

Abstract
Cordycepin (3′-deoxyadenosine) is one of the major bioactive substances produced by Cordyceps militaris, a traditional medicinal mushroom. Cordycepin possesses several biological activities, including both pro-apoptotic and anti-apoptotic properties. In the present report, we investigated an effect of cordycepin on the survival of cells exposed to tumour necrosis factor (TNF)-α. We found that subtoxic doses of cordycepin increased susceptibility of cells to TNF-α-induced apoptosis. It was associated with suppression of nuclear factor-κB (NF-κB), a major prosurvival component involved in TNF-α signalling. The adenosine transporter and A3 adenosine receptor, but not A1 and A2 adenosine receptors, mediated both anti-NF-κB and pro-apoptotic effects. We found that cordycepin had the potential to phosphorylate eukaryotic translation initiation factor 2α (eIF2α) and that activation of eIF2α mimicked the suppressive effect of cordycepin on the NF-κB pathway. Furthermore, activation of eIF2α sensitized cells to TNF-α-induced apoptosis. To identify molecular events downstream of eIF2α, the role of mammalian target of rapamycin complex 1 (mTORC1) was examined. Selective activation of 3eIF2α, as well as treatment with cordycepin, caused phosphorylation of mTORC1. Rapamycin, an inhibitor of mTORC1, significantly reversed the suppressive effects of eIF2α on NF-κB. These results suggest that cordycepin sensitizes cells to TNF-α-induced apoptosis, at least in part, via induction of the eIF2α–mTORC1 pathway and consequent suppression of NF-κB.
Keywords: cordycepin, eIF2α, mTORC1, NF-κB, TNF-α
Introduction
Tumour necrosis factor (TNF)-α is a potent proinflammatory cytokine that plays an important role in the regulation of inflammation, cell proliferation, differentiation and apoptosis. TNF-α induces apoptosis via formation of the death-inducing signalling complex consisting of TNF-receptor (TNFR), Fas-associated death domain protein (FADD) and caspase-8. Activation of caspase-8 leads to cleavage of downstream caspases, including caspase-3, resulting in apoptotic cell death [1]. In addition to this pro-apoptotic signalling, TNF-α also induces prosurvival signalling mediated by nuclear factor-κB (NF-κB). Activation of the NF-κB pathway counteracts TNF-α-induced apoptosis via induction of anti-apoptotic genes, including TNFR-associated factors (TRAF1 and TRAF2), inhibitor of apoptosis proteins (cIAP-1 and cIAP-2) and caspase-8 homologue FLICE-inhibitory protein (cFLIP) [2]. In this process, TNFR1-associated death domain (TRADD) recruits TRAF2 and receptor-interacting protein 1 (RIP1) into the TNFR1 signalling complex. TRAF2 then recruits IκB kinase (IKK) into the TNFR1 complex, leading to the activation of IKK and consequent phosphorylation of IκBα. Phosphorylated IκBα is then degraded via the ubiquitin–proteasome pathway, allowing NF-κB to translocate to the nucleus and to initiate transcription of pro-apoptotic genes [3].
Cordycepin (3′-deoxyadenosine) is one of the major bioactive substances produced by Cordyceps militaris, a parasitic fungus used for traditional Chinese medicine. It possesses a wide range of biological effects, including anti-thrombotic, anti-cancer, anti-viral, anti-fungal and anti-inflammatory activities [4]–[9]. Previous reports have suggested pro-apoptotic effects of cordycepin on several types of cancer cells [10]–[12]. However, we have reported recently that, under some particular stress conditions, cordycepin facilitated cell survival. For example, cordycepin selectively attenuated endoplasmic reticulum (ER) stress-induced apoptosis in renal tubular cells [13]. Currently, it is unclear whether cordycepin regulates cellular fate under other stress conditions. In particular, it is undetermined whether and how cordycepin affects cytokine-triggered apoptosis. In the present investigation, we aimed to examine an effect of cordycepin on the survival of cells exposed to TNF-α. Our results suggest that cordycepin sensitizes cells to TNF-α-induced apoptosis via induction of particular stress signalling and consequent suppression of NF-κB.
