2).
We performed BrdU neuron birthdating at E16.5, after the birth of layer V neurons (Angevine and Sidman, 1961) to confirm that the CUX1+ ectopic neurons observed in deep cortical layers were being born concomitant with superficial cell identity and their presence was not due to changes in cell fate. Sections of E16.5 labeled animals were analyzed at P7, when cortical lamination is essentially complete. As shown in Figure 2C, the distribution of BrdU+ cells is significantly different between WT and KO littermates, the later showing a shortage of cells in upper cortical layers that are redistributed to lower cortical layers. These data indicate that CNTNAP2 is necessary for the normal migration of cortical projection neurons.
Reduced number of interneurons in Cntnap2−/− mice
Since the identification of CNTNAP2 (CASPR2) in 1999 (Poliak et al., 1999), its expression has been reported mainly in excitatory pyramidal cells, at the axon initial segment (Inda et al., 2006) and myelinated axons (Poliak et al., 2003, Horresh et al., 2008). The embryonic expression of Cntnap2 in the ganglionic eminence, where GABAergic interneurons arise, led us to analyze the expression of CNTNAP2 in interneurons. We analyzed available microarray data from single glutamatergic and GABAergic neuronal populations isolated from adult mice (Sugino et al., 2006) using weighted gene co-expression network analysis (WGCNA) (Zhang and Horvath, 2005). WGCNA groups functionally related genes into modules based on expression, in a way that genes showing similar expression patterns across samples are grouped together. Modules are likely to represent biological pathways in a way that they are usually co-regulated or interact (Oldham et al., 2008). Interestingly, we found that Cntnap2 is part of a module with enriched expression in inhibitory relative to excitatory neurons (Figure S3A, Table S2). In addition, three of the most highly connected genes within the module, which are indicative of module function, are Gad1, Gad2 and Slc32a1 (VGAT), genes known to be necessary for GABAergic transmission (Figure 3A), suggesting the possibility of CNTNAP2 involvement in interneuron functioning.
Figure 3. Reduced number of GABAergic interneurons in Cntnap2−/− mice.

(A) Network plot showing the top 300 connections within the Cntnap2 module (B) Expression of Gad1 at P14 shows a reduced number of GABAergic interneurons in somatosensory cortex of KO mice. Scale bar: 50µm (C) Expression of the interneuron markers PAVLB, CALB2, and NPY at P14. Scale bar: 50µm (D) Expression of the GABAergic interneurons PAVLB and NPY in striatum. Scale bar: 100µm. Interneuron quantification is shown as % of WT. *p<0.05, **p<0.01, ***p<0.001. For (B), (C), (D) and (E) n=4 mice/genotype. Data are presented as mean ± S.E.M. See also Table S2 and Figure S3.
Since the potential relationship of CNTNAP2 to GABAergic interneuron function had not been previously explored and interneuron dysfunction has been associated with autism and epilepsy (Levitt et al., 2004), we analyzed the number and distribution of interneurons in Cntnap2−/− mice. GAD1 immunostaining showed that Cntnap2−/− mice have a reduced number of GABAergic interneurons in all laminae (Figure 3B). To test whether the reduction in interneurons was subtype specific, we analyzed the number and distribution of the largely non-overlapping subgroups of interneurons in rodents, parvalbumin (PAVLB), calretinin (CALB2), and neuropeptide Y (NPY) (Wonders and Anderson, 2006). We observed that PAVLB+ interneurons were the most affected (Figure 3C). Because striatal interneurons are also born in the ganglionic eminence (Marin et al., 2001), we next examined the number of interneurons in the striatum. We observed a reduction of striatal GABAergic interneurons in mutants (Figure 3D), without a change in cholinergic interneurons (Figure S3B). PAVLB+ interneurons were also reduced in hippocampus (Figure S3C). Together, these data indicate a role for CNTNAP2 in GABAergic interneuron development.
Cntnap2−/− mice show reduced cortical neuronal synchrony
The abnormal positioning and migration of excitatory principal neurons and reduction in inhibitory interneurons suggested that neuronal network activity might be abnormal. This was particularly important as GABAergic interneurons are recognized to play a crucial role in the precise timing of neuronal activity (Sohal et al., 2009) and there is increasing evidence of abnormal neural synchrony as a pathophysiological mechanism in ASD (Uhlhaas and Singer, 2006; Belmonte et al. 2004). In vivo two-photon calcium imaging of layer II/III neurons from somatosensory cortex (Figure 4A) indicated that the neuronal firing pattern of Cntnap2−/− mice was highly asynchronous relative to WT mice (Figure 4B). The mean correlation coefficient of the firing timing between all cell pairs of neurons over the distance range analyzed was significantly lower in mutant mice (Figure 4C). In addition, neither the average firing amplitude (Figure 4D), nor the average firing rate (Figure 4E) changed significantly between genotypes, suggesting that the asynchronous firing observed in mutant animals is likely due to a network dysfunction rather than to abnormalities in neuronal activity or conduction per se. This is consistent with previous data that show no alterations in peripheral and central nerve conductance in Cntnap2−/− mice (Poliak et al. 2003).
