

Abstract
Action potentials generated in the sinoatrial node (SAN) dominate the rhythm and rate of a healthy human heart. Subsequently, these action potentials propagate to the whole heart via its conduction system. Abnormalities of impulse generation and/or propagation in a heart can cause arrhythmias. For example, SAN dysfunction or conduction block of the atrioventricular node can lead to serious bradycardia which is currently treated with an implanted electronic pacemaker. On the other hand, conduction damage may cause reentrant tachyarrhythmias which are primarily treated pharmacologically or by medical device-based therapies, including defibrillation and tissue ablation. However, drug therapies sometimes may not be effective or are associated with serious side effects. Device-based therapies for cardiac arrhythmias, even with well developed technology, still face inadequacies, limitations, hardware complications, and other challenges. Therefore, scientists are actively seeking other alternatives for antiarrhythmic therapy. In particular, cells and genes used for repairing cardiac conduction damage/defect have been investigated in various studies both in vitro and in vivo. Despite the complexities of the excitation and conduction systems of the heart, cell and gene-based strategies provide novel alternatives for treatment or cure of cardiac arrhythmias. This review summarizes some highlights of recent research progress in this field.
Keywords: cell therapy, gene therapy, conduction repair, arrhythmia
1. Introduction
Cardiac arrhythmias are common and affect millions of people. The sinoatrial node (SAN) in the right atrium is the natural pacemaker and is responsible for initiation of rhythmic action potentials in a normal heart.[1],[2] Each impulse generated in the SAN normally propagates to the atria and then to the ventricles via a special conduction system. SAN malfunction or complete atrioventricular node (AVN) conduction block can lead to bradycardia (< 60 beats/min).[3] When persistent symptomatic bradycardia, such as syncope, occurs, implantation of a permanent electronic pacemaker is necessary.[4],[5] The signs and symptoms of other cardiac arrhythmias can range from completely asymptomatic to sudden cardiac death. The most common complaints in patients with a variety of arrhythmias are dizziness, quivering, shortness of breath, chest discomfort, heart fluttering or pounding, and forceful or painful extra beats.
In recent decades, biomedical sciences and medical device technologies have been advanced significantly. As a result, many new options have become available as antiarrhythmic therapies. Cardiac arrhythmias can be treated with medications (antiarrhythmic drugs), catheter ablations, and implantation of pacemakers or implantable cardioverter defibrillators (ICD). Antiarrhythmic medications modify myocardial electrical activities of the heart and thus suppress or prevent arrhythmias. However, antiarrhythmic drugs can cause side effects which may be serious.[6]–[8] Their side effects can be similar to those of non-antiarrhythmic drugs, such as dizziness, allergies, insomnia, and gastrointestinal disturbances. At the same time, some antiarrhythmic drugs can be proarrhythmic and cause cardiac arrhythmias rather than eliminating them. Unfortunately, the arrhythmias caused by antiarrhythmic drugs are sometimes fatal. In addition, antiarrhythmic drugs may interact with other medications. When this happens, the risk of side effects can increase and the therapeutic effects of one or both of the drugs can be altered.
The development of medical devices has provided great opportunities for the treatment of many patients with cardiac arrhythmias. Electronic cardiac pacemakers and defibrillators effectively improve the quality of life and protect patients from fatal arrhythmias. However, implantable electronic devices are expensive and have some limitations.[9],[10] For example, an implanted pacemaker may be sometime malfunction. Furthermore, the battery energizing the pacemaker has limited longevity.[11] Hence, multiple surgeries to replace the battery or the entire implantable pacemaker may be required in a patient who is otherwise healthy. Other possible acute and chronic risks include electrode fracture or damage to insulation, infection, pulmonary collapse, and vein thrombosis. In addition, implantable pacemakers have very limited or no capacity for directly responding to body needs during physiological and emotional activities (e.g., exercise, stress, rest, or sleep). In children, especially in newborn babies, initial size mismatch and growth affecting the position of leads and pulse generators are also serious issues.[12] Extraction of lead(s) from inside the heart is sometimes needed under certain circumstance. While this procedure is generally simple and very safe, there are risks including internal bleeding and tear of a vein or the heart.[13] Catheter ablation is a common treatment for supraventricular tachycardias and sometimes also for ventricular arrhythmias.[14]–[16] This technique is an effective method to ablate an arrhythmic circuit which terminates some cardiac arrhythmias.[17] The common side effects associated with catheter ablation include bleeding, infection, blood clot in the vessel, subcutaneous hematoma, and perforation of the wall of the heart. In addition, accidental surgical damage of the normal conduction system of the heart may require a permanent electronic pacemaker if the damage causes severe bradycardia.
As cardiac arrhythmias are a serious public health issue and current treatments still have shortfalls, research to develop other alternatives to supplement or replace the current conventional therapies is needed. A biological approach to treating cardiac arrhythmias has been developing for many years. Currently, biological therapy for cardiac arrhythmias is at an early research stage and no products are available for clinical applications yet. However, research data suggest that biological approaches for treating cardiac arrhythmias are potentially attainable (see other recent reviews[18]–[22]). While devices, such as electronic pacemakers, are often used as palliative therapy for severe bradycardia, cell and gene therapies may repair damaged conduction and cure associated arrhythmias. In addition, cell and gene therapies may avoid the device-associated shortfalls mentioned above.
Impaired conduction can delay or block impulse propagation in the heart. Conduction abnormalities can underlie both bradyarrhythmias (e.g., AVN block) and reentrant tachyarrhythmias (e.g., fibrosis-related reentry). Conventional therapies for cardiac arrhythmias include medications, catheter ablation, and implantable devices. Unfortunately, these interventions hardly improve cardiac conduction properties and do not repair conduction defects. Cell and gene therapy offers a new, novel approach to treating arrhythmias. In this area, most studies focus on biological pacemakers that can potentially treat severe bradycardia due to SAN dysfunction or complete AV block. Experimental conduction damage and block mostly used in previous studies were mechanically created in cardiomyocyte cultures (Figure 1) or in animal models via ischemia or ablation method (Figure 2). Recreation of biological pacemaker is via functional and structural replacement of the native SAN function and has been reviewed recently in several other articles.[18]–[23] This review focuses on the highlights of using cells and genes to repair damaged cardiac conduction. Research data accumulated in this field have shown that cell transplantation and/or gene transfer can improve or repair impulse conduction in vitro and in experimentally injured animal hearts in vivo (Table 1). The strategy of using biological approach to treating bradycardia and tachycardia is very similar, but cell types and genes used may differ. For bradycardia, cell or gene therapy alone or a combination of cells and genes targets on enhancing tissue automaticity or repairing damaged conduction, such as AV block, to restore a normal rhythmic rate of the heart. In contrast, cell and gene therapy for tachycardia is to reduce myocardial excitability or repair conduction damage to break a reentrant circuit or silence arrhythmic sites. More detail about the strategy of using biological approach to manipulate myocardial excitability and conduction for the treatment of bradycardia and tachycardia has been discussed in a recent review.[24] In the meantime, the potential challenges facing translation of biological alternatives for antiarrhythmic therapy from pre-clinic to clinic are briefly mentioned.
Figure 1. Creation of cardiac conduction block in vitro. (A): Mouse atrial cells from a HL-5 cell line (purchased from Dr. William C. Claycomb, LSU Health Sciences Center, LA, USA) were seeded and cultured on the 60 electrodes in a microelectrode array (MEA) dish; (B): Synchronized spontaneously beating cardiomyocytes were verified by extracellular electrograms recorded on the 3rd day after cell seeding and culture; (C): One approximately 600 μm channel in the middle of the array was abraded by using a 200 µL pipette tip; (D): Such abrasion created conduction block which was evidenced by the electrogram recordings of two independently beating fields with different rates.
