

Abstract
Families with nonsyndromic dentinogenesis imperfecta (DGI) and the milder, dentin dysplasia (DD), have mutations in one allele of the dentin sialophosphoprotein (DSPP) gene. Because loss of a single Dspp allele in mice (and likely, humans) causes no dental phenotype, the mechanism(s) underling the dominant-negative effects were investigated. DSPP mutations occur in three classes. (The first class, the mid-leader missense mutation, Y6D, was not investigated in this report.) All other 5' mutations of DSPP result in changes/loss in the first three amino acids (IPV) of mature DSPP or, for A15V, some retention of the hydrophobic leader sequence. All of this second class of mutations caused mutant DSPP to be retained in the rER of transfected HEK293 cells. Trafficking out of the rER by co-expressed normal DSPP was reduced in a dose-responsive manner, probably due to formation of Ca2+-dependent complexes with the retained mutant DSPP. IPV-like sequences begin many secreted Ca2+-binding proteins, and changing the third amino acid to the charged aspartate (D) in three other acidic proteins also caused increased rER accumulation. Both the leader-retaining A15V and the long string of hydrophobic amino acids resulting from all known frameshift mutations within the 3'-encoded Ca2+-binding repeat domain (third class of mutations) caused retention by association of the mutant proteins with rER membranes. More 5' frameshift mutations result in longer mutant hydrophobic domains but the milder phenotype, DD, probably due to lower effectiveness of the remaining, shorter Ca2+-binding domain in capturing normal DSPP protein within the rER. This study presents evidence of a shared underlying mechanism of capturing of normal DSPP by two different classes of DSPP mutations and offers an explanation for the mild (DD-II) versus severe (DGI-II & III) nonsyndromic dentin phenotypes. Evidence is also presented that many acidic, Ca2+-binding proteins may use the same IPV-like receptor/pathway for exiting the rER.
Keywords: Dentinogenesis Imperfecta, Dentin Dysplasia, Dentin Sialophosphoprotein, DSPP, Endoplasmic Reticulum Trafficking, DMP1, Osteopontin, SPP1
Introduction
Dentinogenesis imperfecta (DGI-II & III) and dentin dysplasia (DD-II) encompass nearly all patients with nonsyndromic autosomal dominant genetic diseases of dentin (1) . [DGI-I is the syndromic dental symptom of osteogenesis imperfecta associated with mutations in the Col1A1/A2 genes or their modifying/processing enzymes (2)]. Patients with DGI typically have amber-brown, opalescent primary and secondary teeth with poorly mineralized dentin that can wear to the gum line after mechanical shedding of the protective enamel. The primary teeth of DD patients are similar to those with DGI but DD is considered milder because permanent teeth appear more normal, retaining their enamel and typically exhibit only mild discoloration and radiographic abnormalities. DGI-II/III and DD-II were first genetically linked to chromosome 4q (3, 4) and then specifically to the largest member of the SIBLING family of small integrin-binding proteins, dentin sialophosphoprotein (DSPP) (5, 6). Indeed, pioneering protein biochemistry work by Takagai and colleagues had shown many years ago that what is now known to be the carboxy-terminal fragment of DSPP, dentin phosphoprotein or phosphophoryn (DPP), appeared to be greatly decreased or even missing in the dentin of patients with DGI (7). While both nonsyndromic DGI and DD involve mutations in DSPP and share some physical/radiographic features, the diagnosis of DD or DGI is always the same in all affected members of extended families.
Originally thought to be specific to odontoblasts (8), DSPP has recently been shown to be expressed at apparently lower levels in bone (9), cartilage (10), and in many metabolically active ductal epithelial cells such as those found in salivary gland, kidney, and sweat gland (11–13) as well as in a number of epithelial tumors (14). DSPP is cleaved into the two most abundant noncollagenous proteins entrapped within the mineralized compartment of dentin (amino-terminal dentin sialoprotein, DSP, and the carboxy-terminal, DPP) by bone morphogenetic protein-1 (BMP1), the furin-activated protease that processes a number of other secreted matrix proteins as well as many bioactive proteins (15, 16). This disordered protein is highly post translationally modified [containing both N- and O-linked oligosaccharides, often at least one glycosaminoglycan chain, as well as many phosphate groups (particularly within its ~250 SSD repeats)], making it very likely the most acidic protein in humans (14). Unlike the other four members of the SIBLING family that conserve their integrin-binding tripeptide, RGD, half of the mammals studied have lost this motif suggesting that DSPP may be the first family member to no longer require interaction with integrins for any of its critical biological functions (17). Recent analysis suggests that DSPP is the most recent SIBLING to appear in evolution as it may have independently evolved in mammals and reptiles from a duplication of the DMP1 gene (18).
Mice lacking both Dspp alleles (−/−) present with DGI (19), but the teeth of heterozygotic mice (+/−) appear normal (Dr. Ashok Kulkarni, personal communication). As we proposed earlier (20), the only nonsense DSPP mutation reported in humans was predicted in silico to result not in the premature termination of the protein (with associated nonsense-mediated mRNA decay) but in the skipping of exon 3, a relatively common mutational event known to be the result of several other distinct splice site mutations in DSPP (20–26). The results of recent minigene experiments showing exon 3-skipping in a model system support this hypothesis (27). Together, the mouse experiments and the lack of any reported true nonsense mutations in humans strongly suggest that loss of a single allele is recessive and that haploinsufficiency does not explain DD or DGI. We also proposed in 2008 (20), that all but one missense mutation [the mid-leader mutation, Y6D, (28)] in the 5' end of DSPP ultimately result in the loss or change in the first three amino acids of the mature protein (isoleucine-proline-valine, IPV) although how these seemingly minor changes result in the dominant negative effect was not explored at that time. Deletions of base(s) that result in a −1 reading frame anywhere within the long (~700 amino acids), Ca2+-binding carboxy-terminal repeat domain of DSPP [causing the remainder of the translated protein to be changed from acidic to hydrophobic (20, 26, 29, 30)], constitute the third class of nonsyndromic DGI/DD mutations. In this report, we show the shared, underlying causes of the dominant negative effects of both the “IPV” and hydrophobic frameshift/A15V mutations. These results also explain how a specific subset of mutations may cause the milder form of the disease, DD.
Methods
Expression Constructs
DSPP: The first 31 amino acids of normal human DSPP (IPV) as well as the same fragment encoding the ISV (5), ITV (21), ILV (hypothetical), IPD (31), IPD3 (exon 3-skipping), IPF (21), and A15V (32) mutations in-frame with a 3' AgeI site were synthesized as oligonuclotides, double stranded by PCR and cloned into Gateway pENTR/D TOPO vector (Invitrogen, Carlsbad, CA). An identical approach was used to make normal and mutant (V-to-D) constructs using the first 31 amino acids of human OPN and DMP1. Because full-length, 4 kb human DSPP is difficult to maintain unmodified in bacteria (20), the remaining DSPP sequence was obtained by high fidelity PCR from the shorter mouse cDNA [gift of Dr. Kulkarni, (15, 19)] including in-frame flanking AgeI sites used to ligate the fragment into the pENTR plasmid. In situ mutagenesis (Quickchange II XL Site-Directed Mutagenesis Kit, Stratagene, La Jolla, CA) introduced FLAG-encoded peptide carboxy-terminal to the RGD motif in both IPV-DSPP (WT) and the IPD-DSPP fusion mutant for detection and affinity purification by anti FLAG antibodies. One IPV-DSPP clone had tandem amplified the FLAG peptide-containing sequence 6 times (6xFLAG). One IPV-DSPP clone that lost approximately half of the normal repeat domain (329 amino acids remaining) by recombination during subcloning was modified by in situ mutagenesis to accept 287 amino acid-encoding DPP repeat domain starting with a −1 frameshift to mimic a human DGI frameshift mutation (DGI-like FS). A second DSPP-FS plasmid lacking DPP repeats served as a DD-like FS protein. All pENTR clones were shuttled into the Gateway pT-REx-DEST 30 eukaryotic expression vector. The IPV-DSPP pENTR clone (i.e. lacking eucaryotic promoter) was used as the “mock” plasmid DNA control.
GFP fusion proteins
The 31 amino acid IPV and mutant sequence pENTR plasmids noted above were fused to GFP by shuttling the sequences into pcDNA 6.2/C-EmGFP-DEST plasmids (Invitrogen). A GFP lacking a leader sequence (MPV-GFP) served as the leaderless protein standard.
