

Abstract
One of the major obstacles in dissecting the mechanism of pathology in human primary biliary cirrhosis (PBC) has been the absence of animal models. Our laboratory has focused on a model in which mice, following immunization with a xenobiotic chemical mimic of the immunodominant autoepitope of the E2 component of pyruvate dehydrogenase complex (PDC-E2), develop autoimmune cholangitis. In particular, following immunization with 2-octynoic acid (a synthetic chemical mimic of lipoic acid-lysine located within the inner domain of PDC-E2) coupled to bovine serum albumin (BSA), several strains of mice develop typical anti-mitochondrial autoantibodies and portal inflammation. The role of innate immune effector cells, such as natural killer (NK) cells and that NK T cells, was studied in this model based on the hypothesis that early events during immunization play an important role in the breakdown of tolerance. We report herein that, following in-vivo depletion of NK and NK T cells, there is a marked suppression of anti-mitochondrial autoantibodies and cytokine production from autoreactive T cells. However, there was no change in the clinical pathology of portal inflammation compared to controls. These data support the hypothesis that there are probably multiple steps in the natural history of PBC, including a role of NK and NK T cells in initiating the breakdown of tolerance. However, the data suggest that adaptive autoimmune effector mechanisms are required for the progression of clinical disease.
Keywords: ELISA, ELISPOT, mouse model, natural killer cells, primary biliary cirrhosis
Introduction
Primary biliary cirrhosis (PBC) is an autoimmune disease of the liver characterized by specific destruction of the small bile ducts and the presence of readily detectable levels of anti-mitochondrial antibodies (AMA) [1]–[3]. Recently, we reported that natural killer (NK) cells are involved in the destruction of cholangiocytes and NK T cells are partly responsible for the exacerbation of disease in PBC [4]–[6]. While these data are consistent with the view that innate immune effector mechanisms serve as a bridge to acquired immunity, and the data imply a major role for innate immune effector mechanisms in the initiation of pathogenesis of human PBC [7],[8], the precise details of how such innate immune effector mechanisms influence the generation of pathogenic acquired immune responses remains poorly understood. To address this issue, we used a xenobiotic model of human PBC to examine the precise role of NK and NK T cells [9]. Results of the studies reported herein show that the in-vivo depletion of NK and NK T cells prior to immunization in this murine model of human PBC markedly delayed the generation of both anti-mitochondrial antibodies (AMA) and autoreactive T cell responses. Despite the reduction in the autoreactive T and B cell responses to mitochondrial autoantigens, the specific degree of portal inflammation was unchanged, emphasizing the lack of an absolute requirement for the NK/NK T-associated innate immune effector mechanisms in the initiation of a breakdown of tolerance and a potential major role of a continued adaptive response in the natural history of disease.
Materials and methods
Murine immunization
Female C57BL/6J (B6) mice aged 8–9 weeks were obtained from Kyudo (Kumamoto, Japan) and maintained in ventilated cages under specific pathogen-free conditions. Each mouse was immunized intraperitoneally with a mixture of 2-octynoic acid-bovine serum albumin (2OA-BSA) conjugate (100 µg/25 µl) incorporated in complete Freund's adjuvant (CFA; Sigma-Aldrich, St Louis, MO, USA) containing 10 mg/ml of Mycobacterium tuberculosis strain H37Ra. The mice subsequently received biweekly booster doses of 2OA-BSA incorporated in incomplete Freund's adjuvant (IFA; Sigma-Aldrich), as reported previously [9]. Groups of these 2OA-BSA-immunized mice were either treated intravenously with 100 µg of NK1·1 antibody (Cedarlane, Alexis, NC, USA) to deplete NK cells or NK T cells (group A, n = 32) or treated with control mouse immunoglobulin (group B, n = 32) every week before 2OA-BSA treatment and up to the time of killing. As negative controls, female B6 mice (group C, n = 12) were immunized with BSA incorporated in CFA (Sigma-Aldrich) and boosted using the same dose and schedule as the experimental mice. Sera and spleens were collected before and at every 6 weeks post-immunization to 24 weeks. Serological AMA was determined by enzyme-linked immunosorbent assay (ELISA) [10] and spleen mononuclear cells were isolated for detection of NK1·1-positive cells by flow cytometry and enzyme-linked immunospot (ELISPOT) assay. In a nested study, liver samples were collected from eight mice from groups A and B and three mice from group C, each at 6, 12, 18 and 24 weeks, and subjected to histological analysis [11]–[13].