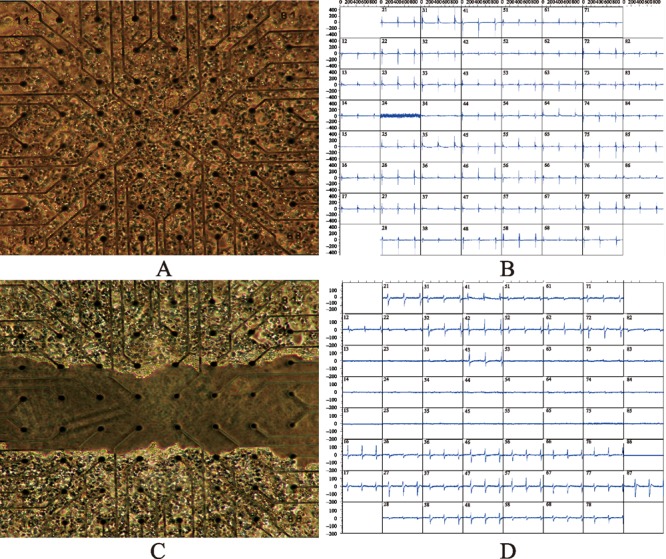
Figure 2. Atrioventricular conduction block after AVN ablation. (A): A schematic diagram of the area usually ablated for creation of complete atrioventricular conduction block; (B): Typical tracings of canine electrocardiograms (Lead II) of the sinus node rhythm; (C) The rhythm of complete atrioventricular block after ablation. The ventricular escape rhythm shows the typical wide QRS complex (C) after AVN ablation with a radio frequency energy catheter. AVN: atrioventricular node.
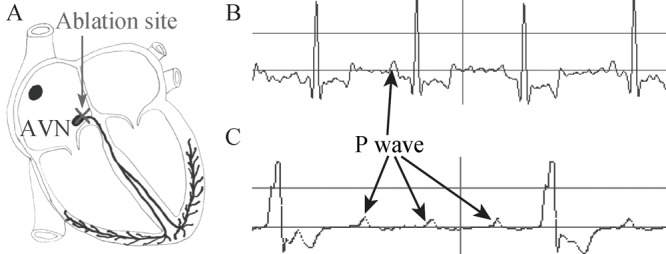
Table 1. Cells and genes for cardiac conduction repair.
| Cell/gene | Cell/Animal model | Result | Ref. |
| hESCs-derived cardiomyocytes | MEA (culture) | Electromechanical integration with rat ventricular myocytes in culture | [25],[26],[85] |
| AV block (pig) | Pacing atrioventricular blocked hearts | [27] | |
| mESCs-derived cardiomyocytes | AV block (mouse) | Restoration of AV conduction | [28],[29] |
| MSCs | MEA (culture) | Repair of conduction damage | [33]–[36] |
| AV block (rat) | Improvement of AV conduction | [37] | |
| Fibroblasts | MEA (culture) | Repair of conduction damage with additional gene transfection | [40],[43]–[46] |
| AV node (dog) | Enhancement of AV conduction with TGF-β1 transfection | [49] | |
| Myoblasts | MEA (culture) | Decrease in arrhythmogenicity with CX43 transduction | [53] |
| AV block (rat) | Repaired AV conduction in 1/3 animals | [54] | |
| Interstitial cells of Cajal | MEA (culture) | Repaired conduction damage | [46] |
| HEK293 cells + Na+ channel gene | MEA (culture) | Resynchronized contractions | [68] |
| Skeletal Na+ channel gene | MEA (culture) | Prevented or disrupted reentrant circuits | [69] |
| Ischemia (mouse, dog) | Increased longitudinal conduction velocity | [70]–[72] | |
h/mESCs: human/mouse embryonic stem cells; MEA: multielectrode array; MSCs: mesenchymal stem cells; AV block: atrioventricular conduction block; HEK293: human embryonic kidney 293 cells.
2. Embryonic stem cells
Myocardial repair, including conduction repair, by transplantation of stem cells has emerged as a novel strategy to treat cardiac diseases in recent years. Cardiomyocytes derived from human embryonic stem cells can electromechanically integrate with primary cultures of neonatal rat ventricular myocytes in vitro.[25],[26] Electrograms recorded simultaneously from 60 electrodes of a multi-electrode array (MEA) showed impulse initiation and conduction within the co-cultures. After adding cardiomyocytes derived from human embryonic stem cells, electrograms demonstrated that tight temporal coupling between the human and rat tissues occurred within one day and lasted continuously to the end of experiment, up to 21 days.[27] In vivo integration was shown in a swine model with complete AV block. Three-dimensional electrophysiological mapping and histopathological examination showed that transplantation of cardiomyocytes derived from human embryonic stem cells could pace an AV blocked heart.[27] These results demonstrate that cardiomyocytes derived from human embryonic stem cells are able to form connections with surrounding host cardiomyocytes.
Connexins (Cx, e.g., Cx43/Cx40) are proteins of the family of the gap junction and particularly important to intercellular communication in heart. Connexins permit ions and small molecules to move between adjacent cells for chemical exchange and electrical propagation. In a mouse AV conduction block model, transplantation of cardiomyocytes derived from mouse embryonic stem cells restored the AV conduction.[28],[29] Substantial amounts of Cx43/Cx40 were verified between embryonic stem cell-derived cardiomyocytes and host cardiomyocytes in the mice who received cell transplantation. In contrast, the non-transplanted mice with AV block showed marked fibrosis and discontinuity of Cx43/Cx40 expression in the AVN region.[28],[29]
3. Mesenchymal stem cells (hMSCs)
In several clinical trials, hMSCs were implanted into the myocardium of the patients with ischemic heart disease.[30]-[32] Regional regeneration of myocardial tissue from implanted cells and improvement of cardiac function were observed. However, the underlying mechanism remains to be elucidated. Recently, hMSCs for cardiac conduction repair have been studied in in vitro experiments.[33],[34] Synchronously beating monolayers of cultured neonatal rat cardiomyocytes in a MEA dish were separated by a mechanically abraded channel to yield two asynchronously beating cardiomyocyte fields. Adding hMSCs to the abraded channel resynchronized the two separated cardiomyocyte fields. Conduction velocity across hMSCs increased progressively after co-culture with cardiomyocytes. Cx43 expression and functional gap junction were formed between hMSCs and cardiomyocytes and such electrical connections were increased following the time after co-cultures.[34] Adding hMSCs to the cultures of acutely isolated canine ventricular myocytes also formed Cx43 and Cx40 connections along the regions of intimate cell-to-cell contact that exhibited cell-to-cell coupling.[35]
Recently, the effects of forced alignment of neonatal rat MSCs with neonatal rat cardiomyocytes on their functional integration were investigated.[36] A laser-dissected channel in a monolayer of originally synchronized beating cardiomyocytes was created in a MEA dish to induce cardiac conduction block. Coatings in the channel were microabraded in a direction parallel or perpendicular to the channel or were unabraded to establish different cell patterns.[36] MSCs added on microabraded coatings resulted in anisotropic cell alignment within the channel. Conduction velocity across MSCs was highest in the perpendicular, intermediate in the isotropic and lowest in the parallel configuration. Alignment-dependent increases in Cx43 expressions were observed. Forced alignment of MSCs affects the time course and the level of functional integration with their surrounding cardiomyocytes.[36]
More recently, another study has shown that local implantation of MSCs into the AVN area improved AV conduction in a rat model with complete AVN block.[37] It is also interesting that the animals with MSC transplantation significantly decreased collagen deposition in the AVN area. The decrease in collagen deposition in MSC-transplanted animals was potentially due to the antifibrotic paracrine effects of implanted MSCs and could improve impulse conduction through the AVN area.[37] However, co-culture of hMSCs with neonatal rat ventricular myocytes decreased conduction velocity and induced reentrant arrhythmias probably due to tissue heterogeneity of inexcitable MSCs with myocytes.[38] To improve the electromechanical integration among MSCs and cardiomyocytes, a recent study showed that infarct rat hearts implanted with cardiogenic cells developed from rat MSCs after treatment with phorbol myristate acetate, a PKC activator, restored conduction velocity via reducing tissue heterogeneity and improved myocardial contractility.[39]
4. Fibroblasts
Myocardial ischemia and infarction can cause cardiomyocyte death. As matured human cardiomyocytes have very limited ability to regenerate, dead myocardium is usually replaced by non-excitable and non-contractile fibrotic scar tissue. Impulse propagation through the scar tissue is altered.[40] Fibroblasts are non-excitable with low levels of connexin expression. Therefore, scar tissue in the myocardium has a very limited capability to conduct electrical impulses and this may slow conduction or even cause conduction block.[33],[41],[42] Consequently, scar tissue in the heart is potentially an arrhythmic substrate. Prevention of its appearance can be therapeutic. Genetic modification of the electrical property of scar tissue may thus have the potential for the treatment of scar-related arrhythmias or myocardial asynchrony.[43]
Cardiac fibroblasts are the predominant cell type of the non-contractile cells in the heart. In a recent study, the feasibility of cardiac conduction repair in vitro was investigated with genetically modified human ventricular scar fibroblasts (hVSFs) by using the MEA technique.[44] Rat cardiomyocytes were cultured in two fields separated by a strip of hVSFs and beat asynchronously. Forced expression of the myocardin (MyoC) gene in the hVSFs improved impulse conduction and resynchronized the cardiomyocytes in two originally separated fields. Also, MyoC-hVSFs showed responsiveness to electrical stimulation.[44] Intracellular recordings demonstrated that MyoC-hVSFs coupled well to surrounding cardiomyocytes and improved their impulse conduction probably via expression of various cardiac ion channels and connexins.