Chromogranin A and osteopontin (OPN)
Full-length human chromogranin A (IOH4766, Invitrogen) and OPN (33) in pENTR were also modified by in situ mutagenesis to produce missense mutants, LPD and IPD respectively. Both wildtype and mutant entry clones were migrated into the pT-REx Dest plasmids for expression.
Antibodies
Rabbit antisera against the DSP portion of mouse DSPP [LF-153, (11)] and against human OPN [LF-166, (34)], were as described. Monoclonal mouse antibody against Green Fluorescence Protein (GFP) was from Covance (Emeryville, CA). Mouse monoclonal antibodies against human chromogranin A and human protein disulphide isomerase (PDI) were from Novus Biologicals (Littleton, CO). Mouse monoclonal anti FLAG M2 antibody was from Agilent Technologies (Santa Clara, CA). IRDye 680 donkey anti rabbit IgG and IRDye 800 donkey anti mouse IgG 2nd antibodies were from LI-COR Biosciences (Lincoln, NE). Alexa Fluor 488 donkey anti mouse IgG and Alexa Fluor 568 donkey anti rabbit IgG (A10042) second antibodies were from Invitrogen. Rabbit antiserum against DPP domain of mouse DSPP (LF-190) peptide: CSRGDASYTSDESSDDDNDSDSH was synthesized, conjugated to activated KLH, and injected into a rabbit at an AAALAC facility (Covance Research Products, Denver, PA) under approved animal protocol.
Quantitative ELISA (FLAG-DSP) sandwich assay
Anti FLAG M2 monoclonal antibody (100 ng/100 µl in PBS) was bound to Greiner High-Binding 96-well microtiter plates (Sigma-Aldrich, St Louis, MO). Wells were blocked with for 1 hour (hr) with 120 µl of Odyssey Blocking Buffer (Li-Cor Biosciences) and washed 3 × 3 minutes (min) with PBS, 0.05% Tween 20 (PBS-T). Conditioned media containing 6xFLAG-labeled DSPP were diluted 1:25 in PBS-T and 100 µl added to wells. After 1 hr, wells were washed, incubated for 1 hr with anti mouse DPP (LF-190), washed 3 times and horseradish peroxidase-conjugated goat anti rabbit IgG (KPL, Gaithersburg, MD) in PBS-T was incubated for 1 hr. After final washes, SureBlue Peroxidase Substrate (KPL) was added and absorbance (490 nm) determined after stopping with dilute acid.
Cell culture and transfection
Human embryonic kidney 293A cell line (HEK293, Invitrogen) was maintained in Dulbecco’s Modified Eagle Medium (Gibco-BRL, Gaithersburg, MD) supplemented with 10% fetal bovine serum (FBS), L-glutamine (2 mM), penicillin (100 IU/ml), and streptomycin (100 µg/ml). The expression constructs were transfected into the cells using Lipofectamine 2000™ reagent (Invitrogen). Briefly, 4 × 105 HEK293 cells/well were plated into 6-well plates pre-coated with poly-L-lysine. After overnight culture, cells were transfected with 1–5 µg of plasmid DNA in Opti-MEM (Gibco-BRL) using Lipofectamine (1µg/2µl). After 6 hr, transfection medium was replaced with Opti-MEM. Transient expression of proteins was analyzed at 24–96 hr post-transfection. For large-scale FLAG-labeled IPD-DSPP protein production, 2.5 ×106 HEK293 cells were seeded into 10-cm plates. After overnight incubation, cells were transfected with 12 µg of plasmid and harvested 96 hr later.
Immunoblotting
Conditioned serum-free media were precipitated in 20% trichloroacetic acid and the pellet rinsed with acetone before being dissolved in PAGE sample buffer. Cells were harvested in lysis buffer (150 mM NaCl, 0.5% Na-deoxycholate, 1% SDS, and 50 mM HEPES, pH 7.5) and then added to PAGE sample buffer. Equal volumes of samples were electrophoresed on NuPAGE 4–12% Bis-Tris PAGE gels (12% Bis-Tris PAGE gels for GFP detection) in MOPS Buffer (Invitrogen), and transferred by electroblotting. Membranes were blocked in Odyssey Blocking Buffer (Li-COR Biosciences), followed by incubation with indicated antibodies over night in 50% LI-COR Blocking Buffer in PBS-T. After 3 × 5 min washes in PBS-T, blots were incubated with the species-specific IRDye-conjugated second antibody. After 3 final washes, proteins were visualized using the LI-COR Odyssey infrared imaging system.
Purification and chondroitinase ABC treatment of cell culture media DSPP/DSP
Serum-free conditioned media were tumbled with 100 µl of Q-Sepharose Fast Flow strong anion exchange beads (GE Healthcare Life Sciences, Piscataway, NJ) and then washed successively with; 1) 10 ml PBS, 2) 5 ml 6 M urea in PBS plus 5 µl 2-mercaptoethanol, 3) 10 ml PBS and finally, 4) 10 ml of PBS containing an additional 100 mM NaCl. The DSPP/DSP proteins were eluted with PBS containing an additional 800 mM NaCl and dialyzed against chondroitinase ABC reaction buffer (50 mM NaCl, 20 mM Tris, pH 7.5). Aliquots were incubated at 37°C for 1 hr alone or with 2.5 mU of chondroitinase ABC (Seikagaku America Inc., Ijamsville, MD). Samples were electrophoresed on NuPAGE 4–12% Bis-Tris gels, followed by the LI-COR Western blot procedure using rabbit anti mouse DSP (LF-153).
Furin inhibitor and endoglycosidase H treatments
HEK293 cells transfected with the WT DSPP DNA construct were switched to serum-free Opti-MEM with or without 10 µM synthetic furin inhibitor dec-Arg-Val-Lys-Arg-chloromethylketone (Alexis Corp., San Diego, CA). Conditioned media were collected 90 hr later and analyzed for DSPP/DSP (LF-153) by Western blot. HEK293 cells transfected for 24 hr with IPD-, ISV- or A15V-DSPP expression constructs were rinsed with PBS and lysed with 50 mM sodium phosphate (pH 7.5), 0.1% SDS and 50 mM 2-mercaptoethanol. The proteins were denatured at 99°C for 5 min and incubated with or without 0.3 mU endoglycosidase H (Glyco, San Leandro, CA) for 3 hr at 37°C and electrophoresed on NuPAGE 4–12% Bis-Tris gels, followed by the Western blot procedure.
Immunocytochemistry
HEK293 cells were plated (1.5 × 105/well) in 12 well-plates on poly-L-lysine-coated glass coverslips and transfected with 1 µg of each plasmid. 24 hr post-transfection cells were fixed for 20 min using 4% paraformaldehyde in PBS, permeabilized with 0.5% saponin in PBS for 15 min and non-specific antibody binding sites blocked with 5% FBS in PBS. Double staining was performed with rabbit antiserum against mouse DSPP (LF-153, 1:300) and monoclonal mouse antibody against human protein disulfide isomerase, PDI, (1:50) in blocking buffer containing 0.2% saponin for 1 hr. Cells were washed 3 times in PBS-T and incubated for 30 min at with Alexa Fluor 488 donkey anti mouse IgG and Alexa Fluor 568 donkey anti rabbit IgG. Images were obtained using Zeiss LSM 700 Confocal microscope (Thornwood, NY).
Precipitation of cell-retained IPD-DSPP with LaCl3
HEK293 cells transfected with FLAG-labeled IPD-DSPP plasmid were harvested 96 hr later and then swelled on ice for 20 min in hypotonic buffer (20 mM Tris, 5 mM EDTA, pH 8.0, Complete Protease Inhibitor Cocktail, Roche, Mannheim, Germany). After lysis with a glass homogenizer, nuclei and debris were removed by centrifugation and microsomes collected by centrifugation for 1 hr at 180,000 × g at 4°C. The supernatant was brought to 0.15 M NaCl and 1% Triton X-100. FLAG-labeled IPD-DSPP was bound to 50 µl M2 anti FLAG Affinity Gel beads (Sigma-Aldrich), washed with 3 × 1.2 ml Tris-buffered saline (TBS) followed by 3 × 1.2 ml TBS containing 1% Tween-20. Protein was eluted with TBS containing FLAG peptide (DYKDDDDK) and dialyzed against TBS. 175 µl aliquots were placed in Beckman Airfuge tubes and 3.5 µl of LaCl3 (10 mM final) or TBS were added. Tubes were centrifuged at ~100,000 × g for 2 hr. One aliquot was immediately removed from the top of both tubes. EDTA was added to the La3+ tube and a second aliquot removed for Western blot analysis.