Figure 4. Reduced neuronal synchronization in Cntnap2−/− mice.

(A) Representative images of the calcium (top) and astrocytic (bottom) signals of a 3 min movie stack. OGB-1, Oregon green-1; SR-101, sulforhodamine-101 (B) Correlation coefficient of neuronal firing for every pair of neurons over cell distance (C) Mean correlation coefficient over the distance range analyzed (240 µm) (D) Firing amplitude presented as the summed fluorescence change across all imaged cells in a 3 minute time window (E) Mean number of firing events per cell in a 3 minute time window. n=4 mice/genotype. Data are presented as mean ± S.E.M. *p<0.05, **p<0.01, ***p<0.001.
Behavioral characterization of Cntnap2−/− mice
Since the diagnostic criteria in ASD are currently based on behavioral symptoms, rather than molecular or neuroanatomical indicators, we performed a complete battery of behavioral tests to examine the potential effect of the absence of CNTNAP2 on behavior. A summary of the tests performed relevant to each behavioral domain related to ASD (Silverman et al., 2010) and the results obtained are presented in Table S3.
Cntnap2−/− mice displayed significantly greater locomotor activity than their WT littermates in the open-field test (Figure S4A-B). This increased activity was also noted when testing for motor coordination and balance with the rotorod, since mutant mice performed significantly better than WT (Figure S4C). Interestingly, several animal models of autism (Kwon et al., 2006; Nakatani et al., 2009) and hyperactivity (Gerlai et al., 2000; Vitali and Clarke, 2004) have been reported to perform better in the rotorod test.
To assess anxiety related responses, we performed the light-dark exploration test and observed no significant differences between genotypes (Figure S4D). Potential sensory deficits were assessed with the hot plate test. As shown, (Figure S4E) Cntnap2−/− mice demonstrated hyper-reactivity to thermal sensory stimuli. To further characterize this sensory deficit we measured the acoustic startle response (Figure S4F) and the degree of pre-pulse inhibition (Figure S4G) and found no significant differences between genotypes. In addition, olfaction analysis did not show any deficit in mutant mice (Figure S4H).
Cntnap2−/− mice show stereotypic motor movements and behavioral inflexibility
Spatial learning and memory was evaluated in the Morris water maze (MWM). Both WT and mutant mice showed similar learning curves, represented as time to locate the platform, in the training phase of the test (Figure 5A). Probe trials were performed the day after the last training trial and an active search for the platform was evaluated. As expected from their learning curves, WT and KO animals performed similarly in the probe test (Figures 5B-C).
Figure 5. Cntnap2−/− show motor stereotypic movements and behavioral inflexibility.

(A-F) Morris water maze (MWM) test. (A-C) Learning, (D-F) Reversal learning (A) Learning curve as indicated by the latency to locate a hidden platform (up to 60s) during a 5 day training period. The average of 4 trials per day is presented (B) Probe test (the platform is removed) showing the percentage of time spent in each pool quadrant (in 60s). Note that both genotypes spend significantly more time in the target quadrant. TA, target; AR, adjacent right; AL, adjacent left; OP, opposite (C) Number of platform site crossings during the probe test (D) Learning curve for the reversal task of the MWM test showing the latency to locate the new platform (E) Probe test. Note that WT but not KO mice spend significantly more time in the target quadrant (F) Number of platform site crossings during the probe test (G) Number of no alternations in the T maze spontaneous alternation test (10 trials) (H) Time spent grooming over a 10 min period. *p<0.05, **p<0.01, ***p<0.001. n=10 mice/genotype. Data are presented as mean ± S.E.M. See also Table S3 and Figure S4.
To assess behavioral flexibility and perseveration we first performed a classic reversal task using the MWM. We found that Cntnap2−/− mice showed impairment in learning the new location of the platform (Figure 5D) and performed poorly in the probe test (Figure 5E-F). To study perseveration in more detail, we performed the spontaneous alternation T maze test. As shown in Figure 5G, Cntnap2−/− mice showed significantly higher number of no alternations in a standard 10 trial test, confirming the perseveration observed in the reversal task on the MWM. In addition to perseveration, repetitive behavior also encompasses motor stereotypies, and both tend to co-occur in children with ASD. Consistent with our other observations, we found that Cntnap2−/− mice spent almost three times more time grooming themselves than their WT littermates (Figure 5H).
Cntnap2−/− mice show communication and social behavior abnormalities
Isolation induced ultrasonic vocalizations (UsVs) are distress calls emitted by pups when separated from their mother, representing an infant-mother vocal communicative behavior that are thought to be relevant to ASD (Crawley, 2007; Silverman et al., 2009). We analyzed the pattern of UsV emission in WT and Cntnap2−/− mice thorough development (P3, P6, P9, P12; Scattoni et al., 2009) and found that Cntnap2−/− mice emitted significantly lower number of ultrasonic calls than WT littermates at all ages (Figure 6A).