Recently, a murine atrial cell HL-5 line was used to assess the effects of cardiac fibroblasts on cardiac conduction damage. HL-5 cells were cultured in a MEA chamber to form a monolayer which beat spontaneously and synchronously.[45] A channel (from 600 µm to 700 µm) in the middle of each array was mechanically created to form two asynchronously beating fields (Figure 1). Human cardiac fibroblasts added to the abraded channel were not able to repair the conduction damage and to resynchronize the two separated asynchronously beating fields.[45],[46] Interestingly, the amplitudes of the field potentials along the abraded channel were significantly reduced after adding cardiac fibroblasts. These results demonstrate that cardiac fibroblasts alone were not able to repair the conduction damage in vitro and even reduced the amplitude of field electrical potentials. Amplitude reduction may slow conduction velocity and cause arrhythmias.
To understand whether fibroblasts could modulate cardiomyocyte excitability in a Cx43-dependent manner, isolated neonatal rat cardiomyocytes were used in a co-culture study system.[40] Cultured cardiomyocytes were grown on monolayers of mouse fibroblasts with genetically altered Cx43 expression. The beating rates of the cardiomyocytes grown on wildtype fibroblasts expressing native levels of Cx43 were significantly lower than those of myocytes grown on fibroblasts devoid of Cx43 (germline knockout) or with dominant-negative functional suppression of Cx43. Transfection of Cx43 to the fibroblasts obtained from Cx43 knockout mice restored cardiomyocyte beating frequency. The beating frequency of cardiomyocytes co-cultured with Cx43-transfected fibroblasts from Cx43 knockout mice were comparable to those observed in co-cultures with wildtype fibroblasts.[40]
It is quite possible that non-cardiac fibroblasts may also be able to conduct electrical impulses after they are genetically manipulated. Human dermal fibroblasts transferred with gene vector encoding MyoD, a skeletal myogenic determination factor, underwent myogenic conversion and showed Ca2+ transients.[43] Membrane excitability was observed in MyoD-induced myotubes after loading with a Ca2+ sensitive dye and electrical stimulation, but there was no electrical coupling among the cells. However, simultaneous transduction of human dermal fibroblasts with MyoD and Cx43 genes resulted in functional coupling which was evidenced via dye transfer from cell to cell.[43] Non-cardiac fibroblasts can thus be genetically modified to act as excitable cells with electrical coupling.
The results from two recent studies have shown that transplantation of genetically engineered fibroblasts with transferred K+ channels (Kv1.3 or Kir2.1) into rat and pig ventricular myocardium reduced cardiac automaticity and prolonged refractoriness due to their coupling with host cardiomyocytes.[47],[48] In vitro experiments and computer model simulations confirmed the in vivo results. Compared with the baseline beating rate of cultured neonatal rat ventricular myocytes and the rate of the neonatal rat ventricular myocytes co-cultured with naive fibroblasts, the rate was significantly reduced in the neonatal rat ventricular myocytes co-cultured with Kv1.3-transfected fibroblasts.[47],[48]
The data mentioned above indicate that non-genetically modified cardiac fibroblasts decrease cardiomyocyte impulse amplitude in vitro and can be arrhythmic substrates due to slowing or blocking electrical signal propagation. However, genetically modified fibroblasts may have therapeutic value by improving electrophysiological responsiveness of myocardial cells to pacing and electrical conduction. One study shows that local injection of fibroblasts into the myocardium can modify AVN function without creation of AV block in dogs.[49] Such effect of injected fibroblasts on AVN conduction is substantially enhanced by pretreatment of fibroblasts with TGF-1.
5. Myoblast cells
Myogenic precursor cells named myoblasts can be harvested from skeletal muscle biopsy. Primary myoblasts have the capability of division and expansion to offer a rich cell source for cell transplantation. Clinically, myoblasts have been transplanted autologously for myocardial repair.[50]-[52] Ventricular tachyarrhythmias and sudden cardiac death potentially related to the failure of forming gap junction have been observed in the clinical trials with myoblast transplantation for heart failure treatment.[50]-[52] Co-cultures of human skeletal myoblasts and rat cardiomyocytes reproduced reentrant arrhythmias that may be similar to the features of ventricular tachycardia observed in patients who received myoblast transplants. The Ca2+ channel blocker nitrendipine, but not the Na+ channel blocker lidocaine, blocked the arrhythmias in culture. Transduction of myoblasts with Cx43 decreased arrhythmogenicity in co-cultures of human skeletal myoblasts and rat cardiomyocytes.[53] A recent study showed that engineered tissue constructs with myoblasts were able to maintain cell-to-cell electrical communication through persistent expression of functional gap junction proteins, Cx43 and Cx45.[54] Implantation of the engineered myoblast tissue constructs into the cardiac AV groove of AVN-ablated rats created an alternative AV conduction pathway. After implantation, myogenic cells in the constructs survived, vascularized, and electromechanically coupled with surrounding host cardiomyocytes. One-third of recipient animals with previous AVN ablation showed a permanent AV conduction through the implanted constructs.[54]
6. Interstitial cells of Cajal (ICC)
A network of interstitial cells, lately named as ICC, in the gut was found and described as neuronal nature by Cajal in 1889.[55] Keith, who discovered the SAN, lately postulated that these cells were “a pacemaker system of the intestinal muscle”.[56] ICC generate spontaneous pacemaker currents for initiation of gastrointestinal motility. Electron microscopy indicates that ICC are specialized myoid cells with two[57] or more[56] sub-populations. The embryological origin of ICC is from the same embryonic germ layer mesoderm[57],[58] as that of heart cells. The source of ICC in mammals is likely to be mesenchymal.[58] Many ion channels in ICC membrane are responsible for the spontaneous rhythm and impulse conduction among ICC and their surrounding cells.[59]–[63] ICC express Cx43 and form conductive networks via Cx43.[57],[64] Gut ICC networks effectively propagate electrical signals which are critical for gut peristalses. Gastrointestinal operation disrupts ICC networks in the surgical region, but after operation ICC can reestablish connections and restore gastrointestinal peristalses within a few days.[65],[66]
Cardiac conduction repair by ICC in vitro was investigated recently.[46] Conduction block was created by abrasion of a synchronously beating monolayer of mouse atrial HL-5 cells cultured in a MEA dish. Abraded channel was about 600 μm to 700 μm in width in the middle of each dish. Field potential recordings showed that abrasion created a complete conduction block and the myocytes on the two sides beat independently. After baseline recording, ICC isolated from porcine ileum (Figure 3) were carefully added to abraded channels and re-cultured for a few days. Synchronous beating of the cardiomyocytes was observed after addition of ICC. Re-synchronous beating was not observed in cardiomyocytes in control abraded MEA dishes or in abraded MEA dishes added with human cardiac fibroblasts.[46] In addition, fluorescence-labeled porcine ICC were injected into the right atrium via an endocardial injection lead to assess if the injected ICC survived in porcine myocardium in vivo. Tissue sections showed that ICC survived in the injected atrial myocardium two weeks after the in vivo injection.[46] These results demonstrated that ICC could be obtained in high numbers from the gut and were able to conduct impulses across damaged cardiomyocyte fields in vitro. Also, ICC survived in porcine atrial myocardium after cell implantation. As ICC show many similarities in electrophysiology to heart cells and as our body is rich in ICC, autologous transplantation will reduce ethical issues and immuno-rejection. However, additional studies are needed to confirm if ICC transplantation can repair cardiac conduction damage in vivo.