Statistical analysis
Statistical analyses were performed using GraphPad Software (San Diego, CA). Pairwise comparisons were carried out by using the non-parametric two-sided t-test for unpaired observations. Results are expressed as mean ±SD.
Results
The signal sequence is removed from “IPV” mutants
Except for the Y6D missense mutation, all of the 5' mutations in DSPP result in loss or changes in the first three amino acids (IPV) of the fully translated protein. These changes include known missense mutations (ISV, ITV, IPD, IPF) as well as all pre-mRNA splice junction motif changes resulting in the skipping of exon 3. The splice junction mutations occur in critical bases of introns 2 or 3 resulting in fusion of exon 2 (ending in IP) to exon 4 (starting with aspartic acid, D). These splicing errors, therefore, also result in the IPV changing to IPD (constructs denoted as IPD3). Similarly, we and others have proposed that what at first seems to be missense/nonsense mutations in the first (V18F) or last (Q45X) codon of exon 3, respectively, actually damage splicing motifs causing significant amounts of IPD3-DSPP to be translated. Because: 1) all of the above mutations (as well as the missense mutation changing the last amino acid of the leader sequence, A15V) are proximal to the site of the co-translational removal of the hydrophobic leader sequence (A/IPV) and 2) the knowledge that leader missense mutation, Y6D causes DD, we first explored this cleavage event. The first 31 amino acids of WT and all 5' mutations of human DSPP were fused to the small protein, green fluorescent protein (GFP), and the relative masses of the newly translated proteins (in HEK293 cells) were analyzed by mobility on SDS polyacrylamide gels. Immunodetected GFP protein bands in Figure 1A show that the leader was removed from all of the IPV-type mutant proteins thus confirming the commonly held theory that even those amino acids immediately carboxy-terminal to the cleavage site are usually not important in directing signal peptide removal. A hypothetical mutation, ILV-GFP, had a leader-like peptide removed at both SignalP 4.0 program’s predicted sites (http://www.cbs.dtu.dk/services/SignalP/) yielding the “normally processed” A/ILV band and a smaller and therefore faster migrating (L/ERH) protein. Replacement of the small amino acid, alanine (A), with the larger, valine (V), resulted in slower mobility on gels consistent with retention of the hydrophobic leader peptide (A15V lane, Figure 1A) although the small amount of this construct that was secreted into the media also suggested removal of the leader carboxy-terminal to the IPV tripeptide sometimes occurred (data not shown). These results were weakly predicted by the low probability score (0.50, only slightly above the program’s cutoff of 0.45) of possessing a leader sequence and then only with a more carboxy-terminal cleavage after amino acid 23 (SK/LER) by the SignalP 4.0 signal protein prediction program. Consequences of the retention of this hydrophobic motif by the A15V-DSPP protein are explored below.
Fig. 1.

Intact IPV motif is not required for leader peptide removal but is critical for trafficking of DSPP in transfected HEK293 cells. All mutations in IPV motif (IPD, exon 3-skipping IPD3, IPF, ISV & ITV) within the first 31 amino acids of DSPP (fused to GFP) do not affect the removal of leader sequence as determined by Western blot (anti GFP) (A). DGI-causing A15V mutation that changes the last amino acid of the leader itself, results in a slightly larger protein consistent with retention of the leader peptide. Hypothetical ILV mutation shows approximately equal amounts of normal and more carboxy-terminal processing of leader. Leaderless construct (MPV-GFP) is used as size control. Wildtype DSPP (IPV) is much more effectively trafficked out of the cell (B) and into the media (C) than each mutant DSPP as detected by anti mouse DSP antibody on Western blots. Cell layer has intact DSPP (B) while same proteins in media (C) have been more fully modified including both some glycosaminoglycan chains (DSP-PG) and nearly complete cleavage to DSP fragment. Frameshift mutation (FS) remains in cell layer (D). All panels are SDS 4–12% NuPAGE (except A that used 12% NuPAGE) electrophoresed in MOPS buffer, transferred to PVDF membranes and immunodetected using primary antibodies noted in Methods and the LI-COR Odyssey infrared imaging system. SeeBlue Plus2 prestained markers were used.
Intracellular retention of “IPV” DSPP mutant proteins
Unlike the Y6D and A15V, the appropriate removal of the leader peptide from all known “IPV” mutation proteins suggests that the dominant negative effects caused by their corresponding DSPP mutant proteins were not related to this co-translational process. Therefore, the same 31 amino acids of human WT (IPV-DSPP) and all corresponding mutant peptides were fused to mouse DSPP to compare post translational events during the biosynthesis of WT and mutant DSPP in cells capable of expressing significant levels of the very acidic DSPP protein. [For reasons that are not clear at this time, many cell lines and even several sources of primary, matrix producing cells (e.g., human bone marrow stromal fibroblasts and skin fibroblasts) that we have shown produce ample levels of other acidic SIBLING proteins such as its close relative, DMP1 (35), do not produce detectable levels of DSPP when transduced with DSPP adenovirus (data not shown). Human embryonic kidney cells (HEK293), perhaps because they are derived from a tissue that can expressed this protein in vivo (12), were found to be able to make and secrete normal mouse DSPP after transfection.] HEK293 cells transfected with IPV-DSPP expression plasmid trafficked this wildtype protein so rapidly out of the cells that intracellular protein was often undetectable in the cell lysates on immunoblots (Figure 1B) and its corresponding DSPP-related protein fragments did accumulate in the culture media (Figure 1C). In contrast, all “IPV” mutations resulted in accumulation of the larger, full-length DSPP protein within the cell (Figure 2B) and correspondingly less in the media (Figure 1C). [As expected, a leaderless MPV-DSPP construct transfected into the HEK293 cells resulted in a faster migrating Western blot band lacking the posttranslational modifications observed in the other mutant DSPP constructs (data not shown).] Based on in silico predictions, we had earlier proposed that the apparent missense, V18F, actually also damages the splicing motif and results in the skipping of exon 3 thereby changing the IPV to IPD3 (20). Furthermore, we would have predicted any IPF-DSPP protein made from this gene (whenever exon 3 was not skipped) to be chemically similar to IPV (hydrophobic-proline-hydrophobic) and, therefore, to be normally trafficked out of the cell. Experimentally, however, the transfection construct directly encoding the IPF-DSPP protein (Figure 1B & 1C) showed that this protein was much more retained within the cell compared to the WT suggesting that both splice variants (IPD3 & IPF) of this mutation would contribute to retention of mutant DSPP within the cell. Notice that although much smaller in amounts, mutant DSPP that was secreted into the media was immunodetected (anti DSP domain) on Western blot in patterns similar to that seen for the WT DSPP, as its broader, BMP1-cleaved DSP fragment. This means that the small amounts of mutant DSPP protein that made it past the initial trafficking block was subsequently modified/processed the same way as WT DSPP. For example, Figure 2A shows that some of the secreted forms of WT and two representative mutant DSPP (IPD and ISV) contained chondroitin/dermatan sulfate glycosaminoglycan (GAG) chains (DSP-PG) that were removed by chondroitinase ABC thereby increasing the intensity of the ~75 kDa DSP fragment bands. That the ~120 kDa band was full-length DSPP is supported by the increase in its intensity when biosynthesized in the presence of a synthetic inhibitor of furin, the protease required for activation of BMP1 (Figure 2B).
Fig. 2.

Any mutant DSPP passing the trafficking block and appearing in the media is modified similarly to the wildtype (IPV) protein. Sharpening of ~70 kDa DSP band by digestion with chondroitinase ABC (cABCase) on Western blots of two representative DSPP mutations (IPD & ISV) show presence of chondroitin/dermatan sulfate chains on some BMP1-processed DSP fragments (DSP-PG, panel A). Addition of synthetic inhibitor of BMP1-activating enzyme (furin) greatly increases the amount of DSPP secreted from HEK293 cells that remains intact (B). Note that the full-length DSPP band is broader than the same protein retained in the cell layer (Fig. 1A) due to abundant Golgi-associated post translational modifications. All panels are SDS 4–12% NuPAGE electrophoresed in MOPS buffer, transferred to PVDF membranes and immunodetected using primary antibodies noted in Methods and the LI-COR Odyssey infrared imaging system. SeeBlue Plus2 prestained markers were used.