Figure 6. Cntnap2−/− mice show communication and social behavior abnormalities.

(A) UsV. Number of calls from pups when separated from their mother at P3, P6, P9 and P12 (5 min) (B) Juvenile play. Time involved in social interaction, as well as repetitive behaviors (grooming and digging) in pairs of mice matched in genotype and sex at age P21 (10 min) (C) Three-chamber social interaction test. Time interacting with either an unfamiliar mouse or an inanimate object (empty cup) in 10 min (D) Nesting behavior. The nesting score represents the amount of nesting material used after a 16 hour period (1, poor; 5, good). n=10 mice/genotype. Data are presented as mean ± S.E.M. *p<0.05, **p<0.01, ***p<0.001. See also Figure S5.
We next analyzed social behavior in pairs of unfamiliar mice at age P21 (juvenile play test). As shown in Figure 6B, Cntnap2−/− mice spent significantly less time interacting with each other and instead showed increased repetitive behaviors such as grooming and digging. We confirmed that the abnormal social behavior was not due to an olfaction defect (Table S3) since Cntnap2 mutant mice actually perform significantly better than WT in the buried food olfaction test (Figure S4H). We also performed a three-chamber social interaction test in adults (Crawley, 2007). As expected for the highly social strain C57/B6J, WT mice showed a highly significant preference for the cup with a mouse, while KO mice did not show a significant preference (Figure 6C). Finally, we examined nest-building behavior, which is relevant to home-cage social behaviors and mediated by dopaminergic function in mice (Szczypka et al., 2001). Cntnap2−/− mice were also significantly impaired in this task (Deacon, 2006), scoring less than half the WT criterion (Figure 6D).
The atypical antipsychotic risperidone rescues the hyperactivity, repetitive behavior/perseveration and nesting deficits in Cntnap2−/− mice
An important goal in developing mouse models of neuropsychiatric diseases is testing therapeutic treatments. Risperidone was the first drug approved by the United States Food and Drug Administration (FDA) for symptomatic treatment of ASD, alleviating hyperactivity, repetitive behavior, aggression, and self-injurious behavior (McDougle et al., 2000, 2008). Cntnap2−/− mice and their WT littermates were treated with risperidone or vehicle for 7–10 days and were tested for improvement in behavior. There were no significant changes in open field activity in treated WT mice, indicating that the dose used for treatment was not sedating (Figure 7A-B). However, risperidone decreased the activity levels of Cntnap2−/− mice to normal WT levels (Figure 7A-B). Consistent with the lack of a sedative effect, treated KO mice improved their nest-building score as well, a task that depends upon sustained activity (Figure 7C). Risperidone also reversed the increased grooming behavior of Cntnap2−/− mice (Figure 7E) and perseveration in the T-maze (Figure 7 F). To assess any effect of risperidone on social behavior, we performed the three chamber social interaction test and found no improvement with treatment (Figure 7G), nor did we find improvement in sensory hypersensitivity (Figure 7D).
Figure 7. Risperidone rescues hyperactivity and repetitive behavior/perseveration in Cntnap2−/− mice.

(A-B) Open field test. Distance travelled (A) and velocity (B) for vehicle (PBS) and drug treated WT and KO mice (20 min) (C) Nesting behavior. Risperidone improves the nesting score of Cntnap2−/− mice (D) Hot plate. Risperidone did not have an effect in the hyper-reactivity to thermal stimuli (E) Grooming behavior is reduced in risperidone treated mutant mice (10 min) (F) Risperidone improved spontaneous alternation of mutant mice (G) Drug treatment did not have an effect in the three-chamber social interaction test. n=10 mice/genotype and treatment condition. Data are presented as mean ± S.E.M. *p<0.05, **p<0.01, ***p<0.001.
DISCUSSION
The development of mouse models of ASD is crucial to study the disorder at the molecular level, gain insight into disease mechanisms, and test potential pharmacological interventions. Here, we show that the consequences of CNTNAP2 deficiency in the mouse resemble many of the behavioral and cognitive features observed in patients with idiopathic autism and of the pathological features observed in patients with recessive CNTNAP2 mutations that cause a Mendelian form of syndromic autism (Strauss et al., 2006). Cntnap2−/− mice have normal anxiety related responses, visual spatial memory, and sensorimotor integration, but show abnormal vocal communication, repetitive and restrictive behaviors, and abnormal social interactions. In addition, they also show hyperactivity and epileptic seizures, both features described in CDFE patients and in many patients with ASD. Heterozygous animals did not show any of the behavioral (Figure S5) or neuropathological (data not shown) abnormalities observed in homozygote knockouts, nor did they develop epileptic seizures, consistent with the recessive nature of the pathology in humans.