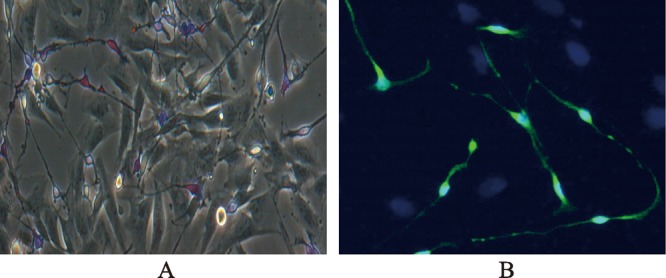
Figure 3. Morphology of interstitial cells of Cajal (ICC) stained with methylene blue (A) and c-kit (B). Intestinal cells were isolated from adult porcine ileum by an enzymatical method. Isolated intestinal cells were cultured and characterized by methylene blue and c-kit staining. Note the morphological differences between methylene blue positive and negative cells. Morphological connections appeared among methylene blue positive cells, and between positive and negative cells (A). ICC were verified by the immunostaining antibody (B) against c-kit (green = ICC) and DAPI (4,6-diamidino-2-phenylindole) (blue = nuclei).
7. Human embryonic kidney cells
Human embryonic kidney 293 (HEK293) cells are derived from a specific cell line originally generated by transformation of HEK cell cultures with sheared adenovirus 5 DNA.[67] In a recent study, genetically engineered HEK293 cells were used as “conducting cables” to repair cardiac conduction damage.[68] An in vitro model of conduction block was established using spatially separated, spontaneously contracting, nonsynchronized human embryonic stem cell–derived cardiomyocytes clusters. Genetically engineered HEK293 cells with expression of the SCN5A-encoded Na+ channel were added to the cultures to assess their ability to electrically couple with cardiomyocytes. MEA and intracellular recordings confirmed that SCN5A-engineered HEK293 cells conducted impulses in the co-cultures and resynchronized contractions between separated clusters.[68] Immunostaining against Cx43 and calcein dye-transfer data showed functional gap junctions between the SCN5A-engineered HEK293 cells and neighboring cardiomyocytes. Therefore, HEK293 cells with transfection of Na+ channels can act as biological conducting cables to connect spatially separated cardiomyocytes. This demonstrates the potential of genetically engineered HEK-293 cells for the treatment of cardiac arrhythmias.[19],[68]
Similar to any other novel therapies at their early stages, cell and gene therapies for cardiac arrhythmias face tremendous challenges. For example, cell implantation for repair of cardiac conduction damage or defect has various limitations and challenges, including cell sources (autologous, allogeneic or xenogeneic), number of injected cells, selection of injection site(s), short and long term cell retention, cell distribution in the targeted and non-targeted tissues, differentiation if using pluripotent cells, proliferation, integration with host cells, immune response to donor and host cells, life span, proarrhythmic properties, and potential for neoplasia. While some success has been observed in in vitro and in vivo studies, the feasibility, efficacy, and safety of using cells to treat conduction damage/defect are largely unknown and have not been tested in any clinical study. The current knowledge regarding cell therapy for conduction repair is primarily derived from in vitro studies of short-term observation (hours or days). There are only a small number of studies in the literature which show some success of cell therapies for conduction repair in animals. Efficacy and safety concerns remain even though various cell types have been implanted into the myocardium for tissue repair in heart failure patients.
8. Gene transfers
Scar formation after myocardial infarction or fibrosis of the myocardium causes a slow conduction which can induce reentrant arrhythmias. Gene transfer to alter the electrical properties of the cells in the area initiating such arrhythmias may improve impulse conduction or silence of the spontaneous depolarization of those cells. Therefore, gene therapy may be an alternative way to treat or cure cardiac arrhythmias. This hypothesis has been examined in several studies by transferring ion channel or non-ion channel genes.
8.1. Skeletal Na+ channel gene
One recent study showed that increased Na+ channel expression by transfection of skeletal Na+ channel isoform (SKM1) into newborn rat ventricular myocyte cultures could speed up electrical conduction and, thus, prevent or disrupt reentrant circuits.[69] However, transfection of the cardiac Na+ channel isoform (Nav1.5) did not eliminate reentry excitation in cardiomyocyte cultures.[69] The explanation is that the channel availability of SKM1 is higher than Nav1.5 when the membrane is partially depolarized by elevated extracellular K+ ions. In addition, in a canine myocardial infarct model, adenovirus SKM1 vectors were injected into the epicardial infarct border zones. Immunohistochemistry confirmed SKM1 expression in the injected area.[70] Expression of SKM1 increased V(max) of depolarized myocardium and reduced the incidence of inducible sustained ventricular tachyarrhythmia/fibrillation. Another study showed that overexpression of SKM1 Na+ channel could preserve normal conduction and decrease arrhythmias during ischemia and reperfusion in murine hearts.[71] Expression of SKM1 Na+ channels in canine myocardium at one week after infarction increases longitudinal but not transverse conduction velocity, consistent with the increased dV/dt(max) and with the cellular localization of SKM1 Na+ channels.[72] These results suggest that gene therapy to normalize excitation or to enhance conduction in diseased myocardium may have antiarrhythmic significance.