Flow cytometric analysis of the cell surface antigens
Two-colour flow cytometry was performed on cell suspensions using a fluorescence activated cell sorter (FACS)Caliber flow cytometer (BD Biosciences, San Jose, CA, USA), as described previously [14]. Cell surface monoclonal antibodies utilized included anti-CD3 and NK1·1 (BD Biosciences). Splenic mononuclear cells (2·5–5·0 × 105) were stained for cell surface antigen expression at 4°C in the dark for 30 min, washed twice in 2 ml phosphate-buffered saline containing 1% bovine serum albumin and 0·01% sodium azide, and were fixed in 200 µl of 1% paraformaldehyde. Isotype-matched control antibodies were used to determine the background levels of staining.
Peptide synthesis
Murine PDC-E2 peptides encompassing the inner lipoyl domain, which correlates with human PBC-specific T cell epitopes [15], were synthesized by fluorenylmethyloxycarbonyl (Fmoc) chemistry and the peptide purity was more than 80%, as determined by high performance liquid chromatography (HPLC). The synthetic peptide sequences were aa 232–246 (GTVQRWEKKVGEKLS), aa 236–250 (RWEKKVGEKLSEGDL), aa 240–254 (KVGEKLSEGDLLAEI), aa 244–258 (KLSEGDLLAEIETDK), aa 248–262 (GDLLAEIETDKATIG), aa 252–266 (AEIETDKATIGFEVQ), aa 256–270 (TDKATIGFEVQEEGY) and aa 260–274 (TIGFEVQEEGYLAKI), all purchased from Genenet (Fukuoka, Japan).
Detection of AMA
AMA was determined by ELISA using the triple-expression hybrid clone, pML-MIT-3 (pML-MIT-3-ELISA) [10],[16],[17]. Briefly, recombinant proteins containing the AMA-reactive immunodominant epitopes localized to the three distinct lipoyl domains of human pyruvate dehydrogenase complex (PDC)-E2 [18], bovine branched chain 2-oxo acid dehydrogenase complex (BCOADC)-E2 [19] and rat 2-oxoglutarate dehydrogenase complex (OGDC)-E2 [10] were cloned and co-expressed in the plasmid vector, pGEX-4T-1 (Pharmacia, Alameda, CA, USA) and the product used as antigen. Serological AMA was determined using serum samples at a 1:250 dilution and the bound antibodies were detected by peroxidase-conjugated goat anti-mouse immunoglobulin (diluted 1:50 and 100 ul/well; Dako, Glostrup, Denmark). The optical density (OD) was determined using a microplate reader at 450 nm.
Evaluation of splenic PDC-E2 peptide-specific T cell responses by ELISPOT
Splenic mononuclear cells were obtained from mice before and at 6, 12, 18 and 24 weeks post-immunization and were treated with either NK1·1 antibody (n = 8 each time) or with control immunoglobulin (n = 8 each time) or negative control (n = 3 each time). A total of 1 × 106 cells were dispensed into each well of a 24-well plate and cultured with murine PDC-E2 synthetic peptides, as mentioned below. After 3 days of culture, viable splenocytes were harvested and ELISPOT assays were performed [RSD ELISPOT set, mouse interferon (IFN)-γ ELISPOT set, Minneapolis, MN, USA]. Briefly, 96-well nitrocellulose plates were coated with an optimized capture monoclonal antibody (mouse anti-IFN-γ) in phosphate-buffered saline (PBS) and incubated overnight at 4°C. Unbound antibody was removed by washing with PBS containing 0·05% Tween (PBS-Tween). Viable cells were added at 3 × 105 cells/well in 100 µl RPMI-1640 in triplicate. The plates were incubated at 37°C, 5% CO2 for 24 h; the plates were then washed, labelled with biotin-labelled anti-IFN-γ and developed by incubation with streptavidin–alkaline phosphatase, followed by incubation with a final substrate solution (BD™ AEC substrate reagent set, San Diego, CA, USA). The reaction was stopped by rinsing the contents with distilled water, and the number of spots was counted by using a KS ELISPOT Reader (Zeiss, Thornwood, NY, USA). Known positive and negative samples were included throughout. In addition, antigen-specific spot-forming cells (SFC) were quantitated as SFC in the presence and absence of antigen and the net SFC calculated. The average number of SFC in the absence of antigen was fewer than 10 (data not shown).