Materials and methods
Reagents
Cordycepin, rapamycin, 1,3-dipropyl-8-sulphophenylxanthine (DPSPX), propyl 6-ethyl-5-ethylsulphanylcarbonyl-2-phenyl-4-propyl-pyridine-3-carboxylate (MRS1523) and nitrobenzylthioinosine (NBTI) were purchased from Sigma-Aldrich Japan (Tokyo, Japan). Human recombinant TNF-α was obtained from R&D Systems (Minneapolis, MN, USA), and salubrinal was obtained from Calbiochem (San Diego, CA, USA). Dehydroxymethylepoxyquinomicin (DHMEQ) [14] was kindly provided by Dr Kazuo Umezawa (Keio University, Tokyo, Japan).
Cell culture
The rat renal tubular epithelial cell line NRK-52E was purchased from American Type Culture Collection (Manassas, VA, USA). Cells were maintained in Dulbecco's modified Eagle's medium/Ham's F-12 (Gibco-BRL, Gaithersburg, MD, USA) supplemented with 5% fetal bovine serum (FBS).
Stable transfection
Using electroporation, NRK-52E cells were stably transfected with pNF-κB-Luc (Panomics, Fremont, CA, USA), which introduces a luciferase gene under the control of the κB sites, and NRK/NF-κB-Luc cells were established.
Transient transfection
Using electroporation, NRK-52E cells were transfected transiently with pIκBα-Fluc or pCMV-Fluc (gifts from Dr David Piwnica-Worms, Washington University) [15], treated with TNF-α in the absence or presence of cordycepin and subjected to luciferase assay. pCMV-Fluc and pIkBa-Fluc introduce firefly luciferase (Fluc) or IkBa fused to Fluc under the control of the cytomegalovirus promoter (CMV). The luciferase activity in pIκBα-Fluc-transfected cells was normalized by the levels of luciferase in pCMV-Fluc-transfected cells. In some experiments, NRK-52E cells were co-transfected with pNF-κB-Luc together with pCMV4 or pCMV4-p65 (Addgene, Cambridge, MA, USA) encoding the p65 NF-κB subunit.
Assessment of cell death
Cells were seeded into 24-well plates and treated with test agents. Both attached and floating cells were harvested and subjected to trypan blue analysis. The percentages of dead cells against total cells were evaluated in individual wells.
Luciferase assay
Luciferase activity was evaluated by the luciferase assay system (Promega, Madison, WI, USA), according to the manufacturer's protocol.
Northern blot analysis
Total RNA was extracted by a single-step method, and Northern blot analysis was performed as described previously [16]. A cDNA for monocyte chemoattractant protein 1 (MCP-1) [17] was used to prepare a radio-labelled probe. Expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control.
Western blot analysis
Western blot analysis was performed using the enhanced chemiluminescence system (Amersham Biosciences, Buckinghamshire, UK). Anti-caspase-3 antibody, anti-phospho eukaryotic translation initiation factor 2α (eIF2α) antibody (Ser51), anti-phospho p70S6 kinase (p70S6K) antibody (Thr389) and anti-p70S6K antibody were purchased from Cell Signaling Technology (Danvers, MA, USA), and anti-eIF2α antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). As a loading control, levels of β-actin were evaluated using anti-β-actin antibody (Sigma-Aldrich Japan). Levels of phosphorylated Akt and total Akt protein were evaluated using PhosphoPlus Akt (Ser473) Antibody Kit (Cell Signaling Technology).
Statistical analysis
In apoptosis assays and reporter assays, experiments were performed in quadruplicate, and data were expressed as means ± standard error (s.e.). Statistical analysis was performed using the non-parametric Mann–Whitney U-test to compare data in different groups. A P-value < 0·05 was considered to indicate a statistically significant difference.