Intracellular accumulation of “IPV” mutants of DSPP hybrids (OPN-DSPP and DMP1-DSPP) as well as full-length OPN and chromogranin A
To see if the amino-terminus of DSPP was unique with respect to trafficking of this highly acidic protein, the first 31 amino acids of two other SIBLINGs, osteopontin (OPN) and dentin matrix protein-1 (DMP1), were fused to the same fragment of mouse DSPP. While these acidic proteins also start with IPV (OPN) or the chemically similar, LPV (DMP1), the remaining N-terminal amino acids of the three proteins’ amino-terminal sequences have no obvious conservation. Figure 3A shows that the IPV-related amino-termini of OPN and DMP1 both permitted the rapid trafficking and secretion of the hybrid proteins out of the cells. However, changing only the third amino acid (V to D) of the hybrid proteins caused cell-retention similar to that of the IPD-mutant derived from human DSPP. Interestingly, changing only the same V to D in both full-length native human OPN as well as human chromogranin A (a glutamic acid-rich neuroendocrine secretory protein whose mature protein starts with LPV) caused these mutant proteins to accumulate in the cell layer as sharper and smaller bands consistent with reduced post translational modifications (Figure 3B and C).
Fig. 3.

Western blot analyses of two hybrid DSPP proteins (OPN-DSPP and DMP1-DSPP) (A) as well as native OPN (B) and chromogranin A (ChrA) (C) shows that changing the third amino acid of the mature proteins from hydrophobic amino acid valine (V) to negatively charged aspartate (D) causes much more protein to be retained in cell layer. Expression plasmids encoding first 31 amino acids of WT human DSPP fused to mouse DSPP (IPV-DSPP) is compared to both 31 amino acids of human OPN (IPV-OPN-DSPP) and human DMP1 (LPV-DMP1-DSPP) shows that the hybrid proteins are as well trafficked out of HEK293 cells as WT DSPP while mutating the IPV-like amino terminus to IPD-like motif interfere with intracellular trafficking (A). OPN and ChrA represent two of many acidic proteins starting with IPV-like sequences and they also traffic less effectively after changing their native valine to aspartic acid (B & C). Notice that the more highly retained mutant proteins are less modified (sharper and smaller bands) than WT proteins (B & C) suggesting trafficking block is early in the secretory pathway. All panels are SDS 4–12% NuPAGE electrophoresed in MOPS buffer, transferred to PVDF membranes and immunodetected using primary antibodies noted in Methods and the LI-COR Odyssey infrared imaging system. SeeBlue Plus2 prestained markers were used.
5′ and frameshift mutations of DSPP cause the mutant proteins to be retained in rER
Compared to the broader, full-length DSPP (~125 kDa) seen in Figure 2B, the very sharp bands of the cell-retained, full-length mutant DSPP bands (~110 kDa, Figure 1B) (as well as the OPN and chromogranin A WT/mutant results above) suggests that the retained mutant proteins lacked Golgi- and post Golgi-associated modifications (e.g., modification of the N-linked oligonucleotides, addition of GAG chains, phosphorylation, BMP1-cleavage) and were, therefore, failing to exit the rER. Figure 4 shows that the cellular DSPP corresponding to three representative 5′ mutants (IPD, ISV and A15V) each contained endoglycosidase-H-sensitive N-linked oligosaccharides, a result consistent with the co-translational, high-mannose type of oligosaccharides found in the rER before modifications performed in Golgi cause the enzyme to become ineffective. Confocal microscopy was performed to verify this subcellular localization. Wildtype DSPP (red color, Figure 5A) appeared as punctate, perinuclear staining and only partially co-localized (yellow) with the rER marker, protein disulfide isomerase (PDI, green). This suggested that normal DSPP was rapidly trafficked out of the rER and what protein was detected appeared most abundant in Golgi-like areas. In contrast, representative mutant DSPP proteins (red) seen in other panels of Figure 5B–E were predominantly co-localized (yellow) with the abundant and more widespread rER marker (green). Notice that the A15V mutant DSPP (Figure 5E) appeared to have both a luminal rER and a membrane-like staining pattern. This suggested that cleavage of the mutation-extended leader motif, whenever it did occur, caused loss of its IPV motif thereby caused retention in the rER’s lumen. However, whenever the leader peptide was retained, its hydrophobic amino acids acted as a transmembrane domain and caused insertion into the rER membranes and even diffusion with time into contiguous membranes (notice red nuclear membranes).
Fig. 4.

Mutant DSPP retained in the HEK293 cell layer is reduced in size by endoglycosidase-H showing that the proteins contain rER-associated high-mannose type N-linked oligosaccharides that are often modified to enzyme-resistant forms in the Golgi network. This result suggests that the mutant proteins fail to exit the rER. SDS 4–12% NuPAGE electrophoresed in MOPS buffer, transferred to PVDF membranes and immunodetected using primary antibodies noted in Methods and the LI-COR Odyssey infrared imaging system. SeeBlue Plus2 prestained markers were used.
Fig. 5.

Confocal microscopy shows mutant DSPP proteins accumulate in the rER. Wildtype (IPV) DSPP (red) is observed predominantly in Golgi-like, punctate, perinuclear staining with limited co-localization (yellow) with rER marker (green, protein disulfide isomerase, PDI) (A). Representative DSPP mutants of the IPV motif (IPD, exon 3-skipping IPD3, and ISV, in red) all show predominant co-localization (yellow) with rER marker (green) (panels B–D). Note the monochrome rER (green) in adjacent, non transfected cells in some panels. A15V mutant (E) shows both rER co-localization (yellow) and some membrane-associated staining (red) suggesting that failure to remove leader sequence causes some protein to co-translationally insert into rER membranes and perhaps migrate into contiguous membranes such as nuclear membranes. Frameshift mutant protein (FS, Panel F) with its long hydrophobic amino acid domain shows predominant membrane-associated localization (red). HEK293 cells on glass coverslips were transfected with noted expression plasmids 24 hr prior to fixation, permeabilization, and cytostaining detected by confocal microscopy with rabbit anti mouse DSP (red) and mouse anti human PDI (green) via appropriate labeled second antibodies. Bars = 10 µm.
The third class of DSPP mutations, −1 frameshifts (FS), always results in predominantly hydrophobic amino acids (A, I and V with some polar but uncharged, T) carboxy-terminal to the mutation (even extending slightly beyond the normal stop codon) (20). Because even short stretches (10–15 amino acids) of hydrophobic amino acids are known to cause a transmembrane protein to insert into the rER membrane co-translationally, it was not surprising to observe strong intracellular membrane-like staining pattern in HEK293 cells expressing a model DGI-like frameshift (FS) mutant protein (Figure 5F). This extremely hydrophobic, cell layer-associated FS-DSPP protein electrophoresed aberrantly on all SDS polyacrylamide gels/buffer systems we tried, always resulting in smeared, very high molecular weight bands on Western blots (Figure 1D). The same figure shows that little, if any, of the FS-DSPP protein was successfully trafficked/processed and secreted into the media.
Secretion of WT DSPP is reduced when co-expressed with mutant DSPP proteins
Thus far we have presented a variety of evidence supporting the hypothesis that all mutations in DSPP cause the mutant protein to fail to efficiently traffic out of the cell, specifically the rER. Next, experiments were performed to see if the dominant negative effects of these same mutations could be due to concurrent loss of the normal DSPP protein and thereby mimicking the results in mice where heterozygotic null Dspp mice (+/−) were normal but mice lacking both alleles (−/−) had DGI-like dentin. HEK293 cells transfected with increasing amounts of plasmid encoding a FLAG-labeled WT IPV-DSPP resulted in the expected increasing amounts of DSPP protein secreted into the media whether detected by antibodies to the DSP domain or FLAG peptide (data not shown). However, when the amount of FLAG-labeled WT IPV-DSPP-encoding plasmid was held constant while increasing the amounts of plasmids encoding the expression of the various mutant DSPP proteins (not FLAG-labeled) by co-transfection, the secretion of total DSPP protein (WT + mutant by immunodetection of DSP domain on Western blots as seen in a representative result in Figure 6A) as well as the WT protein itself (FLAG-capturing sandwich ELISA assays, Figure 6B) each decreased in a dose-dependent manner. Compared to WT DSPP alone, co-expression of equal amounts of the mutant ISV-DSPP protein, for example, caused a ~40% loss in secreted WT DSPP and this increased to a >70% loss at the modest dose of 4:1 (mutant:WT). Mutations in the “IPV” domain (ISV, ITV, IPD, IPD3) as well as the A15V, and −1 frameshift mutations all caused similar dose-response decreases in co-expressed WT DSPP (representative results in Figure 6B). Secretion of FLAG-labeled mutant IPD-DSPP was not increased by co-transfection with WT DSPP (data not shown).
Fig. 6.