Histological analysis of murine liver sections
Immediately after killing, liver was harvested, cut into small fragments and fixed in 10% buffered formalin, embedded in paraffin, and cut into 5-µm sections. Liver sections were deparaffinized, stained with haematoxylin and eosin and evaluated under light microscopy by a ‘blinded’ qualified pathologist; the degree of liver inflammation, portal inflammation, bile duct damage, parenchymal inflammation and granuloma was scored as described previously [20]–[22]. Briefly, each section (except for those that showed bile duct damage or granuloma) was scored as either 0 = no significant change, 1 = minimal, 2 = mild, 3 = moderate or 4 = severe pathology. The sections that showed bile duct damage and granuloma were scored as either 0 = no significant observation, 1 = low frequency observed or 2 = frequently observed.
Statistical analysis
All experiments were performed in triplicate and the data points shown are means of these triplicate analyses. The data are expressed as mean ± standard deviation (s.d.), and the significant differences between samples was determined using Student's t-test. All analyses were two-tailed and P-values < 0·05 were considered significant. Statistical analyses were performed using Intercooled Stata 8·0 (Stata Corp, College Station, TX, USA).
Results
Depletion of NK and NK T cells
To evaluate the role of NK and NK T cells, we depleted NK and NK T cells by administering NK1·1 antibody. This treatment was confirmed to be effective due to the marked reduction in the frequency of NK1·1-positive NK cells or NK T cells by Stanford flow cytometry (Fig. 1).
Fig. 1.
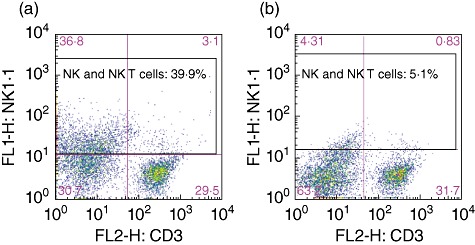
Effect of natural killer (NK)1·1 antibody deletion. Cell surface markers were determined in spleen mononuclear cells by flow cytometry. Splenic mononuclear cells contained 30·7% CD3–NK1·1– cells, 29·5% CD3+NK1·1– cells, 36·8% CD3–NK1·1+ cells and 3·1% CD3+NK1·1+ cells (a). After NK1·1 antibody treatment, NK1·1-positive cells decreased from 39·9% to 5·1% (b).
Anti-mitochondrial antibodies
At both 6 and 12 weeks post-immunization, serum AMA were decreased significantly in the NK1·1-depleted mice immunized with 2OA-BSA (n = 8) compared to sera from control mice immunized with 2OA-BSA. Interestingly, however, after 18 weeks there was no significant difference in AMA titres in the two groups of animals (Fig. 2). As expected, there were no detectable AMA in BSA control mice.
Fig. 2.