Results
Sensitization of cells to TNF-α-induced apoptosis by cordycepin
We reported recently that cordycepin selectively inhibits endoplasmic reticulum (ER) stress-induced apoptosis in NRK-52E cells [13]. To examine whether and how cordycepin regulates cellular fate under other stress conditions, especially under inflammatory stimuli, NRK-52E cells were exposed to TNF-α in the absence or presence of a non-toxic dose (5 µg/ml) of cordycepin. Microscopic analysis showed that TNF-α alone did not induce significant cell injury. However, co-treatment with cordycepin sensitized cells to TNF-α-induced cellular damage (Fig. 1a). Quantitative analysis showed that the percentage of dead cells increased significantly by the treatment with cordycepin only in TNF-α-exposed cells (Fig. 1b). Western blot analysis also showed that exposure to cordycepin caused cleavage of procaspase-3 in TNF-α-treated cells, but not in TNF-α-untreated cells (Fig. 1c).
Fig. 1.

Sensitization of cells to tumour necrosis factor (TNF)-α-induced apoptosis by cordycepin. (a) Normal rat kidney epithelial (NRK-52E) cells were treated with 5 µg/ml cordycepin in the absence or presence of 10 ng/ml TNF-α for 48 h and subjected to phase-contrast microscopy. (b) The percentages of dead cells against total cells were evaluated by trypan blue analysis. Assays were performed in quadruplicate, and data are presented as means ± standard error. *Statistically significant difference (P < 0·05); NS, not statistically significant. (c) Cells were treated with indicated agents for 48 h and subjected to Western blot analysis of caspase-3. The level of β-actin is shown at the bottom as a loading control.
Sensitization to apoptosis by cordycepin through blockade of NF-κB
TNF-α triggers both pro-apoptotic and prosurvival signalling, and NF-κB plays a crucial role in its prosurvival effect [2]. We speculated that cordycepin may suppress activation of NF-κB, leading to sensitization to TNF-α-induced apoptosis. Indeed, a reporter assay showed that cordycepin inhibited TNF-α-triggered activation of NF-κB in a dose-dependent manner (Fig. 2a). This effect was not ascribed to its non-specific effects on transcription or translation, because it did not suppress the activity of luciferase driven by viral promoters and housekeeping gene promoters (data not shown). Consistent with this result, cordycepin also blunted induction of a NF-κB-dependent gene MCP-1 (Fig. 2b). To confirm that the pro-apoptotic effect of cordycepin is indeed ascribed to suppression of NF-κB, the effects of DHMEQ, an inhibitor of NF-κB [14], were tested. As shown in Fig. 2c,d, treatment with DHMEQ mimicked the pro-apoptotic effect of cordycepin in TNF-α-exposed cells. Furthermore, concomitant treatment with TNF-α and DHMEQ caused substantial cleavage of procaspase-3 (Fig. 2e).
Fig. 2.
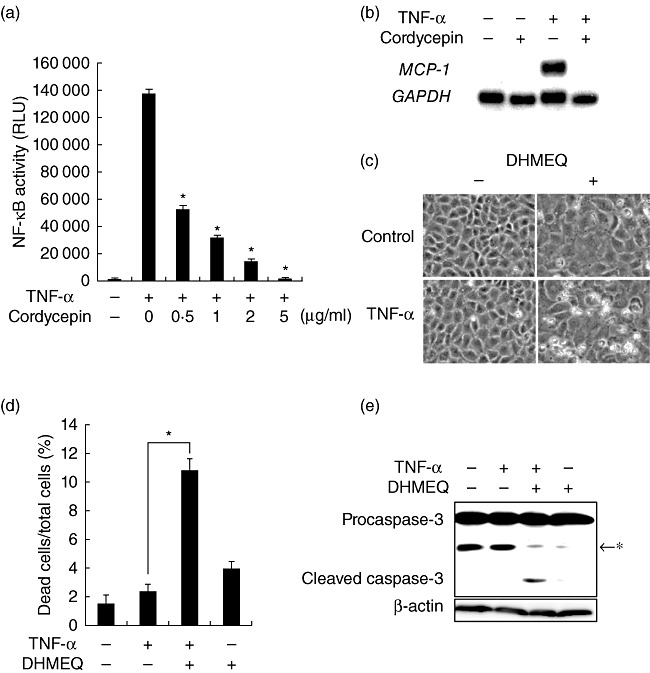
Sensitization to apoptosis by cordycepin through blockade of nuclear factor (NF)-κB. (a) NRK/NF-κB-Luc cells were exposed to tumour necrosis factor (TNF)-α in the presence of serial concentrations of cordycepin for 9 h and subjected to luciferase assay to evaluate NF-κB activity; RLU: relative light unit. (b) Cells were treated with TNF-α in the absence or presence of cordycepin for 6 h and subjected to Northern blot analysis of monocyte chemoattractant protein 1 (MCP-1). The level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is shown as a loading control. (c,d) Cells were treated with TNF-α in the absence or presence of 5 µg/ml dehydroxymethylepoxyquinomicin (DHMEQ) and subjected to phase-contrast microscopy (c) and assessment of cell death (d). (e) Cells were treated with indicated agents and subjected to Western blot analysis of caspase-3; *non-specific band.