8.2. K+ channel genes
The potential to ablate ventricular arrhythmias by transfer of a K+ channel gene was evaluated recently in a porcine model of myocardial infarction.[73] Focal transfer of a gene encoding a dominant-negative version of K+ channel Kv11.1 (normally conducts delayed rectifier K+ current, IKr) to the infarct scar border zone eliminated all ventricular arrhythmias via increased refractoriness to prevent reentry circuits. In addition, no proarrhythmia or other negative effects were observed in the gene-transferred animals.[73] Another in vitro study showed that electrotonic application of inward rectifier current from non-cardiomyocyte cells expressing Kir2.1 channels could inhibit cardiomyocyte automaticity in cultures.[74]
8.3. G-protein genes
Gene transfer with adenovirus constructs encoding β-galactosidase (β-gal), wildtype Gαi2 (wtGi), or constitutively active mutant (cGi) to the AVN area was recently conducted in a swine model with persistent atrial fibrillation and severe heart failure.[75],[76] Heart rates in awake, alert animals were not altered by β-gal or wtGi. Cardiomyopathy worsened over time in the β-gal group and the condition was improved slightly in the wtGi group. However, cGi transfer decreased the heart rate by 15% to 25% and maintained ejection fraction close to the normal level at the end of the study. cGi overexpression in the AVN may physiologically control the ventricular rate during persistent atrial fibrillation.[75] In another study, local gene transfer with the ras-related small G-protein Gem to the heart acted locally as a genetic Ca2+ channel blocker. Expression of Gem markedly decreased the L-type Ca2+ currents and action potential duration in ventricular myocytes isolated from Gem-infected guinea pigs. Gem expression also resulted in a significant shortening of the electrocardiographic QTc interval and a reduction of left ventricular systolic function. Furthermore, Gem expression in the AVN area significantly slowed AVN conduction in a swine study, which effectively reduced the ventricular rate during atrial fibrillation.[77]
8.4. Connexin genes
Modification of electrical conduction may modify the arrhythmia substrate and prevent or inhibit certain cardiac arrhythmias (e.g., ventricular tachycardia). Transfer of mutated connexin genes to reduce gap junctional intercellular communication was investigated in a recent study.[78] Transduction of cultured neonatal rat ventricular myocytes with lentiviral vectors encoding Cx43 internal loop mutants showed a significant reduction of inter-myocyte communication and delayed Ca2+ transients. Compared with non-transduced cardiomyocytes, optical mapping of action potential propagation in gene-modified cardiomyocyte monolayers revealed a 3-fold slowing of conduction velocity. These results demonstrate that transfer of the Cx43 mutant gene reduces electrical conduction among the cultured cardiomyocytes via modification of their electrical coupling. This effect may have therapeutic potentials for atrial or ventricular tachycardia. In contrast, transfer of the gene encoding the pH-insensitive gap junction channel protein Cx32 prevented conduction slowing during ischemia and decreased the incidence and duration of ventricular tachycardia in a mouse ischemia-reperfusion model.[71]
9. Summary
Morbidity and mortality of cardiac arrhythmias are high, especially in the geriatric population. Conduction damage or defect can slow or block electrical impulse propagation and lead to either tachycardia or bradycardia. Current clinical therapies have limited capability to improve cardiac conduction and do not treat anatomically impaired conduction. Biological strategy, either with cell or gene alone or with a combination of cell and gene, has the potential to restore an impaired conduction. Several studies in vitro and in vivo have shown the potentials of their antiarrhythmic effects (Table 1). This article reviews the progress in biological alternatives potentially for future antiarrhythmic therapy. Recent clinical studies to assess the safety and efficacy of gene therapies and gene profiles of cardiovascular diseases are listed in Table 2. Some of the studies have shown certain positive outcomes.[79],[80] However, there is a lack of clinical trials using genes to treat cardiac arrhythmias. Compared with traditional therapies, such as medication, device and surgical approaches, no products related to cell and gene therapies for cardiac arrhythmias are currently available for clinical use. Gene therapy for cardiac arrhythmias faces the same significant obstacles as other gene therapies, such as gene selection, dosage, carrier selection, focal delivery and distribution, longevity, toxicity, inflammation, and risk of immunogenicity/carcinogenicity. In addition, some cells (e.g., stem cells and fibroblasts) proliferate quickly. As a result, these cells may quickly lose the phenotype of the targeted gene which is originally transferred for the therapy. To obtain a stable long-term effect, such as biological pacemaker activity, an optimal dosage and a long-term expression gene carrier may be required.[81],[82] A low dose may be ineffective and a high dose may lead to serious side effects (cell death probably due to intracellular Ca2+ overload).[83] Repeated gene therapy to the same subject can be dangerous because this may stimulate host immune responses. Gene vectors used in current gene therapy studies have the limitation of gene sizes incorporated and the potential to cause diseases. In addition, single gene transfer to treat a disease due to a single gene defect is relatively simpler than trying to correct multiple gene defects for the therapy of multifactorial disorders.
Table 2. Recent clinical studies related to gene therapies and gene profiles of heart diseases.
| Name | Institution | Status | Targeted disease | Gene | Phase |
| VEGF-D gene therapy for the treatment of severe coronary heart disease | Kuopio University Hospital, Finland.CTI: CT01002430 | Recruiting | Angina pectoris, myocardial infarction (“no option-patients”) | VEGF-Da | Phase I |
| SERCA gene therapy trial | Imperial College London, United KingdomCTI: CT00534703 | Not yet recruiting | Advanced heart failure and heart failure received a left ventricular assist device | SERCA2ab | Phase I/II |
| Gene therapy for the treatment of chronic stable angina | ViroMed Co., Ltd.; Northwestern Memorial Hospital Chicago, USA.CTI: CT01002495 | Not yet recruiting | Chronic refractorymyocardial ischemia | Genetic: VM202c | Phase I/II |
| AC6 gene transfer for congestive heart failure | National Heart, Lung, and Blood Institute (NHLBI).CTI: CT00787059 | Recruiting | Congestive heart failure | AC6d | Phase I/II |
| Safety & efficacy study of rAAV1-CB-hAAT for alpha-1 antitrypsin deficiency | Applied Genetic Technologies Corp.CTI: CT01054339 | Active, but notrecruiting | Alpha-1 antitrypsin deficiency | AAT | Phase II |
| Efficacy and safety study of genetically targeted enzyme replacement therapy for advanced heart failure (CUPID) | Celladon Corporation, USA.CTI: CT00454818 | Active, but not recruiting | Heart failure, congestive ilated cardiomyopathy | Gene transfer agent MYDICAR®f | Phase I/II |
| Gene expression profiling in subjects with postoperative atrial fibrillation after cardiac surgery | Brigham and Women'sHospital, Boston, USA.CTI: CT00833313 | Recruiting | Atrial fibrillation after heart surgery (germline variation) | Gene expression profiling/atrial biopsy | NA |
| CRP gene variants and CAD in a Chinese Han population | Fudan University, Shanghai, ChinaCTI: CT00780221 | Recruiting | Circulating CRP levels and CAD risk | CRP gene and protein/blood sample | NA |
Studies listed in the table are from the website: http://clinicaltrials.gov. Reader can find more details about each trial from the website if needed. CTI: ClinicalTrials.gov Identifier; NA: not applied; VEGF-D: Endocardial vascular endothelial growth factor D; AC6: Adenylyl cyclase type 6; CRP: C-Reactive protein; CAD: coronary artery disease.
aVEGF-D is a vascular endothelial growth factor that is encoded by the FIGF gene in humans. VEGF-D is one of the placenta growth factors. This protein can bind and activate VEGFR-2 and VEGFR-3 receptors to induce angiogenesis, lymphangiogenesis, and endothelial cell growth.
bSERCA is the acronym of the sarcoplasmic reticulum (SR) Ca2+ ATPase which is a membrane protein that catalyzes the ATP-dependent transport of Ca2+ from the cytosol to the SR. SERCA2a is a SERCA isoform which is the main cytosolic Ca2+ regulator in the heart. Deficiency of SERCA2a often occurs in advanced heart failure and may facilitate progressive systolic and diastolic dysfunction. SERCA2a gene transfer may results in the restoration of SERCA2a and improvement of failed heart function.[79]
cThe information about VM202 is not publically available.
dAdenylyl cyclase, also called 3′,5′-cyclic AMP synthetase, is a membrane-associated enzyme that catalyzes the formation of the secondary messenger cyclic adenosine monophosphate (cAMP) from adenosine triphosphate (ATP). AC6 is adenylate cyclase type 6 that in humans is encoded by the ADCY6 gene. Animal data showed that increased amounts of AC6 protein in heart cells appeared to make the heart pump more vigorously.
eAlpha-1 antitrypsin (AAT) is a protein made in the liver and then released to the bloodstream. Alpha-1 antitrypsin deficiency (AATD) is an inherited disease due to mutations in the SERPINA1 gene on chromosome 14. This gene codes for AAT which protects the lungs from damage and make them work normally. AATD has low levels of, or no, AAT in the blood and causes lung and/or liver disease. This study attempts to make patient's cells to produce enough AAT via transferring the healthy gene to the cells.
fSERCA2a gene is used in this study (see the noteb above).