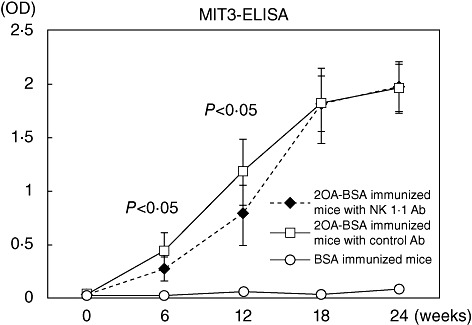
Antibody to mitochondrial antigens. Quantification of anti-mitochondrial antibody (AMA) in the sera by enzyme-linked immunosorbent assay (ELISA) at 6-week intervals after immunization shows significant increases in optical density (OD). Sera obtained from mice before and at 6, 12, 18 and 24 weeks post-immunization were treated with either natural killer (NK)1·1 antibody (n = 8 each time) or with control immunoglobulin (n = 8 each time) or negative control (n = 3 each time). In immunized mice, NK1·1 antibody-treated mice had lower OD than control immunoglobulin-treated mice (P < 0·05) within 12 weeks.
Detection of IFN-γ-secreting T cells recognizing mouse PDC-E2 peptides
We evaluated T cell responses to PDC-E2 at 6, 12, 18 and 24 weeks using our ELISPOT assay in individual NK1·1-depleted and control 2OA-BSA immunized mice (Fig. 3). As noted, the numbers of IFN-γ-secreting T cells from the control 2OA-BSA-immunized mice both at 6 and 12 weeks were significantly higher than the 2OA-BSA-immunized NK1·1-depleted group. However, the mean number of such IFN-γ-secreting T cells was similar in both groups at 18 and 24 weeks.
Fig. 3.
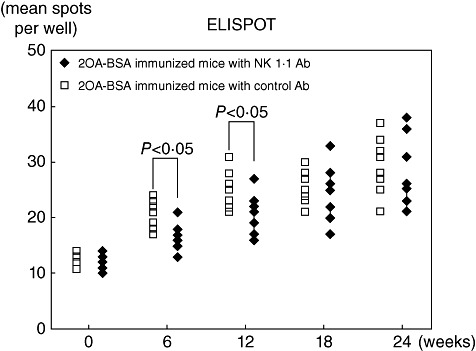
Enzyme-linked immunosospot assay (ELISPOT) assay [interferon (IFN)-γ] of 2-octynoic acid-bovine serum albumin (2OA-BSA)-immunized mice with natural killer (NK)1·1 antibody and with control immunoglobulin. Cytokine production of splenic mononuclear cells in response to pyruvate dehydrogenase complex (PDC)-E2 synthetic peptides using ELISPOT. The y-axis indicates the number of IFN-γ-producing T cells reactive to PDC-E2 synthetic peptides/3 × 105 splenic mononuclear cells. Note that NK1·1 antibody-treated mice had lower frequencies of autoreactive T cells than those with control immunoglobulin (P < 0·05) within 12 weeks.
Histological studies
The coded series of liver tissues from the various groups of mice were studied by a pathologist blinded to the groupings of the donor mice. As seen in Fig. 4, there were no major differences in the degree of lymphoid cell infiltration in tissues from mice treated with the NK1·1 antibody compared with tissues from the control mice at 24 weeks. Both the levels of bile duct lesions and lymphoid cell infiltration appear to be mild in the NK1·1-depleted and control mice. These data suggest that NK1·1 depletion initiated prior to immunization does not appear to influence the degree of lymphoid cell infiltrates and/or severity of the ductal lesions (data not shown).
Fig. 4.
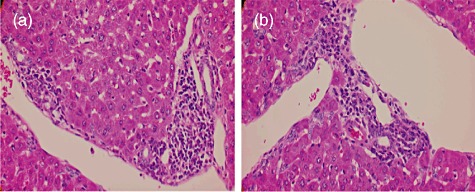
Microscopic examination of liver tissue from 2-octynoic acid-bovine serum albumin (2OA-BSA) with natural killer (NK)1·1 antibody and control immunoglobulin. (a) Light microscopy (×400) with NK1·1 antibody shows mild infiltration of lymphocytes in portal tracts, particularly surrounding damaged intralobular bile ducts. (b) Light microscopy (×400) with control immunoglobulin demonstrates similar findings to those shown in (a).