Roles of adenosine receptors and adenosine transporter
A previous report suggested that cordycepin inhibited tumour cell growth through binding to the A3 adenosine receptor [18]. We also showed that cordycepin suppressed ER stress-induced apoptosis through the A3 adenosine receptor and adenosine transporter [13]. We tested the effects of adenosine receptor antagonists and an adenosine transport inhibitor on the pro-apoptotic action of cordycepin. Microscopic analysis showed that the cellular damage caused by cordycepin in TNF-α-treated cells was attenuated by the adenosine transport inhibitor NBTI. The A3 receptor antagonist MRS1523 also modestly inhibited the pro-apoptotic effect (Fig. 3a,b). In contrast, the A1/A2 receptor antagonist DPSPX did not reverse the effect of cordycepin.
Fig. 3.

Roles of adenosine receptors and adenosine transporter. (a, b) Cells were treated with cordycepin and tumour necrosis factor (TNF)-α in the presence of 10 µM nitrobenzylthioinosine (NBTI), 5 µM MRS1523 (MRS) or 10 nM 1,3-dipropyl-8-sulphophenylxanthine (DPSPX) and subjected to phase-contrast microscopy (a) and assessment of cell death (b). (c) NRK/NF-κB-Luc cells were stimulated with indicated agents and subjected to luciferase assay.
We further tested whether and how adenosine receptors and adenosine transporter are involved in the regulation of NF-κB by cordycepin. Reporter assay showed that, in the presence of NBTI or MRS1523, the suppressive effect of cordycepin on NF-κB was reversed significantly (Fig. 3c). Consistent with the results on apoptosis, the effect of NBTI was stronger than that of MRS1523. In contrast, treatment with DPSPX did not affect the suppressive effect of cordycepin.
Inhibition of NF-κB by cordycepin through activation of eIF2α
We reported recently that cordycepin has the potential to induce activation of eIF2α[13], a putative regulator of NF-κB [19],[20]. We examined whether or not eIF2α is involved in the regulation of NF-κB by cordycepin. As shown in Fig. 4a, cordycepin rapidly induced phosphorylation of eIF2α, and it was sustained for at least 24 h. To examine the role of eIF2α in the suppression of NF-κB by cordycepin, the effect of salubrinal, a selective inhibitor of eIF2α dephosphorylation [21], was tested. Induction of eIF2α phosphorylation by salubrinal was confirmed in NRK-52E cells by Western blot analysis (data not shown). Reporter assay showed that salubrinal significantly reduced activation of NF-κB by TNF-α (Fig. 4b). Northern blot analysis also showed that activation of eIF2α blunted NF-κB-dependent induction of MCP-1 in TNF-α-stimulated cells (Fig. 4c). Reporter assay showed that salubrinal significantly reversed degradation of IκBα in response to TNF-α (Fig. 4d). Furthermore, activation of NF-κB by over-expression of p65 was also inhibited by blockade of eIF2α (Fig. 4e).