Mutant DSPP suppresses wildtype DSPP trafficking/secretion in a dose-dependent manner using multivalent cations. Panel A shows a representative HEK293 cell experiment where increasing doses of mutant DSPP (IPD3) causes the constant amount of expressed WT DSPP to be correspondingly less well secreted into the media as measured by Western blot of total DSP accumulation in media in 48 hr. Co-transfection of FLAG-labeled WT DSPP with increasing doses of six different mutant DSPP (not FLAG-labeled) shows that each of these mutations cause significant decreases in the secretion of WT DSPP into the media as measured by FLAG-anchored sandwich ELISA assay (representative experiment in Panel B). FS mutant DSPP used in panel B had DGI-like number of normal number of SSD repeats amino-terminal to the site of the frameshift. Panel C shows that frameshift mutant DSPP with few normal SSD repeats (FS DD-like) was less effective at reducing the amount of WT DSPP released into the media than a FS with many more normal SSD repeats (FS DGI-like) at each dose of mutation-encoding plasmid in their co-transfection of HEK293 cells. Shown are the FLAG-anchored DPP ELISA results from 24 hr conditioned media (median +/− standard deviation, n=4 experiments with P<0.05 = * and P<0.01 = **). For all experiments above, plasmid containing WT DSPP DNA sequence but lacking eukaryotic promoter (mock DNA) was added to a total 5 µg per transfection with Lipofectamine 2000. FLAG-antibody affinity purified mutant IPD-DSPP (FLAG) by itself remains in the supernatant (detected on Western blot with anti-FLAG antibody, panel D, lane: DSPP) while addition of 10 mM La3+ to duplicate tube caused complete loss of DSPP in supernatant by 100,000 × g centrifugation for 2 hr (lane: DSPP+La3+). The IPD-DSPP could be recovered from the La3+-induced pellet by subsequent addition of excess EDTA to the La3+ tube (lane: DSPP+La3+ → EDTA).
Our hypothesis of the mechanism by which mistrafficked mutant DSPP causes retention of the co-expressed WT DSPP is that, in the high Ca2+ environment of the rER, the accumulating mutant DSPP form multivalent cation-dependent complexes through its ~200 negatively charged aspartate amino acids (D) found within its serine-serine-aspartate (SSD) repeat domain. Because only trace amounts of rER-retained mutant IPD-DSPP (FLAG-labeled) protein could be affinity purified from transfected HEK293 cells, we were unable to have enough DSPP to directly mimic the relatively high protein concentrations found in the rER microenvironment and cause stable Ca2+–based precipitates in vitro. However, addition of the more energetically favorable La3+ trivalent ion caused all of the trace DSPP to be pelleted at 100,000 × g (Figure 6D). (This La3+-DSPP complex was completely resolubilized by subsequent chelation of the trivalent ion with EDTA.) This strongly suggests that retention and buildup of critical amounts of mutant DSPP favors multivalent cation-dependent gels/precipitates in the rER and that co-expressed WT DSPP would then become entrapped through its own negatively charged repeat domain.
Figure 7A summarizes DD/DGI phenotypes associated with the locations of all of the known 5' mutations and approximate locations of the reported FS mutations. [Precise localization of each FS mutation within a single representative DSPP sequence is not possible due to the large diversity in the SSD domain’s repeat lengths within human DSPP alleles (20)]. With perhaps one as yet unexplained exception, all of the DD families have more 5' FS mutations than any FS causing DGI. Therefore, we hypothesize that the ability of the mutant DSPP proteins to capture the WT DSPP within the rER decreases when the number of remaining wildtype Ca2+-binding motifs (i.e., 5' to the mutation) drop below a critical number corresponding to a point somewhere within the region currently separating the DD-associated and DGI-associated FS mutations (Figure 7A). To test this hypothesis, the dose-response of a DGI-like FS-DSPP construct described above was compared to a DD-like FS-DSPP construct (lacking essentially all of the SSD repeats and therefore is similar to that of the most 5' FS DD) in their relative abilities to suppress the secretion of a fixed amount of co-transfected WT DSPP in the FLAG-based quantitative ELISA assay. As is likely the case in human DD, the DD-like FS construct did suppress the amount of WT DSPP secreted from the HEK293 cells, but not to the extent that the mutant protein expressing the DGI-like number of Ca2+-binding repeats did at each dose (Figure 6C).
Fig. 7.

DSPP mutations that cause nonsyndromic DD/DGI and models illustrating why they are dominant negative. Panel A shows the locations of DSPP missense/nonsense mutations above the wildtype protein sequence. The leader peptide is underlined. IPV motif (plus D beginning exon 4 that result in IPD3 motif when exon 3 is skipped) are shown in a larger font. Splice site-relevant sequences from introns 2 and 3 (plus bases for first and last codons of exon 3) are shown in lower case. Grey highlights denote DD-associated mutations (e.g., 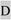 and
and  for amino acid changes and
for amino acid changes and  for base changes leading to skipping of exon 3) and dark highlights (e.g.,
for base changes leading to skipping of exon 3) and dark highlights (e.g., 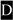 ,
, 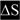 , and with
, and with  ,)respectively) for DGI-associated mutations. The ΔS and ΔD represent locations of frameshift mutations. Letters with asterisk (F* and X*) are missense and nonsense codons respectively, that result in skipping of exon 3. Note that the frameshift mutations causing DD cluster in the 5′ region of repeat domain while DGI are more 3′. References for mutations are cited in the text. The rER of WT odontoblasts produce and traffic the full complement of normal DSPP protein (+/+ in Panel B). Complete loss of one allele (DSPP −/+) results in only half the amount of normal DSPP exiting the rER, but the phenotype remain normal. Expression of a DD-like mutant protein from one allele causes some WT DSPP to also be retained within the rER (DSPP, DD-like) thereby causing the mild DD. In contrast, other mutant alleles produce a DSPP protein that poorly exits the rER and more effectively captures the WT DSPP (DSPP, DGI-like) causing the more severe DGI (B). Unblocked arrow (⇩) denotes successful trafficking of WT DSPP (IPV) out of the rER. Changes in the “IPV” motif (▼) results in a mutant proteins (grey and lacking the ▼ in panel C) which cannot interact with its proposed receptor and form calcium (+) bridged complexes that then adsorb WT DSPP, thereby also keeping the normal protein from trafficking out of the rER. Retention of the hydrophobic leader sequence by A15V causes the mutant protein to become embedded within the rER membrane as a single-pass transmembrane protein where it entraps WT DSPP through the same Ca2+ bridges (D). Similarly, the mutant hydrophobic domains of both DD-FS and DGI-FS DSPP become embedded in rER membrane (E). DD-FS, having shorter normal Ca2+-binding domains remaining, are less efficient at entrapping WT DSPP than the DGI-FS with its much longer negatively charged SSD repeat domain (E).
,)respectively) for DGI-associated mutations. The ΔS and ΔD represent locations of frameshift mutations. Letters with asterisk (F* and X*) are missense and nonsense codons respectively, that result in skipping of exon 3. Note that the frameshift mutations causing DD cluster in the 5′ region of repeat domain while DGI are more 3′. References for mutations are cited in the text. The rER of WT odontoblasts produce and traffic the full complement of normal DSPP protein (+/+ in Panel B). Complete loss of one allele (DSPP −/+) results in only half the amount of normal DSPP exiting the rER, but the phenotype remain normal. Expression of a DD-like mutant protein from one allele causes some WT DSPP to also be retained within the rER (DSPP, DD-like) thereby causing the mild DD. In contrast, other mutant alleles produce a DSPP protein that poorly exits the rER and more effectively captures the WT DSPP (DSPP, DGI-like) causing the more severe DGI (B). Unblocked arrow (⇩) denotes successful trafficking of WT DSPP (IPV) out of the rER. Changes in the “IPV” motif (▼) results in a mutant proteins (grey and lacking the ▼ in panel C) which cannot interact with its proposed receptor and form calcium (+) bridged complexes that then adsorb WT DSPP, thereby also keeping the normal protein from trafficking out of the rER. Retention of the hydrophobic leader sequence by A15V causes the mutant protein to become embedded within the rER membrane as a single-pass transmembrane protein where it entraps WT DSPP through the same Ca2+ bridges (D). Similarly, the mutant hydrophobic domains of both DD-FS and DGI-FS DSPP become embedded in rER membrane (E). DD-FS, having shorter normal Ca2+-binding domains remaining, are less efficient at entrapping WT DSPP than the DGI-FS with its much longer negatively charged SSD repeat domain (E).