Discussion
Recent work has emphasized that the unique destruction of biliary cells requires the triad of macrophages from patients with PBC, biliary epithelial cell apotopes and AMAs; this leads to a burst of proinflammatory cytokines [23]. In addition, there is evidence that NK cells are involved in biliary cell cytotoxicity, and in this respect it is noteworthy that there is considerable heterogeneity among the NK and perhaps also the NK T cell lineages [24],[25]. Thus, previous dogmas with regard to NK cells require re-examination, particularly with regard to function, as there is now evidence for NK cell memory and a regulatory function has also been ascribed to NK cells [25]. One of the strongest cases for NK cell heterogeneity comes from studies of the phenotypical and functional differences of the NK cell lineages that reside within the gut compared with the blood and lymph nodes [26],[27]. Thus, while organ-resident NK cells control the magnitude of organ inflammation, they also have a role concurrently in influencing the generation of autoimmunity and pathology [28],[29]. Peripherally derived NK cells have an impact upon autoimmune responses which are manifested by their ability to synthesize cytokines rapidly that, in turn, influence the quality and quantity of acquired immune responses [30]–[34]. While the CD1d-deficient mouse [35]–[38] and the use of α-GalCer to activate NK T cells [39]–[41] are both available to perform standard addition/subtraction experiments in efforts to define a role for the NK T cell lineage, reagents are not readily available for a similar study of the role of NK cells. This is due to the fact that the use of the classical NK1·1 monoclonal antibody (mAb) to deplete NK cells also deletes NK T cells, because the latter lineage also expresses NK1·1. As NK T cells have been shown to contribute to the exacerbation of disease in PBC [5],[6], results of the findings reported herein indicate that the depletion of both NK cells and NK T cells prior to immunization has a minimal role in the overall breakdown of tolerance. Thus, and as shown herein, while depletion of NK1·1 cells appeared to delay significantly the generation of autoimmune-specific acquired humoral and cellular responses, the data indicate clearly that depletion of the NK1·1 lineage did not lead to any detectable differences in the pathology seen in the NK1·1-depleted versus control mice.
It is well known that liver contains NK cell subsets which have reduced effector function [42],[43], but under appropriate inflammatory conditions become potent killers [44]. NK cells sense normal or abnormal cells with their inhibitory or activating receptors [32]. Thus, under normal circumstances, NK cells will not damage autologous cells due to the engagement of inhibitory receptors. Nevertheless, NK cells have the capacity to spontaneously kill autologous immature dendritic cells [45], stromal cells or fibroblasts [46], and under special conditions such as the presence of restricted innate immune stimulation NK cells destroy autologous biliary epithelial cells [24]. The natural history of autoimmune cholangitis in this model requires, first, the loss of tolerance to PDC-E2 and secondly, the inflammatory portal infiltrates in liver. Our data imply that there are different phases to the natural history of disease, a theme which is similar to our previously published work [47],[48]. In other words, one factor which can facilitate the onset of autoimmunity is NK and NK T cell populations. However, once tolerance is initiated, the disease will be perpetuated via other mechanisms, again highlighting the promiscuous nature of autoimmunity and the involvement of multiple effector pathways.
Acknowledgments
Financial support was provided by a Grant-in-Aid for Scientific Research (C) (Kakenhi 22590739) and partially by the Research Program of Intractable Disease provided by the Ministry of Health, Labor, and Welfare of Japan; NIH grant no. DK067003.
Disclosure
The authors have no conflicts of interest to declare.