Autism and epilepsy are neurodevelopmental syndromes with a high frequency of co-occurrence (Geschwind, 2009), which suggests shared underlying mechanisms. At a molecular level, CNTNAP2 is a single pass transmembrane protein with a short cytoplasmic region involved in clustering K+ channels at juxtaparanodes in myelinated axons (Horresh et al., 2008), and a long extracellular region that forms a neuron-glia cell adhesion complex with contactin 2 (TAG-1), which is necessary for the proper localization of K+ channels in this structure (Poliak et al., 2003). Thus, defects in myelination and K+ channel mislocalization at the nodes of Ranvier could theoretically lead to epilepsy in Cntnap2−/− mice. However, this is not likely the case, since both light microscopic and ultra-structural analysis using electron microscopy in the peripheral and central nerves in CNTNAP2 deficient mice (Poliak et al., 2003) showed that nodal morphology and myelination were normal. In addition, electrophysiological investigation of nerve conductance revealed no abnormalities in conduction velocity, refractory period or excitability (Poliak et al., 2003). In the current study, neuropathological analysis of Cntnap2−/− mice revealed two major mechanisms that have been shown to lead to epilepsy in humans, cortical neuronal migration abnormalities and a reduction in the number of GABAergic interneurons. Whereas neuronal migration abnormalities might have been expected based on observations in patients with CDFE syndrome, the reduction in GABAergic neurons was unexpected, as CNTNAP2 has not been previously associated with GABAergic neuronal function and no such deficit has been demonstrated in CDFE. These data suggest that assessment of interneurons in patients with CDFE would be worthwhile. Further, whether this reduction in interneurons is also due to a migration defect or rather to a defect in neurogenesis, differentiation and/or survival, remains to be elucidated in future work. Nevertheless, the embryonic expression of the gene in the ganglionic eminences and in migrating interneurons supports a role for CNTNAP2 in the early development and migration of these cells.
The large extracellular domain of CNTNAP2 is composed of a number of protein-protein interaction domains common to cell adhesion molecules including laminin G, EGF repeats, and discoidin-like domains (Poliak et al., 1999). During myelination, CNTNAP2 localizes to the developing nodes of Ranvier where, as previously mentioned, it interacts extracellularly with TAG-1. Interestingly, TAG-1 is also expressed embryonically and blockade of its function results in migration abnormalities of cortical pioneer neurons and GABAergic interneurons (Denaxa et al., 2001; Morante-Oria et al., 2003), although normal numbers of interneurons were reported in TAG-1 deficient mice, likely due to compensatory mechanisms (Denaxa et al., 2005). Thus, analysis of CNTNAP2 interactors, including TAG-1, during embryogenesis could provide insight to the role of CNTNAP2 in neuronal migration.
One physiological consequence of the observed neuropathology caused by Cntnap2 knockout is significantly reduced neuronal synchronization. It is generally accepted that most cognitive functions are based on the coordinated interactions of large neuronal ensembles within and across different specialized brain areas (Uhlhaas and Singer, 2006). Synchronization determines the pattern of neuronal interactions in a way that effective neuronal connectivity would diminish when synchronization is less precise (Womelsdorf et al., 2007). A number of functional neuroimaging studies have reported reduced connectivity in ASD (Just et al., 2004; Villalobos et al., 2005; Cherkassky et al., 2006; Damarla, 2010), supporting the notion that the deficits in cognition and behavior associated with ASD are most likely the result of a developmental disconnection (Geschwind and Levitt, 2007). Interestingly, we have recently associated common genetic-risk variants in CNTNAP2 with abnormal functional brain connectivity in humans (Scott-Van Zeeland et al., 2010). Our observations of migration abnormalities and reduced number of GABAergic interneurons in Cntnap2−/− mice, together with the normal global neuronal activity, as measured by the firing rate and amplitude, suggest an abnormal neuronal circuit architecture as the cause of the asynchronous firing pattern, rather than abnormalities in neuronal function per se. There are a number of factors that contribute to the precise timing of neural activity (reviewed in Wang, 2010). Interestingly, GABAergic interneurons, in particular PAVLB+, have been reported to play a crucial role in the rhythmic pacing of cortical neuronal activity (Sohal et al., 2009). Therefore, further studies analyzing the structure of neuronal networks and interneuron function in Cntnap2−/− mice may have important implications both for the potential understanding and treatment of ASD.
The ultimate goal of understanding the pathophysiology of the disorder is to develop therapeutic interventions that improve or restore normal brain activity and, ultimately, the associated cognitive and behavioral deficits. Recent studies in mouse models are very encouraging in this regard, including Rett syndrome (Guy et al., 2007), fragile X syndrome (Dolen et al., 2007), neurofibromatosis type 1 (Costa et al., 2002), Down syndrome (Fernandez et al., 2007) and tuberous sclerosis (Ehninger et al., 2008). Here, we have shown that risperidone efficiently reduces hyperactivity, motor stereotypies and perseveration in Cntnap2−/− mice, while having no effect on sociability. This observation provides evidence that different pathways lead to the ASD associated core domains of social and repetitive behavior observed in this mouse model, parallel to the situation in humans, and supports the validity of this mouse model for testing new pharmacological treatments.