Discussion
The last few years have seen an increase in the number of DSPP mutations found in patients with nonsyndromic DD and DGI. For several years it was commonly hypothesized that haploinsufficiency (loss of half of the DSPP protein) would explain the diseases but two observations suggest that this is not the case. First, mice with only one intact Dspp allele (+/−) have normal teeth (Dr. Kulkarni, personal communication) while littermates lacking both alleles (−/−) have poorly mineralized dentin similar to human DGI-II (19). Second, among the many DSPP mutations in humans, the only reported nonsense mutation actually involves the splice-critical last base of an exon (Q45X) thereby causing the skipping of the same exon 3 frequently shown to cause other cases of DD or DGI (20, 27). This suggests that humans carrying simple, true nonsense mutations are probably non symptomatic much like the heterozygotic null (+/−) Dspp mice. We continue to predict that researchers will eventually find the rare families with recessively inherited nonsyndromic DGI that have these simple nonsense mutations (including −2 frameshifts) or null alleles in DSPP.
In this report we have undertaken the tasks of deciphering the mechanism(s) giving rise to dominant negative effects caused by two of the three classes of nonsyndromic DSPP mutations and to explain why some mutations cause DD while others cause the more severe, DGI. [We have verified the previously published results (28) showing that the first class of mutations (the mid signal peptide missense mutation, Y6D) caused mutant protein to be directly synthesized in the cytosol and not to significantly enter the rER. Therefore, the Y6D mutation causes its own dominant negative effect in DD using a mechanism other than retention in the rER and is the focus of a future study.] In 2008 we proposed that all other 5' mutations in DSPP (including several mutations that lead to skipping of exon 3) could be considered to result in simple changes in the first three amino acids (IPV) of the protein (after co-translational removal of its 15 amino acid leader peptide). Whether these simple changes were involved in signal peptide cleavage (and therefore were mechanistically related to the Y6D mutation) or some subsequent trafficking event, however, remained unexplored at that time. In this report, we first showed that the commonly held assumption that amino acids carboxy-terminal to the cleavage site do not influence co-translational removal of the leader sequence held true for all of the “IPV” mutations when fused to GFP, a protein sufficiently small to experimentally observe the presence or absence of a few amino acids. For the A15V mutation (in which the last and critically small amino acid, alanine, of the signal peptide itself was replaced by the much larger valine), the IPV motif was weakly predicted in silico to be lost when the signal peptidase cleaves several amino acids more carboxy-terminal. While we did observe a small amount of the more carboxy-terminal cleavage of the signal peptide in the media, the cell-associated A15V-GFP protein remained higher in molecular weight suggesting that its signal peptide was often not removed. Retention of this hydrophobic leader sequence could result in the protein becoming membrane bound via this single pass-like transmembrane domain, the consequences of which will be discussed later.
After observing that the changes in the proximal IPV tripeptide did not interfere with removal of the leader sequence from the “IPV” mutant constructs (ISV, ITV, IPD, etc.), the trafficking of WT and mutant DSPP during biosynthesis and secretion was explored next. Unlike the WT IPV-DSPP (which was rapidly trafficked out of the rER, Golgi-modified, and then secreted out of the cell), all 5' mutations were significantly retained within the cells. The IPV-like amino-terminal peptides (31 amino acids) of two other acidic proteins, osteopontin (IPV) and DMP1 (LPV), were equally effective in trafficking the DSPP out of the cell but changing their valine to the charged aspartic acid (IPD and LPD respectively) caused cell retention of both hybrid proteins, a result similar to that seen for mutant DSPP protein constructs. This suggests that DSPP is not unique in using the IPV motif for rapidly trafficking most of these proteins out of the rER of cells. Indeed, many secreted, acidic, Ca2+-binding proteins start with IPV-like hydrophobic-proline-hydrophobic (or hydrophobic-proline-polar, but not charged) tripeptides suggesting a common pathway for trafficking such proteins involves this motif. These include many other tooth and bone-related Ca2+-binding proteins (amelogenin, ameloblastin, enamelin, amelotin, MEPE, SPARC, SPARCL1, SCPPPQ1, etc) as well as serum (fetuin), digestive (trypsinogen) and neuroendocrine (chromogranins A and B) proteins. We tested this hypothesis by seeing if changing the IPV-like amino-termini of native osteopontin (IPV) and chromogranin A (LPV) to the mutant tripeptide with a charged amino acid, aspartate, (IPD or LPD) also changed the trafficking of these native proteins. In each case, the mutant protein was more highly retained within the cells and usually as smaller, less modified proteins. Thus, the study of the trafficking of the exceptionally acidic protein, DSPP, in DD and DGI has opened the door to exploring the role of the IPV-like amino-termini in assisting the trafficking of many secreted calcium-binding proteins. [This process may be evolutionally conserved as far back as echinoderms as a major protein in the sea urchin tooth, the highly acidic (pI = 3.7) phosphodontin, starts with the IPV-like tripeptide, APV.] Note that we observed at least some trafficking/processing/secretion of each acidic protein containing mutations within the IPV-like motif. However, an intact IPV-like motif appeared to aid in the speed/efficiency of monomeric proteins exiting the rER, suggesting the presence of an “IPV receptor” that assists the movement of the proteins within the rER or, more likely, in the packaging of the proteins within exit vesicles. Acidic proteins that fail to interact with this hypothetical “IPV receptor” can likely still exit the rER by “bulk flow” or other mechanisms. We hypothesize, however, that once mutant proteins accumulate to critical concentrations, dimers, oligomers, or perhaps higher complexes of acidic proteins like DSPP form. Once these higher complexes form, they become unable to use the “IPV receptors” to exit the rER and other, rER-stress related processes, are likely activated.
Biochemical and confocal microscopy evidence suggests that the frameshift mutant DSPP proteins are also retained in the rER. A model frameshift DSPP mutant containing a DGI-like number of normal repeats followed by a −1 frameshift-encoded hydrophobic domain of similar length, was completely retained in the cell layer. This protein pelleted in the 100,000 × g microsome membrane fraction (data not shown) and was microscopically observed associated with rER-like membranes. Both the frameshift and A15V mutants also appeared in membranes contiguous with the rER (e.g., nuclear membrane) in the experimentally transfected HEK293 cells but it is not known if such diffusion happens in the highly polarized odontoblasts of DD and DGI patients.
As discussed above, haploinsufficiency is unlikely to explain nonsyndromic DGI or the less severe disease, DD. Therefore, DD is hypothesized to occur when mutant odontoblasts successfully secrete an amount less than half of the DSPP of normal cells, but some critical amount more than the odontoblasts in DGI patients (Figure 7B). Classically, dominant negative effects are often observed when a mutant protein forms a dysfunctional dimer (or higher complexes) with the normal allele’s protein. As it is translated into the rER, DSPP with its >250 negatively charged aspartic acids (isoelectric point, pI <3.5) would immediately interact with the abundant (0.5–2 mM) Ca2+ found within this organelle. When two or more such large, Ca2+-coated proteins come into contact with each other, it is reasonable to hypothesize that the divalent calcium ions would form entropy-driven, energy-releasing bridges between the two proteins. In fact, it has been shown that three acidic, Ca2+-binding proteins, OPN, DMP1 and bone sialoprotein (BSP), each form (reversible) multivalent cation (Ca2+ and La3+) dependent complexes that can store/dissipate significant amounts of energy as measured by atomic force microscopy (36, 37). Although we were unable to affinity purify sufficient amounts of cell-associated IPD-DSPP mutant protein to mimic the protein-rich environment of the mutant cell’s rER and cause Ca2+-dependent precipitates to form in volumes necessary for ultracentrifugation (100,000 × g) experiments, we were able to precipitate all of this protein with the representative but energetically more favorable trivalent cations (La3+). When such DSPP becomes sufficiently abundant in the rER, dimers followed by oligomers and then possibly large gels or precipitates would form. Once such mistrafficked mutant DSPP proteins begin to form these complexes, co-translated WT DSPP would unavoidably become incorporated into them through their own polyanionic domains and would also be retained within the rER (Figure 7C). The exact fate of these complexes is the focus of another project but it is well established that aggregates of misfolded proteins induce ER stress responses including autophagy. For example, rER-associated aggregates of specific mutations of type I collagen can induce autophagy which results in the isolation and subsequent digestion of both the mutant and trimer-associated normal collagen chains (38). (Most if not all syndromic DD and DGI are caused by mutations in either type I collagen genes or in the enzymes/chaperone proteins associated with their processing.)