References
- 1.Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–73. doi: 10.1056/NEJMra043898. [DOI] [PubMed] [Google Scholar]
- 2.Lleo A, Invernizzi P, Mackay IR, Prince H, Zhong RQ, Gershwin ME. Etiopathogenesis of primary biliary cirrhosis. World J Gastroenterol. 2008;14:3328–37. doi: 10.3748/wjg.14.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu H, Norman GL, Shums Z, et al. PBC screen: an IgG/IgA dual isotype ELISA detecting multiple mitochondrial and nuclear autoantibodies specific for primary biliary cirrhosis. J Autoimmun. 2010;35:436–42. doi: 10.1016/j.jaut.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Kita H, Naidenko OV, Kronenberg M, et al. Quantitation and phenotypic analysis of natural killer T cells in primary biliary cirrhosis using a human CD1d tetramer. Gastroenterology. 2002;123:1031–43. doi: 10.1053/gast.2002.36020. [DOI] [PubMed] [Google Scholar]
- 5.Wu SJ, Yang YH, Tsuneyama K, et al. Innate immunity and primary biliary cirrhosis: activated invariant natural killer T cells exacerbate murine autoimmune cholangitis and fibrosis. Hepatology. 2011;53:915–25. doi: 10.1002/hep.24113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chuang YH, Lian ZX, Yang GX, et al. Natural killer T cells exacerbate liver injury in a transforming growth factor beta receptor II dominant-negative mouse model of primary biliary cirrhosis. Hepatology. 2008;47:571–80. doi: 10.1002/hep.22052. [DOI] [PubMed] [Google Scholar]
- 7.Selmi C, Lleo A, Pasini S, Zuin M, Gershwin ME. Innate immunity and primary biliary cirrhosis. Curr Mol Med. 2009;9:45–51. doi: 10.2174/156652409787314525. [DOI] [PubMed] [Google Scholar]
- 8.Berg PA. The role of the innate immune recognition system in the pathogenesis of primary biliary cirrhosis: a conceptual view. Liver Int. 2011;31:920–31. doi: 10.1111/j.1478-3231.2011.02457.x. [DOI] [PubMed] [Google Scholar]
- 9.Wakabayashi K, Lian ZX, Leung PS, et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology. 2008;48:531–40. doi: 10.1002/hep.22390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moteki S, Leung PS, Dickson ER, et al. Epitope mapping and reactivity of autoantibodies to the E2 component of 2-oxoglutarate dehydrogenase complex in primary biliary cirrhosis using recombinant 2-oxoglutarate dehydrogenase complex. Hepatology. 1996;23:436–44. doi: 10.1002/hep.510230307. [DOI] [PubMed] [Google Scholar]
- 11.Bioulac-Sage P. Primary biliary cirrhosis: a new histological staging and grading system proposed by Japanese authors. Clin Res Hepatol Gastroenterol. 2011;35:333–5. doi: 10.1016/j.clinre.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Nakanuma Y, Zen Y, Harada K, et al. Application of a new histological staging and grading system for primary biliary cirrhosis to liver biopsy specimens: interobserver agreement. Pathol Int. 2010;60:167–74. doi: 10.1111/j.1440-1827.2009.02500.x. [DOI] [PubMed] [Google Scholar]
- 13.Ludwig J, Dickson ER, McDonald GS. Staging of chronic nonsuppurative destructive cholangitis (syndrome of primary biliary cirrhosis) Virchows Arch A Pathol Anat Histol. 1978;379:103–12. doi: 10.1007/BF00432479. [DOI] [PubMed] [Google Scholar]
- 14.Wakabayashi K, Lian ZX, Moritoki Y, et al. IL-2 receptor alpha(–/–) mice and the development of primary biliary cirrhosis. Hepatology. 2006;44:1240–9. doi: 10.1002/hep.21385. [DOI] [PubMed] [Google Scholar]
- 15.Shimoda S, Nakamura M, Ishibashi H, Hayashida K, Niho Y. HLA DRB4 0101-restricted immunodominant T cell autoepitope of pyruvate dehydrogenase complex in primary biliary cirrhosis: evidence of molecular mimicry in human autoimmune diseases. J Exp Med. 1995;181:1835–45. doi: 10.1084/jem.181.5.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gershwin ME, Mackay IR, Sturgess A, Coppel RL. Identification and specificity of a cDNA encoding the 70 kd mitochondrial antigen recognized in primary biliary cirrhosis. J Immunol. 1987;138:3525–31. [PubMed] [Google Scholar]