Repetitive behavior is recognized to reflect a disruption of the coordinated function of the cortico-striatal circuit (Albin et al., 1989). Briefly, two main pathways compose this system: the direct pathway, which promotes motor behavior, and the indirect pathway, which inhibits it (Gerfen et al., 1990). In general, stereotypies indicate an unbalanced activity of this network favoring the direct pathway (Lewis et al. 2007). At the neuronal level, the ultimate firing output to the direct and indirect pathways is determined by excitatory inputs from cortex and thalamus, and inhibitory inputs from local interneurons within the striatum (Kreitzer and Malenka, 2008). Interestingly, PALVB+ fast spiking interneurons, which are the main source of inhibitory input in the cortico-striatal circuit, have recently been shown to target mainly the direct pathway (Gittis et al., 2010). Therefore, the reduced number of this type of interneuron in Cntnap2−/− mice likely results in over-activation of this pathway, leading to hyperactivity and repetitive behavior. Indeed, risperidone is known to potentiate the indirect pathway, which likely re-balances the activity of this network alleviating these behaviors.
Nest-building has also been shown to be related to the dopaminergic pathway (Szczypka et al., 2001), and has been reported disrupted in mice with hyperactivity (Kwon et al., 2006; Zhou et al., 2010). That risperidone also normalized the nesting ability in KO mice provides additional evidence for abnormal striatal dopaminergic function in this model Exploration of cortico-striatal function in Cntnap2−/− mice will provide a better understanding of the neural basis of repetitive behavior in ASD. In addition, the dissociation between repetitive and social behavior with regards to treatment response, suggests that Cntnap2−/− mice will be useful for dissecting the distinct circuitries involved in these core components of autistic related abnormal behavior. Finally, since understanding of CNTNAP2 function was previously focused primarily on postnatal development, these data set a new direction for investigation of CNTNAP2’s role during development and in the formation and function of neuronal circuits.
EXPERIMENTAL PROCEDURES
Mice
Cntnap2 mutant and WT mice were obtained from heterozygous crossings and were born with the expected Mendelian frequencies. The day of vaginal plug detection was designated as E0.5 and the day of birth as P0. The three obtained genotypes were housed together with 3–4 same sex mice per cage. They were kept in 12h light/12h dark cycle and had ad-lib access to food and water. All procedures involving animals were performed in accordance with the UCLA animal research committee.
Western blot, in situ hybridization, immunohistochemistry
Western blot, in situ hybridization and immunohistochemistry were performed using standard methods. See Supplemental Experimental Procedures section for details.
BrdU labeling
Timed pregnant mice (E16.5) received a single i.p. injection of BrdU (50 mg/kg body weight, Sigma).
Imaging and Cell Counts
Images were acquired with a Zeiss LSM-510 laser scanning confocal microscope. Eight anatomically matched sections from at least 3 mice per genotype were selected for cell counting. For cortical images cells were counted in matched areas of fixed size (1.2 mm wide) in each hemisphere of somatosensory cortex. The boundaries of the different cortical layers were determined by counterstaining each section with DAPI.
Weighted Gene Coexpression Network Analysis (WGCNA)
The original microarray dataset was deposited by Sugino et al. (2006) in the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/projects/geo/index.cgi) with accession number GSE2882. WGCNA was performed as previously described (Winden et al., 2009). More detailed conditions are included in the Supplemental Experimental Procedures section.
Electroencephalographic (EEG) recordings
Mice were deeply anesthetized with isoflurane and microelectrodes (0.005-inch diameter) were implanted over right frontal and parietal cortices. Mice were allowed to recover for 48h and EEG activity was recorded daily for up to 2 weeks.
In vivo two-photon calcium imaging
Neuronal activity was imaged using a calcium indicator injected in layer II/III of somatosensory cortex in young adult mice (2–4 months of age). See the Supplemental Experimental Procedures section for details.
Behavioral tests
Ten mice per genotype were evaluated for each behavioral test. Mice were tagged with either an ear tag number or a toe tattoo (in the case of pups). Experimenters were blinded to the genotype during testing. Behavioral tests were performed in the UCLA behavioral test core and analyzed with TopScan (Clever Sys, Inc) automated system. UsV were analyzed with Avisoft sound analysis and synthesis software for laboratory animals.
Morris water maze
The MWM test was performed as described elsewhere (Vorhees and Williams, 2006). Briefly, mice were trained to locate a hidden platform based on distal visual cues to escape from the pool. Mice received four training trials per day (with different start points) for five consecutive days. On day 6 the platform was removed and a probe test was performed. The next day, the platform was moved to the opposite quadrant and the reversal task of the test was started. Mice received again four training trials per day to locate the new platform. A probe test was performed on day 10.