Fig. 4.
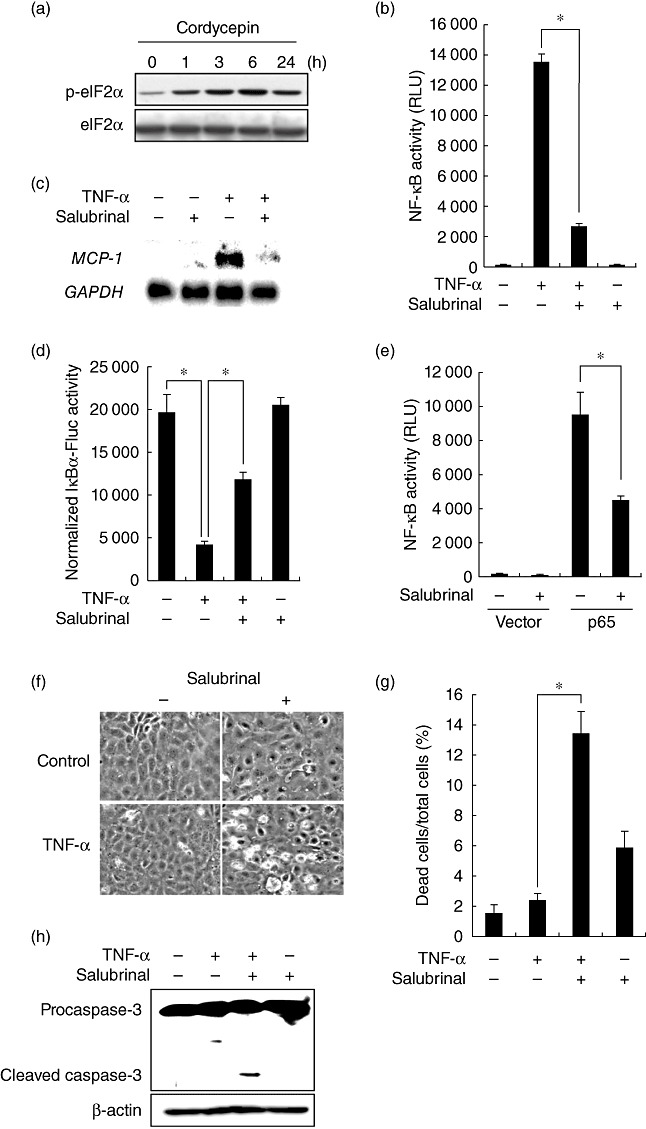
Inhibition of nuclear factor (NF)-κB by cordycepin through activation of eukaryotic translation initiation factor 2α (eIF2α). (a) Cells were treated with cordycepin for the indicated time-periods and subjected to Western blot analysis of phosphorylated eIF2α (p-eIF2α). Total protein level of eIF2α is shown at the bottom as a loading control. (b) NRK/NF-κB-Luc cells were stimulated with tumour necrosis factor (TNF)-α in the absence or presence of 50 µM salubrinal and subjected to luciferase assay. (c) Cells were treated with the indicated agents and subjected to Northern blot analysis. (d) Cells were transfected with pIκBα-Fluc or pCMV-Fluc, treated with TNF-α in the absence or presence of salubrinal and subjected to luciferase assay. The luciferase activity in pIκBα-Fluc-transfected cells was normalized by the levels of luciferase in pCMV-Fluc-transfected cells. (e) Cells were transfected with NF-κB-Luc together with empty vector or p65, treated with or without salubrinal and subjected to luciferase assay. (f,g) Cells were treated with TNF-α in the absence or presence salubrinal and subjected to phase-contrast microscopy (f) and assessment of cell death (g). (h) Cells were exposed to the indicated agents and subjected to Western blot analysis of caspase-3.
