

The concept underlying much of cancer therapy is that patients with specific types and stages of cancer should be treated according to standardized, predetermined protocols. However, recent advances in drug development, pharmacogenomics, and the molecular characterization of tumors — including genomewide RNA expression, DNA copy-number and sequence analyses, and micro-RNA and proteomic profiling — have the potential to allow individualized selection of treatment as determined by the characteristics of the patient and the tumor.
In principle, targeted therapies attack cancer-cell–specific attributes that are essential for growth or survival (“oncogene addiction”) and avoid the potentially severe side effects of conventional cytotoxic treatments. Successful examples of targeted therapy include imatinib for the treatment of BCR-ABL–positive chronic myeloid leukemia and the reversible tyrosine kinase inhibitors, erlotinib and gefitinib, for blocking the gene that encodes epidermal growth factor receptor (EGFR) in subgroups of patients with non–small-cell lung cancer.
Initially, the basis for the selective response to tyrosine kinase inhibitors in patients with non–small-cell lung cancer was unknown. Subsequently, the identification of mutations in the tyrosine kinase domain of EGFR that resulted in ligand-independent gene activation provided a seemingly rational basis for the response. The vast majority of EGFR mutations are either a deletion of a conserved sequence in exon 19 or a single point mutation in exon 21 (L858R). These activating mutations result in ligand-independent tumor-cell dependence on EGFR signaling1 and simultaneously provide the means to inhibit the tumor.
It soon became obvious, however, that the as- sociation between mutations and responsiveness to tyrosine kinase inhibitors was much more complex than had been envisioned. Although mutations correlated well with responsiveness and tumor-free survival, overall survival appeared to correlate better with increased gene copy number and possibly with the copy number of HER2, another member of the EGFR family.2,3 Not all EGFR mutations are equal, and some, especially insertion mutations in exon 20, confer intrinsic resistance on tumor cells. A further confounder was the finding that activating mutations in the down- stream KRAS gene (usually mutually exclusive with EGFR mutations) also conferred intrinsic resistance. Almost all non–small-cell lung cancers that respond initially to tyrosine kinase inhibitors even- tually relapse and resist further treatment. Known mechanisms of secondary resistance include a specific EGFR mutation (T790 M) and amplifica- tion or overexpression of the MET gene.
Two articles in this issue of the Journal report the use of tyrosine kinase inhibitors for the initial treatment of subgroups of patients with non– small-cell lung cancer.4,5 These studies combined molecular analyses with therapy using gefitinib or erlotinib in subgroups of patients who were selected from more than 3300 patients of East Asian or European origin who had non–small- cell lung cancer. Both studies showed significant increases in rates of response and progression-free survival in patients whose tumors had EGFR mutations. In both studies, toxic effects associated with therapy were modest and well tolerated.
The nonrandomized European study by Ro-sell et al.4 shows the feasibility of large-scale screening for EGFR mutations in patients with advanced non–small-cell lung cancer for selection for erlotinib therapy. Overall, the rate of survival of patients carrying an EGFR mutation who were treated with the tyrosine kinase inhibitor was relatively high, as compared with that of historical control subjects who were treated with chemotherapy. In the study by Mok et al.,5 patients of East Asian origin with adenocarcinoma who had little or no exposure to tobacco smoke were treated with either gefitinib or standard double chemotherapy. Responses to the tyrosine kinase inhibitor were almost entirely limited to the mutation-positive group, whereas mutation-negative patients benefited from chemotherapy. However, overall survival, perhaps the most important end point of cancer treatment, was not improved by gefitinib, for reasons not discussed by the authors.
Other reports have indicated that the use of tyrosine kinase inhibitors may improve overall survival of certain subgroups of patients with lung cancer, independently of the EGFR mutation status.3 It has been suggested that EGFR mutations represent favorable prognostic markers of survival and are predictive of tumor shrinkage, but the evidence that they can predict a differential effect of tyrosine kinase inhibitors on survival is incomplete.6 Several questions remain to be answered before we can extrapolate the results of the randomized East Asian trial to a Western population. Is erlotinib a suitable substitute for ge-fitinib? Because there was no improvement in survival in the randomized trial, is it necessary to perform mutational analyses in selected subpopulations, or can they be treated empirically? The lack of a difference in overall survival suggests that the order of drug administration may not be important, as long as patients who do not have a response can cross over to the other study group. Despite these unanswered questions, the studies by Rosell et al. and Mok et al. and other reports suggest that first-line tyrosine kinase therapy should be considered for carefully selected subgroups of patients of East Asian and non–East Asian origin who have non–small-cell lung cancer.
EGFR signaling occurs through a complex, multidimensional pathway, and activation of the pathway results in downstream events stimulating five of the six hallmarks of cancer (independence of growth signals, insensitivity to growth-inhibitory signals, resistance to programmed cell death, angiogenesis, and metastasis); the sixth hallmark, which is not activated in this pathway, is a limitless potential to replicate7 (Fig. 1). Thus, inhibition of EGFR signaling, the poster child for personalized medicine, presents multiple opportunities for testing a wealth of targeted therapies. The pathway is deregulated in the majority of non–small-cell lung tumors and cell lines.8 In a few years, an oncologist may be able to order a molecular analysis of the EGFR and other pathways in the patient’s tumor cells (including key mutations, copy-number changes of genes, and expression and activation of genes) for a modest cost. It may be possible to perform such assays without the necessity of invasive procedures.9 Individualized therapy would then be selected from a battery of highly active therapies after integrating the genomic or proteomic profile with clinical and demographic data. Targeting of the EGFR pathway may have the additional benefit of overcoming the radioresistance associated with increased EGFR expression, whereas EGFR mutations abrogate such radioprotection.10
Figure 1. Targeting the Epidermal Growth Factorb Receptor (EGFR) Signaling Pathway.