- 17.Miyakawa H, Tanaka A, Kikuchi K, et al. Detection of antimitochondrial autoantibodies in immunofluorescent AMA-negative patients with primary biliary cirrhosis using recombinant autoantigens. Hepatology. 2001;34:243–8. doi: 10.1053/jhep.2001.26514. [DOI] [PubMed] [Google Scholar]
- 18.Cha S, Leung PS, Coppel RL, Van de Water J, Ansari AA, Gershwin ME. Heterogeneity of combinatorial human autoantibodies against PDC-E2 and biliary epithelial cells in patients with primary biliary cirrhosis. Hepatology. 1994;20:574–83. [PubMed] [Google Scholar]
- 19.Leung PS, Chuang DT, Wynn RM, et al. Autoantibodies to BCOADC-E2 in patients with primary biliary cirrhosis recognize a conformational epitope. Hepatology. 1995;22:505–13. [PubMed] [Google Scholar]
- 20.Dhirapong A, Lleo A, Yang GX, et al. B cell depletion therapy exacerbates murine primary biliary cirrhosis. Hepatology. 2011;53:527–35. doi: 10.1002/hep.24044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moritoki Y, Zhang W, Tsuneyama K, et al. B cells suppress the inflammatory response in a mouse model of primary biliary cirrhosis. Gastroenterology. 2009;136:1037–47. doi: 10.1053/j.gastro.2008.11.035. [DOI] [PubMed] [Google Scholar]
- 22.Yang GX, Lian ZX, Chuang YH, et al. Adoptive transfer of CD8(+) T cells from transforming growth factor beta receptor type II (dominant negative form) induces autoimmune cholangitis in mice. Hepatology. 2008;47:1974–82. doi: 10.1002/hep.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lleo A, Bowlus CL, Yang GX, et al. Biliary apotopes and anti-mitochondrial antibodies activate innate immune responses in primary biliary cirrhosis. Hepatology. 2010;52:987–98. doi: 10.1002/hep.23783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimoda S, Harada K, Niiro H, et al. Interaction between Toll-like receptors and natural killer cells in the destruction of bile ducts in primary biliary cirrhosis. Hepatology. 2011;53:1270–81. doi: 10.1002/hep.24194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature. 2011;481:394–8. doi: 10.1038/nature10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrews DM, Estcourt MJ, Andoniou CE, et al. Innate immunity defines the capacity of antiviral T cells to limit persistent infection. J Exp Med. 2010;207:1333–43. doi: 10.1084/jem.20091193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alter G, Heckerman D, Schneidewind A, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature. 2011;476:96–100. doi: 10.1038/nature10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hao J, Liu R, Piao W, et al. Central nervous system (CNS)-resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. J Exp Med. 2010;207:1907–21. doi: 10.1084/jem.20092749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brauner H, Elemans M, Lemos S, et al. Distinct phenotype and function of NK cells in the pancreas of nonobese diabetic mice. J Immunol. 2010;184:2272–80. doi: 10.4049/jimmunol.0804358. [DOI] [PubMed] [Google Scholar]
- 30.Yokoyama WM, Plougastel BF. Immune functions encoded by the natural killer gene complex. Nat Rev Immunol. 2003;3:304–16. doi: 10.1038/nri1055. [DOI] [PubMed] [Google Scholar]
- 31.Raulet DH. Interplay of natural killer cells and their receptors with the adaptive immune response. Nat Immunol. 2004;5:996–1002. doi: 10.1038/ni1114. [DOI] [PubMed] [Google Scholar]