Using cells and/or genes to modify cardiac conduction or repair conduction damage/defect is novel and has made some progress in experimental research, but at its early research stage. The biological approach to treating arrhythmias still faces many challenges and obstacles as discussed in other articles,[18]–[23],[84] including proarrhythmia.[38] However, such biological approaches have shown significant promises at various levels of preclinical development. Further extensive investigation and effort in this field are needed to facilitate crossing the bridge between preclinical research and patient application.
Acknowledgments
I would like to thank Alena Nikolskaya, Lepeng Zeng, Xiaohong Qiu, Eric S. Richardson, and Professor Paul A. Iaizzo for their collaborations. I also express my gratitude to Professor Joseph J. McArdle for his helpful comments on the manuscript.
References
- 1.Boyett MR, Dobrzynski H. The sinoatrial node is still setting the pace 100 years after its discovery. Circ Res. 2007;100:1543–1545. doi: 10.1161/CIRCRESAHA.107.101101. [DOI] [PubMed] [Google Scholar]
- 2.Monfredi O, Dobrzynski H, Mondal T, et al. The anatomy and physiology of the sinoatrial node—a contemporary review. Pacing Clin Electrophysiol. 2010;33:1392–1406. doi: 10.1111/j.1540-8159.2010.02838.x. [DOI] [PubMed] [Google Scholar]
- 3.Dobrzynski H, Boyett MR, Anderson RH. New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation. 2007;115:1921–1932. doi: 10.1161/CIRCULATIONAHA.106.616011. [DOI] [PubMed] [Google Scholar]
- 4.Kantrowitz A. Implantable cardiac pacemakers. Ann N Y Acad Sci. 1964;111:1049–1067. doi: 10.1111/j.1749-6632.1964.tb53173.x. [DOI] [PubMed] [Google Scholar]
- 5.Kantrowitz A. Complete heart block treated with an implanted, controllable cardiac pacemaker. Ann N Y Acad Sci. 1964;118:113–118. doi: 10.1111/j.1749-6632.1964.tb33966.x. [DOI] [PubMed] [Google Scholar]
- 6.Hall MC, Todd DM. Modern management of arrhythmias. Postgrad Med J. 2006;82:117–125. doi: 10.1136/pgmj.2005.033654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roden DM. Risks and benefits of antiarrhythmic therapy. N Engl J Med. 1994;331:785–791. doi: 10.1056/NEJM199409223311207. [DOI] [PubMed] [Google Scholar]
- 8.Chaudhry GM, Haffajee CI. Antiarrhythmic agents and proarrhythmia. Crit Care Med. 2000;28:N158–N164. doi: 10.1097/00003246-200010001-00008. [DOI] [PubMed] [Google Scholar]
- 9.Hauser RG, Hayes DL, Kallinen LM, et al. Clinical experience with pacemaker pulse generators and transvenous leads: an 8-year prospective multicenter study. Heart Rhythm. 2007;4:154–160. doi: 10.1016/j.hrthm.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Bernstein AD, Parsonnet V. Survey of cardiac pacing and implanted defibrillator practice patterns in the United States in 1997. Pacing Clin Electrophysiol. 2001;24:842–855. doi: 10.1046/j.1460-9592.2001.00842.x. [DOI] [PubMed] [Google Scholar]
- 11.Senaratne J, Irwin ME, Senaratne MP. Pacemaker longevity: are we getting what we are promised? Pacing Clin Electrophysiol. 2006;29:1044–1054. doi: 10.1111/j.1540-8159.2006.00497.x. [DOI] [PubMed] [Google Scholar]
- 12.Friedman RA, Fenrich AL, Kertesz NJ. Congenital complete atrioventricular block. Pacing Clin Electrophysiol. 2001;24:1681–1688. doi: 10.1046/j.1460-9592.2001.01681.x. [DOI] [PubMed] [Google Scholar]
- 13.Kay GN, Brinker JA, Kawanishi DT, et al. Risks of spontaneous injury and extraction of an active fixation pacemaker lead: report of the accufix multicenter clinical study and worldwide registry. Circulation. 1999;100:2344–2352. doi: 10.1161/01.cir.100.23.2344. [DOI] [PubMed] [Google Scholar]
- 14.Richardson AW, Callans DJ, Josephson ME. Electrophysiology of postinfarction ventricular tachycardia: a paradigm of stable reentry. J Cardiovasc Electrophysiol. 1999;10:1288–1292. doi: 10.1111/j.1540-8167.1999.tb00306.x. [DOI] [PubMed] [Google Scholar]
- 15.Richardson AW, Josephson ME. Ablation of ventricular tachycardia in the setting of coronary artery disease. Curr Cardiol Rep. 1999;1:157–164. doi: 10.1007/s11886-999-0075-z. [DOI] [PubMed] [Google Scholar]
- 16.Roberts-Thomson KC, Lau DH, Sanders P. The diagnosis and management of ventricular arrhythmias. Nat Rev Cardiol. 2011;8:311–321. doi: 10.1038/nrcardio.2011.15. [DOI] [PubMed] [Google Scholar]
- 17.Wijnmaalen AP, Zeppenfeld K. Ventricular tachycardia ablation: indications and techniques. Minerva Cardioangiol. 59:149–169. [PubMed] [Google Scholar]
- 18.Prystowsky EN, Camm J, Lip GY, et al. The impact of new and emerging clinical data on treatment strategies for atrial fibrillation. J Cardiovasc Electrophysiol. 2010;21(8):946–958. doi: 10.1111/j.1540-8167.2010.01770.x. [DOI] [PubMed] [Google Scholar]
- 19.Gepstein L. Cell and gene therapy strategies for the treatment of postmyocardial infarction ventricular arrhythmias. Ann N Y Acad Sci. 2010;1188:32–38. doi: 10.1111/j.1749-6632.2009.05080.x. [DOI] [PubMed] [Google Scholar]
- 20.Blank AC, van Veen TA, Jonsson MK, et al. Rewiring the heart: stem cell therapy to restore normal cardiac excitability and conduction. Curr Stem Cell Res Ther. 2009;4:23–33. doi: 10.2174/157488809787169066. [DOI] [PubMed] [Google Scholar]
- 21.Cho HC, Marban E. Biological therapies for cardiac arrhythmias: can genes and cells replace drugs and devices? Circ Res. 2010;106:674–685. doi: 10.1161/CIRCRESAHA.109.212936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosen MR, Danilo P, Robinson RB. Gene and cell therapy for life-threatening cardiac arrhythmias. Dialog Cardiovasc Med. 2009;14:44–51. [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao YF, Sigg DC. Biological approaches to generating cardiac biopacemaker for bradycardia. Sheng Li Xue Bao. 2007;59:562–570. [PubMed] [Google Scholar]
- 24.Gepstein L, Feld Y, Yankelson L. Somatic gene and cell therapy strategies for the treatment of cardiac arrhythmias. Am J Physiol Heart Circ Physiol 2004; 286. 2004;286:H815, H822. doi: 10.1152/ajpheart.00962.2003. [DOI] [PubMed] [Google Scholar]
- 25.Kehat I, Gepstein A, Spira A, et al. High-resolution electrophysiological assessment of human embryonic stem cell-derived cardiomyocytes: a novel in vitro model for the study of conduction. Circ Res. 2002;91:659–661. doi: 10.1161/01.res.0000039084.30342.9b. [DOI] [PubMed] [Google Scholar]
- 26.Itzhaki I, Maizels L, Huber I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 27.Kehat I, Khimovich L, Caspi O, et al. Electromechanical integration of cardiomyocytes derived from human embryonic stem cells. Nat Biotechnol. 2004;22:1282–1289. doi: 10.1038/nbt1014. [DOI] [PubMed] [Google Scholar]
- 28.Lee JK YD, Iwase M. A novel mouse model of sudden cardiac death with acquired long QT and Torsade de Pointes secondary to complete AV block. Biophys J. 2005;88:472A. [Google Scholar]