From our results, a corollary hypothesis can be developed; the milder disease, DD, may be caused in cases where either 1) less than the normal amount of mutant DSPP protein is made from the mutant allele thereby limiting the amount of initial complex formation (plus, more than half of the normal amount of WT DSPP is made in these cases) or 2) the mutant DSPP itself is less effective at forming the Ca2+-dependent complexes with the WT DSPP. Both events could lead to larger amounts of the WT allele’s DSPP being successively trafficked out of the rER and into the dentin matrix thereby causing a less severe mineralization defect. The splicing mutation, c.53–6T>G, represents an example of the first scenario, whereby, only about half of its mutant allele’s transcribed precursor-mRNA appears to be spliced without exon 3, while the other half of the message is normally spliced (23, 27). This differential splicing event would result in both 1) reduced amounts of mutant IPD3 (~25% of total DSPP) available to form the primary rER aggregates and 2) an increase in the amount of normal DSPP (~75%) available to attempt to exit the organelle via the proposed “IPV receptor”. This hypothetical case could result in ~40% of the WT protein being secreted; less than the 50% needed for normal dentin but perhaps more than what causes the more severe DGI phenotype. The strikingly differential location of the DD versus DGI frameshift mutations supports our second hypothesis. We noted in 2008 that FS mutations that occurred earlier in the repeat domain were associated with DD and more 3' mutations caused DGI (20), and this trend has been strongly reinforced with the reports from several additional affected patients/families (26, 29, 30) (Figure 7A). At first this seems counterintuitive because more 5' mutations directly result in the translation of many more mutant hydrophobic amino acids in a protein/domain that is hydrophillic and probably completely disordered (39). Logically, this would seem to be worse for the odontoblasts and, therefore, the patients. However, we propose that it is more important that the more 5' the FS mutation, the shorter the remaining length of normal, negatively charged SSD repeats that can entrap the WT DSPP through Ca2+ bridges (Figure 7E). In vitro, we were able to show that a DD-like FS construct (lacking essentially all SSD repeats), was less effective at trapping WT DSPP compared to the DGI-like FS DSPP protein that continued to express many calcium-binding SSD repeats. The normal portion of the DPP domains in the actual FS DD patients (counting from BMP1 cleavage site) was ~100–230 while the DGI patients’ FS mutations leave ~300–800 unchanged amino acids amino-terminal to their mutations (Figure 7A). All FS-DSPP mutant proteins (irrespective of the length of their mutant hydrophobic domain) would be membrane-associated and entrapped within the rER, so the length of the mutant hydrophobic domain itself may be irrelevant. (Interestingly, it can be proposed that any extended family discovered in the future to have some members with DD and others with the DGI phenotype, will likely be found to have −1 FS mutations within the domain currently between the DD and DGI associated mutations summarized in Figure 7A.) Finally, any A15V-DSPP protein which retains its short hydrophobic leader sequence and becomes rER membrane-associated, has its full complement of Ca2+-binding repeats to capture WT DSPP and cause the more severe DGI, similar to that of the more 3' FS mutations.
In conclusion, we have presented evidence that all nonsyndromic DD and DGI-associated mutations in DSPP (except Y6D) cause retention of the mutant protein within the cell, probably within the rER. This can happen because either: 1) one of the first three amino acids of the mature protein (IPV) are mutated by a variety of mutational events and fail to interact with a proposed “IPV receptor” that may aid in efficient trafficking of many different acidic proteins out of rER; or 2) mutant leader sequence (A15V) or any −1 frameshift within the repeat domain result in hydrophobic domains that anchor the mutant protein within the rER membrane. The dominant negative effects are due to the carboxy-terminal acidic repeat domain of the retained mutant proteins forming entropy-driven, multivalent cation (Ca2+) bridges with each other and then entrapping WT DSPP through its own calcium-binding domain. The milder DD appears to be caused by either 1) significantly fewer mutant proteins (differential splicing of the mutant allele) available to participate in the primary rER mutant IPD3-DSPP complexes or 2) shorter, less entrapping Ca2+-binding domains of frameshift mutant DSPPs, both resulting in sufficiently more WT DSPP being secreted out of the cell and aiding in a more normally mineralized dentin matrix although the actual role that DSPP plays in this process remains essentially unexplained.
Acknowledgements
This work was supported by the Intramural Research Program of the NIH, NIDCR. The authors wish to thank Dr. Ashok Kulkarni (NIDCR) for the mouse DSPP cDNA.
Footnotes
Disclosures
All authors state that they have no conflicts of interest.
Authors’ roles: Study design, LWF, ZvM. Study conduct, data collection & analyses: LWF, ZvM, DAM, SM & MDP. Data interpretation: LWF, ZvM. Drafting manuscript: LWF, ZvM, SM. Approving final version of manuscript: LWF, ZvM, DAM, SM & MDP. LWF & ZvM take responsibility for the integrity of the data analyses.
References
- 1.Shields ED, Bixler D, el-Kafrawy AM. A proposed classification for heritable human dentine defects with a description of a new entity. Arch Oral Biol. 1973 Apr;18(4):543–553. doi: 10.1016/0003-9969(73)90075-7. [DOI] [PubMed] [Google Scholar]
- 2.Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. 2011;7(9):540–557. doi: 10.1038/nrendo.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ball SP, Cook PJ, Mars M, Buckton KE. Linkage between dentinogenesis imperfecta and Gc. Ann Hum Genet. 1982 Jan 1;46(Pt 1):35–40. doi: 10.1111/j.1469-1809.1982.tb00693.x. [DOI] [PubMed] [Google Scholar]
- 4.Dean JA, Hartsfield JK, Jr, Wright JT, Hart TC. Dentin dysplasia, type II linkage to chromosome 4q. J Craniofac Genet Dev Biol. 1997 Oct-Dec;17(4):172–177. [PubMed] [Google Scholar]
- 5.Hart PS, Hart TC. Disorders of human dentin. Cells Tissues Organs. 2007;186(1):70–77. doi: 10.1159/000102682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacDougall M, Simmons D, Luan X, Gu TT, DuPont BR. Assignment of dentin sialophosphoprotein (DSPP) to the critical DGI2 locus on human chromosome 4 band q21.3 by in situ hybridization. Cytogenet Cell Genet. 1997;79(1–2):121–122. doi: 10.1159/000134697. [DOI] [PubMed] [Google Scholar]
- 7.Takagi Y, Veis A, Sauk JJ. Relation of mineralization defects in collagen matrices to noncollagenous protein components. Identification of a molecular defect in dentinogenesis imperfecta. Clin Orthop Relat Res. 1983 Jun;(176):282–290. [PubMed] [Google Scholar]
- 8.MacDougall M, Simmons D, Luan X, Nydegger J, Feng J, Gu TT. Dentin phosphoprotein and dentin sialoprotein are cleavage products expressed from a single transcript coded by a gene on human chromosome 4. Dentin phosphoprotein DNA sequence determination. J Biol Chem. 1997 Jan 10;272(2):835–842. doi: 10.1074/jbc.272.2.835. [DOI] [PubMed] [Google Scholar]
- 9.Qin C, Brunn JC, Cadena E, Ridall A, Tsujigiwa H, Nagatsuka H, Nagai N, Butler WT. The expression of dentin sialophosphoprotein gene in bone. J Dent Res. 2002 Jun;81(6):392–394. doi: 10.1177/154405910208100607. [DOI] [PubMed] [Google Scholar]
- 10.Zhang R, Chen FM, Zhao SL, Xiao MZ, Smith AJ, Feng JQ. Expression of dentine sialophosphoprotein in mouse nasal cartilage. Arch Oral Biol. 2011 Nov 14; doi: 10.1016/j.archoralbio.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 11.Ogbureke KU, Fisher LW. Expression of SIBLINGs and their partner MMPs in salivary glands. J Dent Res. 2004 Sep;83(9):664–670. doi: 10.1177/154405910408300902. [DOI] [PubMed] [Google Scholar]