T maze spontaneous alternation
Mice were placed on the base of a T maze and were given the choice to explore either the right or left arm of the maze for 10 consecutive trials. A choice was assumed to be made when the mice stepped with the four paws into an arm. At that moment the gate to that arm was closed and the animal was allowed to explore the arm for 5 seconds.
Ultrasonic Vocalization (UsV)
Pups were removed from the dam and placed in individual heated sound proof chambers equipped to record UsV for 5 minutes. To avoid potential confounding effects due to temperature, the room was maintained at 21°C and body temperature was measured with a rectal probe after the 5 minutes of the test at P6 (~35°C in both genotypes).
Juvenile play
Mice at age P21 were placed in a cage (previously habituated to it) with an unfamiliar mouse matched in genotype and sex for 10 minutes. The time mice were engaged in social interaction (nose to nose sniffing, nose to anus sniffing, following or crawling on/under each other) and the time mice spent engaged in repetitive behaviors (grooming and digging) was measured by a human observer (Silverman et al., 2010).
Three chamber social interaction test
The social interaction test was performed as previously described (Silverman et al., 2010) Briefly, after habituation, a mouse was placed in the central chamber of a clear Plexiglas box divided into three interconnected chambers and was given the choice to interact with either an empty wire cup (located in one side chamber) or a similar wire cup with an unfamiliar mouse inside (located in the opposite chamber). Time sniffing each cage was measured.
Drug administration
Risperidone (0.2 mg/kg, Sigma) was administered by a daily i.p. injection in a volume of 10 ml/kg for 7 consecutive days. Behavioral tests were performed on days 8, 9 and 10. Mice also received drug treatment during these days approximately 1h prior to testing.
Statistical analyses
All results are expressed as mean ± SEM. For cell quantifications and neuronal synchrony comparisons between groups a one way ANOVA was used. To compare cell distributions within the cortical layers we used two-way ANOVA. For behavioral tests either one or two way ANOVA with repeated measures followed by Bonferroni-Dunn post-hoc tests, when applied, were used.
Supplementary Material
HIGHLIGHTS.
The Cntnap2−/− mouse model of ASD shows high construct, face and predictive validity
CNTNAP2 affects the development of neuronal circuits including GABAergic neurons
Our results support a role for neuronal synchrony in the pathophysiology of ASD
This model permits dissociation of the circuitries involved in ASD core behaviors
ACKNOLEDGMENTS
We thank the UCLA behavioral testing core and its supervisor, Dr. Ravi Ponnusamy, for assistance with behavioral testing. Also, we thank Dr. Alcino Silva, co-director of the core, for his critical discussions about mouse behavioral testing; Dr. Stephanie White for the UsV equipment and software; Dr. Carolyn Houser for assistance with GAD1 IHC; and Dr. William Yang and Dr. Istvan Mody for helpful discussions. We would also like to thank Jamee Bomar and Dr. Asami Oguro-Ando for help with floating IHC and helpful discussions, Greg Osborn for help with UsV analysis, Clark Rosensweig for help with mouse genotyping, Dr. Irina Voineagu for critically reading the manuscript and Dr. Eric Wexler for useful discussions on drug treatment. This work was supported by grants NIH/NIMH R01 MH081754-02R to D.H.G.; CART/Autism Center of Excellence project II to D.H.G; NIH/NS50220 to E.P. and Dr. Miriam and Sheldon G. Adelson Medical Research Foundation to D.H.G. and E.P. E.P. is the Incumbent of the Hanna Hertz Professorial Chair for Multiple Sclerosis and Neuroscience.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abrahams BS, Tentler D, Perederiy JV, Oldham MC, Coppola G, Geschwind DH. Genome-wide analyses of human perisylvian cerebral cortical patterning. Proc. Natl. Acad. Sci. 2007;104(45):17849–17854. doi: 10.1073/pnas.0706128104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, Sebat J, Wigler M, Martin CL, Ledbetter DH, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 2008;82(1):150–159. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- Angevine JB, Sidman RL. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature. 1961;192:766–768. doi: 10.1038/192766b0. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Washington, DC: 2000. [Google Scholar]
- Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, Rea A, Guy M, Lin S, Cook EH, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 2008;82(1):160–164. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala R, Shu T, Tsai L. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128:29–43. doi: 10.1016/j.cell.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Bakkaloglu B, O'Roak BJ, Louvi A, Gupta AR, Abelson JF, Morgan TM, Chawarska K, Klin A, Ercan-Sencicek AG, Stillman AA, et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 2008;82(1):165–173. doi: 10.1016/j.ajhg.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte MK, Allen G, Beckel-Mitchener A, Boulanger LM, Carper RA, Webb SJ. Autism and abnormal development of brain connectivity. J. Neurosci. 2004;24(42):9228–9231. doi: 10.1523/JNEUROSCI.3340-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadman KK, Yang M, Crawley JN. Criteria for validating mouse models of psychiatric diseases. Am. J. Med. Genet. 2009;150B(1):1–11. doi: 10.1002/ajmg.b.30777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherkassky VL, Kana RK, Keller TA, Just MA. Functional connectivity in a baseline resting-state network in autism. Neuroreport. 2006;17(16):1687–1690. doi: 10.1097/01.wnr.0000239956.45448.4c. [DOI] [PubMed] [Google Scholar]
- Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, Kucherlapati R, Jacks T, Silva AJ. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415:526–530. doi: 10.1038/nature711. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol. 2007;17(4):448–459. doi: 10.1111/j.1750-3639.2007.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damarla SR, Keller TA, Kana RK, Cherkassky VL, Williams DL, Minshew NJ, Just MA. Cortical underconnectivity coupled with preserved visuospatial cognition in autism: Evidence from an fMRI study of an embedded figures task. Autism Res. 2010;3(5):273–279. doi: 10.1002/aur.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon RM. Assessing nest building in mice. Nat. Protoc. 2006;1(3):1117–1119. doi: 10.1038/nprot.2006.170. [DOI] [PubMed] [Google Scholar]
- Denaxa M, Chan CH, Schachner M, Parnavelas JG, Karagogeos D. The adhesion molecule TAG-1 mediates the migration of cortical interneurons from the ganglionic eminence along the corticofugal fiber system. Development. 2001;128:4635–4644. doi: 10.1242/dev.128.22.4635. [DOI] [PubMed] [Google Scholar]
- Denaxa M, Kyriakopoulou K, Theodorakis K, Trichas G, Vidaki M, Takeda Y, Watanabe K, Karagogeos D. The adhesion molecule TAG-1 is required for proper migration of the superficial migratory stream in the medulla but not of cortical interneurons. Dev. Biol. 2005;288:87–99. doi: 10.1016/j.ydbio.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat. Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez F, Morishita W, Zuniga E, Nguyen J, Blank M, Malenka RC, Garner CC. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat. Neurosci. 2007;10:411–413. doi: 10.1038/nn1860. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Sibley DR. D1 and D2 dopamine receptor–regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- Gerlai R, Pisacane P, Erickson S. Heregulin, but not ErbB2 or ErbB3, heterozygous mutant mice exhibit hyperactivity in multiple behavioral tasks. Behav. Brain Res. 2000;109(2):219–227. doi: 10.1016/s0166-4328(99)00175-8. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr. Opin. Neurobiol. 2007;17(1):103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Geschwind DH. Advances in autism. Annu. Rev. Med. 2009;60:367–380. doi: 10.1146/annurev.med.60.053107.121225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Nelson AB, Thwin MT, Palop JJ, Kreitzer AC. Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. J. Neurosci. 2010;30(6):2223–2234. doi: 10.1523/JNEUROSCI.4870-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459(7246):569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horresh I, Poliak S, Grant S, Bredt D, Rasband MN, Peles E. Multiple molecular interactions determine the clustering of Caspr2 and Kv1 channels in myelinated axons. J. Neurosci. 2008;28(52):14213–14222. doi: 10.1523/JNEUROSCI.3398-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inda MC, DeFelipe J, Muñoz A. Voltage-gated ion channels in the axon initial segment of human cortical pyramidal cells and their relationship with chandelier cells. Proc. Natl. Acad. Sci. 2006;103(8):2920–2925. doi: 10.1073/pnas.0511197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just MA, Cherkassky VL, Keller TA, Minshew NJ. Cortical activation and synchronization during sentence comprehension in high- functioning autism: evidence of underconnectivity. Brain. 2004;127:1811–1821. doi: 10.1093/brain/awh199. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron. 2008;60(4):543–554. doi: 10.1016/j.neuron.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt P, Eagleson KL, Powell EM. Regulation of neocortical interneuron development and the implications for neurodevelopmental disorders. Trends Neurosci. 2004;27(7):400–406. doi: 10.1016/j.tins.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Lewis MH, Tanimura Y, Lee LW, Bodfish JW. Animal models of restricted repetitive behavior in autism. Behav. Brain Res. 2007;176(1):66–74. doi: 10.1016/j.bbr.2006.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín O, Yaron A, Bagri A, Tessier-Lavigne M, Rubenstein JL. Sorting of striatal and cortical interneurons regulated by semaphorin-neuropilin interactions. Science. 2001;293(5531):872–875. doi: 10.1126/science.1061891. [DOI] [PubMed] [Google Scholar]
- McDougle CJ, Scahill L, McCracken JT, Aman MG, Tierney E, Arnold LE, Freeman BJ, Martin A, McGough JJ, Cronin P, et al. Research Units on Pediatric Psychopharmacology (RUPP) Autism Network. Background and rationale for an initial controlled study of risperidone. Child Adolesc. Psychiatr. Clin. N. Am. 2000;9(1):201–224. [PubMed] [Google Scholar]