- 29.Lee JK TY. Small animal models for arrhythmia studies. In: Sigg DS, Iaizzo PA, Xiao YF, et al., editors. Cardiac Electrophysiology Methods and Models. Springer; London: 2010. pp. 261–280. [Google Scholar]
- 30.Katritsis DG, Sotiropoulou PA, Karvouni E, et al. Transcoronary transplantation of autologous mesenchymal stem cells and endothelial progenitors into infarcted human myocardium. Catheter Cardiovasc Interv. 2005;65:321–329. doi: 10.1002/ccd.20406. [DOI] [PubMed] [Google Scholar]
- 31.Glikson M, Lipchenca I, Viskin S, et al. Long-term outcome of patients who received implantable cardioverter defibrillators for stable ventricular tachycardia. J Cardiovasc Electrophysiol. 2004;15:658–664. doi: 10.1046/j.1540-8167.2004.03344.x. [DOI] [PubMed] [Google Scholar]
- 32.Katritsis DG, Sotiropoulou P, Giazitzoglou E, et al. Electrophysiological effects of intracoronary transplantation of autologous mesenchymal and endothelial progenitor cells. Europace. 2007;9:167–171. doi: 10.1093/europace/eul184. [DOI] [PubMed] [Google Scholar]
- 33.Beeres SL, Atsma DE, van der Laarse A, et al. Human adult bone marrow mesenchymal stem cells repair experimental conduction block in rat cardiomyocyte cultures. J Am Coll Cardiol. 2005;46:1943–1952. doi: 10.1016/j.jacc.2005.07.055. [DOI] [PubMed] [Google Scholar]
- 34.Pijnappels DA, Schalij MJ, van Tuyn J, et al. Progressive increase in conduction velocity across human mesenchymal stem cells is mediated by enhanced electrical coupling. Cardiovasc Res. 2006;72:282–291. doi: 10.1016/j.cardiores.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 35.Valiunas V, Doronin S, Valiuniene L, et al. Human mesenchymal stem cells make cardiac connexins and form functional gap junctions. J Physiol. 2004;555:617–626. doi: 10.1113/jphysiol.2003.058719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pijnappels DA, Schalij MJ, Ramkisoensing AA, et al. Forced alignment of mesenchymal stem cells undergoing cardiomyogenic differentiation affects functional integration with cardiomyocyte cultures. Circ Res. 2008;103:167–176. doi: 10.1161/CIRCRESAHA.108.176131. [DOI] [PubMed] [Google Scholar]
- 37.Yokokawa M, Ohnishi S, Ishibashi-Ueda H, et al. Transplantation of mesenchymal stem cells improves atrioventricular conduction in a rat model of complete atrioventricular block. Cell Transplant. 2008;17:1145–1155. doi: 10.3727/096368908787236594. [DOI] [PubMed] [Google Scholar]
- 38.Chang MG, Tung L, Sekar RB, et al. Proarrhythmic potential of mesenchymal stem cell transplantation revealed in an in vitro coculture model. Circulation. 2006;113:1832–1841. doi: 10.1161/CIRCULATIONAHA.105.593038. [DOI] [PubMed] [Google Scholar]
- 39.Song H, Hwang HJ, Chang W, et al. Cardiomyocytes from phorbol myristate acetate-activated mesenchymal stem cells restore electromechanical function in infarcted rat hearts. Proc Natl Acad Sci USA. 2011;108:296–301. doi: 10.1073/pnas.1015873107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kizana E, Ginn SL, Smyth CM, et al. Fibroblasts modulate cardiomyocyte excitability: implications for cardiac gene therapy. Gene Ther. 2006;13:1611–1615. doi: 10.1038/sj.gt.3302813. [DOI] [PubMed] [Google Scholar]
- 41.Gaudesius G, Miragoli M, Thomas SP, et al. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ Res. 2003;93:421–428. doi: 10.1161/01.RES.0000089258.40661.0C. [DOI] [PubMed] [Google Scholar]
- 42.Miragoli M, Gaudesius G, Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ Res. 2006;98:801–810. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 43.Kizana E, Ginn SL, Allen DG, et al. Fibroblasts can be genetically modified to produce excitable cells capable of electrical coupling. Circulation. 2005;111:394–398. doi: 10.1161/01.CIR.0000153812.64956.EF. [DOI] [PubMed] [Google Scholar]
- 44.Pijnappels DA, van Tuyn J, de Vries AA, et al. Resynchronization of separated rat cardiomyocyte fields with genetically modified human ventricular scar fibroblasts. Circulation. 2007;116:2018–2028. doi: 10.1161/CIRCULATIONAHA.107.712935. [DOI] [PubMed] [Google Scholar]
- 45.Xiao YF. Multi-channel system for analysis of cardiac rhythmicity and conductivity in vitro. In: Sigg DS, Iaizzo PA, Xiao YF, et al., editors. Cardiac Electrophysiology Methods and Models. Springer; London: 2010. pp. 395–418. [Google Scholar]
- 46.Xiao YF, Nikolskaya A, Richardson EC, et al. The use of gut interstitial cells of cajal for cardiac conduction repair. Heart Rhythm. 2010;7(5):S412–S413. [Google Scholar]
- 47.Feld Y, Melamed-Frank M, Kehat I, et al. Electrophysiological modulation of cardiomyocytic tissue by transfected fibroblasts expressing potassium channels: a novel strategy to manipulate excitability. Circulation. 2002;105:522–529. doi: 10.1161/hc0402.102661. [DOI] [PubMed] [Google Scholar]
- 48.Yankelson L, Feld Y, Bressler-Stramer T, et al. Cell therapy for modification of the myocardial electrophysiological substrate. Circulation. 2008;117:720–731. doi: 10.1161/CIRCULATIONAHA.106.671776. [DOI] [PubMed] [Google Scholar]
- 49.Bunch TJ, Mahapatra S, Bruce GK, et al. Impact of transforming growth factor-beta1 on atrioventricular node conduction modification by injected autologous fibroblasts in the canine heart. Circulation. 2006;113:2485–2494. doi: 10.1161/CIRCULATIONAHA.105.570796. [DOI] [PubMed] [Google Scholar]
- 50.Smits PC, van Geuns RJ, Poldermans D, et al. Catheter-based intramyocardial injection of autologous skeletal myoblasts as a primary treatment of ischemic heart failure: clinical experience with six-month follow-up. J Am Coll Cardiol. 2003;42:2063–2069. doi: 10.1016/j.jacc.2003.06.017. [DOI] [PubMed] [Google Scholar]
- 51.Menasche P, Hagege AA, Vilquin JT, et al. Autologous skeletal myoblast transplantation for severe postinfarction left ventricular dysfunction. J Am Coll Cardiol. 2003;41:1078–1083. doi: 10.1016/s0735-1097(03)00092-5. [DOI] [PubMed] [Google Scholar]
- 52.Menasche P, Alfieri O, Janssens S, et al. The myoblast autologous grafting in ischemic cardiomyopathy (MAGIC) trial: first randomized placebo-controlled study of myoblast transplantation. Circulation. 2008;117:1189–1200. doi: 10.1161/CIRCULATIONAHA.107.734103. [DOI] [PubMed] [Google Scholar]
- 53.Abraham MR, Henrikson CA, Tung L, et al. Antiarrhythmic engineering of skeletal myoblasts for cardiac transplantation. Circ Res. 2005;97:159–167. doi: 10.1161/01.RES.0000174794.22491.a0. [DOI] [PubMed] [Google Scholar]
- 54.Choi YH, Stamm C, Hammer PE, et al. Cardiac conduction through engineered tissue. Am J Pathol. 2006;169:72–85. doi: 10.2353/ajpath.2006.051163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cajal RS. Histologie du systeme nerveux de l'Homme et des Vertebres. 1911;2:891–942. Grand Sympathique. Grand Sympathique Paris, Malorine. [Google Scholar]