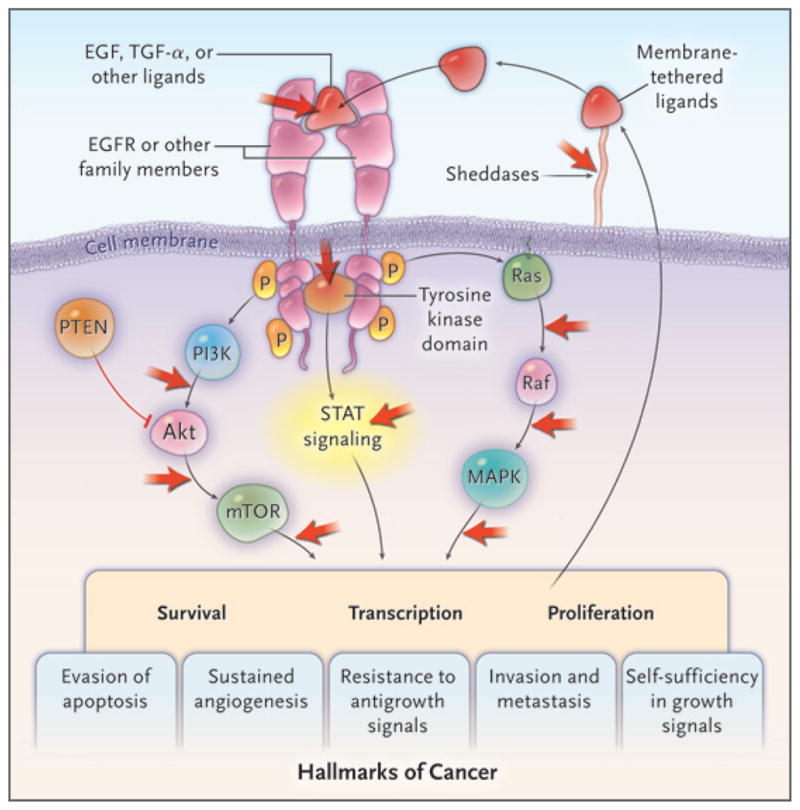
The family of surface-receptor tyrosine kinases encoded by EGFR consists of four members that form homodimers or heterodimers after ligand binding. Dimerization results in the activation of tyrosine kinases, which is followed by stimulation of three major signaling pathways, eventually leading to the activation of five of the six hallmarks of cancer (with the exception of limitless replication).7 The classic mutations of the EGFR kinase domain result in ligand-independent activation of the pathway. Tyrosine kinase inhibitors, such as erlotinib and gefitinib, interfere with the kinase activity of the gene and prevent downstream signaling. Activation of EGFR signaling also leads to an autocrine loop resulting from the formation and release of ligands. The ligands require release from their membrane-bound precursor forms by the activity of sheddase proteins. Only nodal points in the pathway are displayed, indicating targets of therapies in current clinical use or in trials (red arrows). Akt denotes protein kinase B, PI3K phosphatidylinositol 3-kinase, PTEN phosphatase and tensin homologue, MAPK mitogen- activated protein kinase, mTOR mammalian target of rapamycin, P phosphorylation, STAT signal transducer and activator of transcription, and TGF-α transforming growth factor α.
Therapeutic applications of radiotherapy began shortly after Wilhelm Roentgen’s discovery of x-rays in 1895, and the realization that cancers could be treated by pharmacologic agents dates back to the 1940s. By contrast, personalized medicine is still in its infancy. Clearly, we have to develop better drugs, learn how to combine individual targeted therapies with others and with cytotoxic agents, and overcome resistance. However, the plethora of targets that are presented by cancer cells and the large number of agents currently in clinical trials or being developed offer the promise of a bright future for cancer therapy.
Footnotes
Dr. Gazdar reports receiving consulting fees from AstraZeneca, Genentech, and Boehringer Ingelheim. No other potential conflict of interest relevant to this article was reported.
References
- 1.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–81. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 2.Cappuzzo F, Varella-Garcia M, Shigematsu H, et al. Increased HER2 gene copy number is associated with response to gefitinib therapy in epidermal growth factor receptor-positive non-small-cell lung cancer patients. J Clin Oncol. 2005;23:5007–18. doi: 10.1200/JCO.2005.09.111. [DOI] [PubMed] [Google Scholar]
- 3.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 4.Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361:958–67. doi: 10.1056/NEJMoa0904554. [DOI] [PubMed] [Google Scholar]
- 5.Mok TS, Wu Y-L, Thongprasert S, et al. Gefitinib or carbo-platin–paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 6.Shepherd FA, Tsao MS. Unraveling the mystery of prognostic and predictive factors in epidermal growth factor receptor therapy. J Clin Oncol. 2006;24:1219–20. doi: 10.1200/JCO.2005.04.4420. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 8.Gandhi J, Zhang J, Xie Y, et al. Alterations in genes of the EGFR signaling pathway and their relationship to EGFR tyrosine kinase inhibitor sensitivity in lung cancer cell lines. PLoS One. 2009;4(2):e4576. doi: 10.1371/journal.pone.0004576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sequist LV, Engelman JA, Lynch TJ. Toward noninvasive genomic screening of lung cancer patients. J Clin Oncol. 2009;27:2589–91. doi: 10.1200/JCO.2008.20.4875. [DOI] [PubMed] [Google Scholar]
- 10.Chen DJ, Nirodi CS. The epidermal growth factor receptor: a role in repair of radiation-induced DNA damage. Clin Cancer Res. 2007;13:6555–60. doi: 10.1158/1078-0432.CCR-07-1610. [DOI] [PubMed] [Google Scholar]