- 32.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu HJ, Sawaya H, Binstadt B, et al. Inflammatory arthritis can be reined in by CpG-induced DC-NK cell cross talk. J Exp Med. 2007;204:1911–22. doi: 10.1084/jem.20070285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feuerer M, Shen Y, Littman DR, Benoist C, Mathis D. How punctual ablation of regulatory T cells unleashes an autoimmune lesion within the pancreatic islets. Immunity. 2009;31:654–64. doi: 10.1016/j.immuni.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hua J, Liang S, Ma X, Webb TJ, Potter JP, Li Z. The Interaction between regulatory T cells and NKT cells in the liver: a CD1d bridge links innate and adaptive immunity. PLoS ONE. 2011;6:e27038. doi: 10.1371/journal.pone.0027038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh KH, Lee C, Lee SW, et al. Activation of natural killer T cells inhibits the development of induced regulatory T cells via IFNgamma. Biochem Biophys Res Commun. 2011;411:599–606. doi: 10.1016/j.bbrc.2011.06.193. [DOI] [PubMed] [Google Scholar]
- 37.Teige A, Bockermann R, Hasan M, Olofsson KE, Liu Y, Issazadeh-Navikas S. CD1d-dependent NKT cells play a protective role in acute and chronic arthritis models by ameliorating antigen-specific Th1 responses. J Immunol. 2010;185:345–56. doi: 10.4049/jimmunol.0901693. [DOI] [PubMed] [Google Scholar]
- 38.Shin Y, Hong C, Lee H, Shin JH, Hong S, Park SH. NKT cell-dependent regulation of secondary antigen-specific, conventional CD4+ T cell immune responses. J Immunol. 2010;184:5589–94. doi: 10.4049/jimmunol.0903121. [DOI] [PubMed] [Google Scholar]
- 39.Simkins HM, Hyde E, Farrand KJ, et al. Administration of alpha-galactosylceramide impairs the survival of dendritic cell subpopulations in vivo. J Leukoc Biol. 2011;89:753–62. doi: 10.1189/jlb.0910480. [DOI] [PubMed] [Google Scholar]
- 40.Patel O, Cameron G, Pellicci DG, et al. NKT TCR recognition of CD1d-alpha-C-galactosylceramide. J Immunol. 2011;187:4705–13. doi: 10.4049/jimmunol.1100794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knothe S, Mutschler V, Rochlitzer S, et al. The NKT cell ligand alphagalactosylceramide suppresses allergic airway inflammation by induction of a Th1 response. Vaccine. 2011;29:4249–55. doi: 10.1016/j.vaccine.2011.03.068. [DOI] [PubMed] [Google Scholar]
- 42.Lassen MG, Lukens JR, Dolina JS, Brown MG, Hahn YS. Intrahepatic IL-10 maintains NKG2A+Ly49- liver NK cells in a functionally hyporesponsive state. J Immunol. 2010;184:2693–701. doi: 10.4049/jimmunol.0901362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maroof A, Beattie L, Zubairi S, Svensson M, Stager S, Kaye PM. Posttranscriptional regulation of II10 gene expression allows natural killer cells to express immunoregulatory function. Immunity. 2008;29:295–305. doi: 10.1016/j.immuni.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burt BM, Plitas G, Zhao Z, et al. The lytic potential of human liver NK cells is restricted by their limited expression of inhibitory killer Ig-like receptors. J Immunol. 2009;183:1789–96. doi: 10.4049/jimmunol.0900541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson JL, Heffler LC, Charo J, Scheynius A, Bejarano MT, Ljunggren HG. Targeting of human dendritic cells by autologous NK cells. J Immunol. 1999;163:6365–70. [PubMed] [Google Scholar]
- 46.Poggi A, Zocchi MR. Antigen presenting cells and stromal cells trigger human natural killer lymphocytes to autoreactivity: evidence for the involvement of natural cytotoxicity receptors (NCR) and NKG2D. Clin Dev Immunol. 2006;13:325–36. doi: 10.1080/17402520600578194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ambrosini YM, Yang GX, Zhang W, et al. The multi-hit hypothesis of primary biliary cirrhosis: polyinosinic-polycytidylic acid (poly I:C) and murine autoimmune cholangitis. Clin Exp Immunol. 2011;166:110–20. doi: 10.1111/j.1365-2249.2011.04453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gershwin ME, Mackay IR. The causes of primary biliary cirrhosis: convenient and inconvenient truths. Hepatology. 2008;47:737–45. doi: 10.1002/hep.22042. [DOI] [PubMed] [Google Scholar]