- 56.Faussone-Pellegrini MS. Interstitial cells of Cajal: once negligible players, now blazing protagonists. Ital J Anat Embryol. 2005;110:11–31. [PubMed] [Google Scholar]
- 57.Takaki M. Gut pacemaker cells: the interstitial cells of Cajal (ICC) J Smooth Muscle Res. 2003;39:137–161. doi: 10.1540/jsmr.39.137. [DOI] [PubMed] [Google Scholar]
- 58.Young HM, Ciampoli D, Southwell BR, et al. Origin of interstitial cells of Cajal in the mouse intestine. Dev Biol. 1996;180:97–107. doi: 10.1006/dbio.1996.0287. [DOI] [PubMed] [Google Scholar]
- 59.Hirst GD, Edwards FR. Role of interstitial cells of Cajal in the control of gastric motility. J Pharmacol Sci. 2004;96:1–10. doi: 10.1254/jphs.crj04002x. [DOI] [PubMed] [Google Scholar]
- 60.Huizinga JD, Golden CM, Zhu Y, et al. Ion channels in interstitial cells of Cajal as targets for neurotransmitter action. Neurogastroenterol Motil 2004; 16 (Suppl. :S106–S111. doi: 10.1111/j.1743-3150.2004.00484.x. [DOI] [PubMed] [Google Scholar]
- 61.Zhu Y, Golden CM, Ye J, et al. ERG K+ currents regulate pacemaker activity in ICC. Am J Physiol Gastrointest Liver Physiol. 2003;285:G1249–G1258. doi: 10.1152/ajpgi.00149.2003. [DOI] [PubMed] [Google Scholar]
- 62.Strege PR, Bernard CE, Ou Y, et al. Effect of mibefradil on sodium and calcium currents. Am J Physiol Gastrointest Liver Physiol. 2005;289:G249–G253. doi: 10.1152/ajpgi.00022.2005. [DOI] [PubMed] [Google Scholar]
- 63.Liu HN, Ohya S, Wang J, et al. Involvement of ryanodine receptors in pacemaker Ca2+ oscillation in murine gastric ICC. Biochem Biophys Res Commun. 2005;328:640–646. doi: 10.1016/j.bbrc.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 64.Li Z, Zhou Z, Daniel EE. Expression of gap junction connexin 43 and connexin 43 mRNA in different regional tissues of intestine in dog. Am J Physiol. 1993;265:G911–G916. doi: 10.1152/ajpgi.1993.265.5.G911. [DOI] [PubMed] [Google Scholar]
- 65.Baig MK, Wexner SD. Postoperative ileus: a review. Dis Colon Rectum. 2004;47:516–526. doi: 10.1007/s10350-003-0067-9. [DOI] [PubMed] [Google Scholar]
- 66.Yanagida H, Yanase H, Sanders KM, et al. Intestinal surgical resection disrupts electrical rhythmicity, neural responses, and interstitial cell networks. Gastroenterology. 2004;127:1748–1759. doi: 10.1053/j.gastro.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 67.Graham FL, Smiley J, Russell WC, et al. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 68.Hofshi A, Itzhaki I, Gepstein A, et al. A combined gene and cell therapy approach for restoration of conduction. Heart Rhythm. 2011;8:121–130. doi: 10.1016/j.hrthm.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 69.Protas L, Dun W, Jia Z, et al. Expression of skeletal but not cardiac Na+ channel isoform preserves normal conduction in a depolarized cardiac syncytium. Cardiovasc Res. 2009;81:528–535. doi: 10.1093/cvr/cvn290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lau DH, Clausen C, Sosunov EA, et al. Epicardial border zone overexpression of skeletal muscle sodium channel SkM1 normalizes activation, preserves conduction, and suppresses ventricular arrhythmia: an in silico, in vivo, in vitro study. Circulation. 2009;119:19–27. doi: 10.1161/CIRCULATIONAHA.108.809301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anyukhovsky EP, Sosunov EA, Kryukova YN, et al. Expression of skeletal muscle sodium channel (Nav1.4) or connexin32 prevents reperfusion arrhythmias in murine heart. Cardiovasc Res. 2011;89:41–50. doi: 10.1093/cvr/cvq284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Coronel R, Lau DH, Sosunov EA, et al. Cardiac expression of skeletal muscle sodium channels increases longitudinal conduction velocity in the canine 1-week myocardial infarction. Heart Rhythm. 2009;7:1104–1110. doi: 10.1016/j.hrthm.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sasano T, McDonald AD, Kikuchi K, et al. Molecular ablation of ventricular tachycardia after myocardial infarction. Nat Med. 2006;12:1256–1258. doi: 10.1038/nm1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.de Boer TP, van Veen TA, Houtman MJ, et al. Inhibition of cardiomyocyte automaticity by electrotonic application of inward rectifier current from Kir2.1 expressing cells. Med Biol Eng Comput. 2006;44:537–542. doi: 10.1007/s11517-006-0059-8. [DOI] [PubMed] [Google Scholar]
- 75.Guder G, Frantz S, Bauersachs J, et al. Low circulating androgens and mortality risk in heart failure. Heart. 96:504–509. doi: 10.1136/hrt.2009.181065. [DOI] [PubMed] [Google Scholar]
- 76.Donahue JK, Heldman AW, Fraser H, et al. Focal modification of electrical conduction in the heart by viral gene transfer. Nat Med. 2000;6:1395–1398. doi: 10.1038/82214. [DOI] [PubMed] [Google Scholar]
- 77.Murata M, Cingolani E, McDonald AD, et al. Creation of a genetic calcium channel blocker by targeted gem gene transfer in the heart. Circ Res. 2004;95:398–405. doi: 10.1161/01.RES.0000138449.85324.c5. [DOI] [PubMed] [Google Scholar]
- 78.Kizana E, Chang CY, Cingolani E, et al. Gene transfer of connexin43 mutants attenuates coupling in cardiomyocytes: novel basis for modulation of cardiac conduction by gene therapy. Circ Res. 2007;100:1597–1604. doi: 10.1161/CIRCRESAHA.106.144956. [DOI] [PubMed] [Google Scholar]
- 79.Lyon AR, Bannister ML, Collins T, et al. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ Arrhythm Electrophysiol. 2011;4:362–372. doi: 10.1161/CIRCEP.110.961615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jaski BE, Jessup ML, Mancini DM, et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009;15:171–181. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xiao YF, Nikolskaya A, Zeng L, et al. Optimal dose of Adv-hHCN4 creates stable and long-lasting biopacing activity in cultured ventricular cardiomyocytes. Circulation. 2008;118:S921. [Google Scholar]
- 82.Zeng L, Qiu X, Urban J, et al. AAV1-HCN4-mediated biological pacemaker paces the canine heart with av block over 7 months and responds well to autonomic challenges [Abstract] Circulation. 2010;122:18147. [Google Scholar]
- 83.Xiao YF, Nikolskaya A, Zeng L, et al. Calcium overload is potentially the main cause of cardiomyocyte death after heavily infecting with Adv-hHCN4 [Abstract] J Am Coll Cardiol. 2010;55:5. [Google Scholar]
- 84.Donahue JK, Kikuchi K, Sasano T. Gene therapy for cardiac arrhythmias. Trends Cardiovasc Med. 2005;15:219–224. doi: 10.1016/j.tcm.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 85.Satin J, Kehat I, Caspi O, et al. Mechanism of spontaneous excitability in human embryonic stem cell derived cardiomyocytes. J Physiol. 2004;559:479–496. doi: 10.1113/jphysiol.2004.068213. [DOI] [PMC free article] [PubMed] [Google Scholar]