- 12.Ogbureke KU, Fisher LW. Renal expression of SIBLING proteins and their partner matrix metalloproteinases (MMPs) Kidney Int. 2005 Jul;68(1):155–166. doi: 10.1111/j.1523-1755.2005.00389.x. [DOI] [PubMed] [Google Scholar]
- 13.Ogbureke KU, Fisher LW. SIBLING expression patterns in duct epithelia reflect the degree of metabolic activity. J Histochem Cytochem. 2007 Apr;55(4):403–409. doi: 10.1369/jhc.6A7075.2007. [DOI] [PubMed] [Google Scholar]
- 14.Bellahcene A, Castronovo V, Ogbureke KU, Fisher LW, Fedarko NS. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): multifunctional proteins in cancer. Nat Rev Cancer. 2008 Mar;8(3):212–226. doi: 10.1038/nrc2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Marschall Z, Fisher LW. Dentin sialophosphoprotein (DSPP) is cleaved into its two natural dentin matrix products by three isoforms of bone morphogenetic protein-1 (BMP1) Matrix Biol. 2010 May;29(4):295–303. doi: 10.1016/j.matbio.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.von Marschall Z, Fisher LW. Decorin is processed by three isoforms of bone morphogenetic protein-1 (BMP1) Biochem Biophys Res Commun. 2010 Jan 15;391(3):1374–1378. doi: 10.1016/j.bbrc.2009.12.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKnight DA, Fisher LW. Molecular evolution of dentin phosphoprotein among toothed and toothless animals. BMC Evol Biol. 2009;9:299. doi: 10.1186/1471-2148-9-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fisher LW. DMP1 and DSPP: evidence for duplication and convergent evolution of two SIBLING proteins. Cells Tissues Organs. 2011;194(2–4):113–118. doi: 10.1159/000324254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sreenath T, Thyagarajan T, Hall B, Longenecker G, D'Souza R, Hong S, Wright JT, MacDougall M, Sauk J, Kulkarni AB. Dentin sialophosphoprotein knockout mouse teeth display widened predentin zone and develop defective dentin mineralization similar to human dentinogenesis imperfecta type III. J Biol Chem. 2003 Jul 4;278(27):24874–24880. doi: 10.1074/jbc.M303908200. [DOI] [PubMed] [Google Scholar]
- 20.McKnight DA, Suzanne Hart P, Hart TC, Hartsfield JK, Wilson A, Wright JT, Fisher LW. A comprehensive analysis of normal variation and disease-causing mutations in the human DSPP gene. Hum Mutat. 2008 Dec;29(12):1392–1404. doi: 10.1002/humu.20783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao S, Yu C, Chou X, Yuan W, Wang Y, Bu L, Fu G, Qian M, Yang J, Shi Y, Hu L, Han B, Wang Z, Huang W, Liu J, Chen Z, Zhao G, Kong X. Dentinogenesis imperfecta 1 with or without progressive hearing loss is associated with distinct mutations in DSPP. Nat Genet. 2001 Feb;27(2):201–204. doi: 10.1038/84848. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, Zhao J, Li C, Gao S, Qiu C, Liu P, Wu G, Qiang B, Lo WH, Shen Y. DSPP mutation in dentinogenesis imperfecta Shields type II. Nat Genet. 2001 Feb;27(2):151–152. doi: 10.1038/84765. [DOI] [PubMed] [Google Scholar]
- 23.Kim JW, Nam SH, Jang KT, Lee SH, Kim CC, Hahn SH, Hu JC, Simmer JP. A novel splice acceptor mutation in the DSPP gene causing dentinogenesis imperfecta type II. Hum Genet. 2004 Aug;115(3):248–254. doi: 10.1007/s00439-004-1143-5. [DOI] [PubMed] [Google Scholar]
- 24.Lee SK, Hu JC, Lee KE, Simmer JP, Kim JW. A dentin sialophosphoprotein mutation that partially disrupts a splice acceptor site causes type II dentin dysplasia. J Endod. 2008 Dec;34(12):1470–1473. doi: 10.1016/j.joen.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holappa H, Nieminen P, Tolva L, Lukinmaa PL, Alaluusua S. Splicing site mutations in dentin sialophosphoprotein causing dentinogenesis imperfecta type II. Eur J Oral Sci. 2006 Oct;114(5):381–384. doi: 10.1111/j.1600-0722.2006.00391.x. [DOI] [PubMed] [Google Scholar]
- 26.Lee KE, Kang HY, Lee SK, Yoo SH, Lee JC, Hwang YH, Nam KH, Kim JS, Park JC, Kim JW. Novel dentin phosphoprotein frameshift mutations in dentinogenesis imperfecta type II. Clin Genet. 2011 Apr;79(4):378–384. doi: 10.1111/j.1399-0004.2010.01483.x. [DOI] [PubMed] [Google Scholar]
- 27.Lee KE, Lee SK, Jung SE, Lee Z, Kim JW. Functional splicing assay of DSPP mutations in hereditary dentin defects. Oral Dis. 2011 Oct;17(7):690–695. doi: 10.1111/j.1601-0825.2011.01825.x. [DOI] [PubMed] [Google Scholar]
- 28.Rajpar MH, Koch MJ, Davies RM, Mellody KT, Kielty CM, Dixon MJ. Mutation of the signal peptide region of the bicistronic gene DSPP affects translocation to the endoplasmic reticulum and results in defective dentine biomineralization. Hum Mol Genet. 2002 Oct 1;11(21):2559–2565. doi: 10.1093/hmg/11.21.2559. [DOI] [PubMed] [Google Scholar]
- 29.Song YL, Wang CN, Fan MW, Su B, Bian Z. Dentin phosphoprotein frameshift mutations in hereditary dentin disorders and their variation patterns in normal human population. J Med Genet. 2008 Jul;45(7):457–464. doi: 10.1136/jmg.2007.056911. [DOI] [PubMed] [Google Scholar]
- 30.Nieminen P, Papagiannoulis-Lascarides L, Waltimo-Siren J, Ollila P, Karjalainen S, Arte S, Veerkamp J, Tallon Walton V, Chimenos Kustner E, Siltanen T, Holappa H, Lukinmaa PL, Alaluusua S. Frameshift mutations in dentin phosphoprotein and dependence of dentin disease phenotype on mutation location. J Bone Miner Res. 2011 Apr;26(4):873–880. doi: 10.1002/jbmr.276. [DOI] [PubMed] [Google Scholar]
- 31.Lee SK, Lee KE, Jeon D, Lee G, Lee H, Shin CU, Jung YJ, Lee SH, Hahn SH, Kim JW. A novel mutation in the DSPP gene associated with dentinogenesis imperfecta type II. J Dent Res. 2009 Jan;88(1):51–55. doi: 10.1177/0022034508328168. [DOI] [PubMed] [Google Scholar]
- 32.Malmgren B, Lindskog S, Elgadi A, Norgren S. Clinical, histopathologic, and genetic investigation in two large families with dentinogenesis imperfecta type II. Hum Genet. 2004 Apr;114(5):491–498. doi: 10.1007/s00439-004-1084-z. [DOI] [PubMed] [Google Scholar]
- 33.Young MF, Kerr JM, Termine JD, Wewer UM, Wang MG, McBride OW, Fisher LW. cDNA cloning, mRNA distribution and heterogeneity, chromosomal location, and RFLP analysis of human osteopontin (OPN) Genomics. 1990 Aug;7(4):491–502. doi: 10.1016/0888-7543(90)90191-v. [DOI] [PubMed] [Google Scholar]
- 34.Fisher LW, Stubbs JT, 3rd, Young MF. Antisera and cDNA probes to human and certain animal model bone matrix noncollagenous proteins. Acta Orthop Scand Suppl. 1995 Oct;266:61–65. [PubMed] [Google Scholar]
- 35.von Marschall Z, Fisher LW. Dentin matrix protein-1 isoforms promote differential cell attachment and migration. J Biol Chem. 2008 Nov 21;283(47):32730–32740. doi: 10.1074/jbc.M804283200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fantner GE, Adams J, Turner P, Thurner PJ, Fisher LW, Hansma PK. Nanoscale ion mediated networks in bone: osteopontin can repeatedly dissipate large amounts of energy. Nano Lett. 2007 Aug;7(8):2491–2498. doi: 10.1021/nl0712769. [DOI] [PubMed] [Google Scholar]
- 37.Adams J, Fantner GE, Fisher LW, Hansma PK. Molecular energy dissipation in nanoscale networks of Dentin Matrix Protein 1 is strongly dependent on ion valence. Nanotechnology. 2008 Sep 24;19(38):384008. doi: 10.1088/0957-4484/19/38/384008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishida Y, Yamamoto A, Kitamura A, Lamande SR, Yoshimori T, Bateman JF, Kubota H, Nagata K. Autophagic elimination of misfolded procollagen aggregates in the endoplasmic reticulum as a means of cell protection. Mol Biol Cell. 2009 Jun;20(11):2744–2754. doi: 10.1091/mbc.E08-11-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cross KJ, Huq NL, Reynolds EC. Protein dynamics of bovine dentin phosphophoryn. J Pept Res. 2005 Aug;66(2):59–67. doi: 10.1111/j.1399-3011.2005.00273.x. [DOI] [PubMed] [Google Scholar]