- McDougle CJ, Stigler KA, Erickson CA, Posey DJ. Atypical antipsychotics in children and adolescents with autistic and other pervasive developmental disorders. J. Clin. Psychiatry. 2008;69(4):15–20. [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 2007;8(6):427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Morante-Oria J, Carleton A, Ortino B, Kremer EJ, Fairén A, Lledo PM. Subpallial origin of a population of projecting pioneer neurons during corticogenesis. Proc Natl Acad Sci U S A. 2003;100(21):12468–12473. doi: 10.1073/pnas.1633692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani J, Tamada K, Hatanaka F, Ise S, Ohta H, Inoue K, Tomonaga S, Watanabe Y, Chung YJ, Banerjee R, et al. Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell. 2009;137(7):1235–1246. doi: 10.1016/j.cell.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat. Neurosci. 2010;13(10):1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nat. Neurosci. 2008;11(11):1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, Trimmer JS, Shrager P, Peles E. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron. 1999;24(4):1037–1047. doi: 10.1016/s0896-6273(00)81049-1. [DOI] [PubMed] [Google Scholar]
- Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, Stewart CL, Xu X, Chiu SY, Shrager P, et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol. 2003;162(6):1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation: II. Motor seizure, Electroencephalogr. Clin. Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Scott-Van Zeeland AA, Abrahams BS, Alvarez-Retuerto AI, Sonnenblick LI, Rudie JD, Ghahremani D, Mumford JA, Poldrack RA, Dapretto M, Geschwind DH, et al. Altered functional connectivity in frontal lobe circuits is associated with variation in the autism risk gene CNTNAP2. Sci. Transl. Med. 2010;2(56):56–80. doi: 10.1126/scitranslmed.3001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scattoni ML, Crawley J, Ricceri L. Ultrasonic vocalizations: a tool for behavioural phenotyping of mouse models of neurodevelopmental disorders. Neurosci. Biobehav. Rev. 2009;33(4):508–515. doi: 10.1016/j.neubiorev.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 2010;11(7):490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, Stephan DA, Morton DH. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N. Engl. J. Med. 2006;354:1370–1377. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat. Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- Szczypka MS, Kwok K, Brot MD, Marck BT, Matsumoto AM, Donahue BA, Palmiter RD. Dopamine production in the caudate putamen restores feeding in dopamine-deficient mice. Neuron. 2001;30(3):819–828. doi: 10.1016/s0896-6273(01)00319-1. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Neural synchrony in brain disorders: Relevance for cognitive dysfunctions and pathophysiology. Neuron. 2006;52:155–168. doi: 10.1016/j.neuron.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Vernes SC, Newbury DF, Abrahams BS, Winchester L, Nicod J, Groszer M, Alarcón M, Oliver PL, Davies KE, Geschwind DH, et al. A functional genetic link between distinct developmental language disorders. N. Engl. J. Med. 2008;359(22):2337–2345. doi: 10.1056/NEJMoa0802828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalobos ME, Mizuno A, Dahl BC, Kemmotsu N, Muller RA. Reduced functional connectivity between V1 and inferior frontal cortex associated with visuomotor performance in autism. NeuroImage. 2005;25:916–925. doi: 10.1016/j.neuroimage.2004.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitali R, Clarke S. Improved rotorod performance and hyperactivity in mice deficient in a protein repair methyltransferase. Behav. Brain Res. 2004;153(1):129–141. doi: 10.1016/j.bbr.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006;1(2):848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ. Neurophysiological and computational principles of cortical rythms in cognition. Physiol. Rev. 2010;90(3):1195–1268. doi: 10.1152/physrev.00035.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Arking DE, Gene Discovery Project of Johns Hopkins & the Autism Consortium. Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461(7265):802–808. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womelsdorf T, Schoffelen JM, Oostenveld R, Singer W, Desimone R, Engel AK, Fries P. Modulation of neuronal interactions through neuronal synchronization. Science. 2007;316(5831):1609–1612. doi: 10.1126/science.1139597. [DOI] [PubMed] [Google Scholar]
- Winden KD, Oldham MC, Mirnics K, Ebert PJ, Swan CH, Levitt P, Rubenstein JL, Horvath S, Geschwind DH. The organization of the transcriptional network in specific neuronal classes. Molecular Systems Biology. 2009;5:291–308. doi: 10.1038/msb.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonders CP, Anderson SA. The origin and specification of cortical interneurons. Nat. Rev. Neurosci. 2006;7(9):687–696. doi: 10.1038/nrn1954. [DOI] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene coexpression network analysis. Stat. Appl. Genet. Mol. Biol. 2005;4:17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- Zhou M, Rebholz H, Brocia C, Warner-Schmidt JL, Fienberg AA, Nairn AC, Greengard P, Flajolet M. Forebrain overexpression of CK1δ leads to down-regulation of dopamine receptors and altered locomotor activity reminiscent of ADHD. Proc. Natl. Acad. Sci. 2010;107(9):4401–4406. doi: 10.1073/pnas.0915173107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.