To investigate whether activation of eIF2α sensitizes cells to TNF-α-induced apoptosis, NRK-52E cells were exposed to TNF-α in the absence or presence of salubrinal. Microscopic analysis showed that salubrinal mimicked the pro-apoptotic effect of cordycepin. That is, salubrinal rendered the cells susceptible to TNF-α-induced apoptosis (Fig. 4f,g). Consistent with this result, Western blot analysis also showed that TNF-α caused cleavage of procaspase-3 only in the presence of salubrinal (Fig. 4h).
Inhibition of NF-κB through the eIF2α–mammalian target of rapamycin complex 1 (mTORC1) pathway
mTORC1 is one of the key regulators for a wide range of cell functions. We reported recently that mTORC1 has the potential to suppress activation of NF-κB by TNF-α[22]. To investigate the mechanisms underlying the suppressive effect of eIF2α on NF-κB, a link between eIF2α and mTORC1 was examined. For this purpose, cells were treated with salubrinal, and the activity of mTORC1 was evaluated by Western blot analysis. Phosphorylation of p70S6K was used as an indicator of mTORC1 activation. As shown in Fig. 5a, activation of mTORC1 was induced following treatment with salubrinal. Activation of mTORC1 was also observed in the cells treated with cordycepin (Fig. 5b). To examine whether activation of mTORC1 contributes to the suppressive effect of eIF2α on NF-κB, an effect of rapamycin (inhibitor of mTORC1) was tested. Reporter assay showed that activation of NF-κB by TNF-α was inhibited in the presence of salubrinal and was reversed partially, but significantly, by treatment with rapamycin (Fig. 5c). A similar effect was also observed by treatment with cordycepin (Fig. 5d). Of note, phosphorylation of eIF2α by cordycepin was not affected by treatment with rapamycin (Fig. 5e), suggesting that activation of eIF2α was an event upstream of mTORC1 activation in cordycepin-treated cells.
Fig. 5.
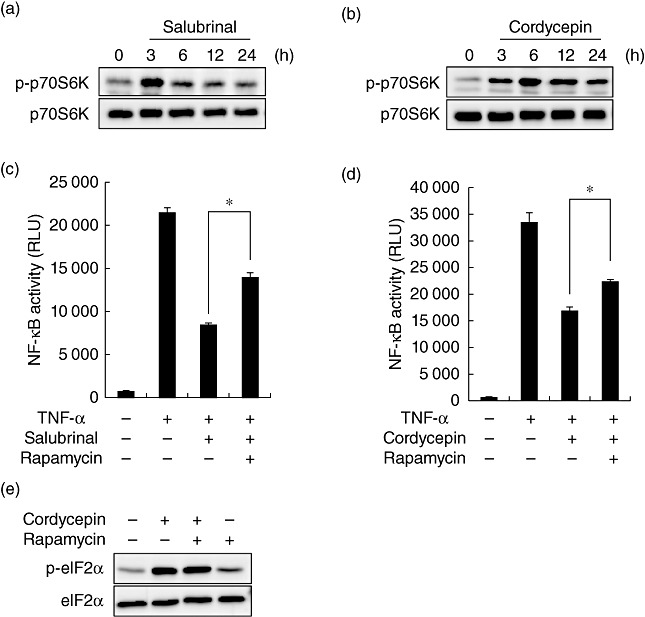
Inhibition of nuclear factor (NF)-κB through the eIF2α–mammalian target of rapamycin complex 1 (mTORC1) pathway. (a,b) Cells were treated with salubrinal (a) or cordycepin (b) for the indicated time-periods, and phosphorylation of p70S6K was evaluated by Western blot analysis. Total p70S6K protein is shown at the bottom as a loading control. (c,d) NRK/NF-κB-Luc cells were stimulated with tumour necrosis factor (TNF)-α in the absence or presence of 200 nM rapamycin and 25 µM salubrinal (c) or 200 nM rapamycin and 0·5 µg/ml cordycepin (d) and subjected to luciferase assay. (e) Cells were treated with the indicated agents for 6 h and subjected to Western blot analysis of eIF2α.
Discussion
In the present report, we demonstrate the pro-apoptotic potential of cordycepin to sensitize cells to TNF-α-induced apoptosis. This effect was ascribed to the suppression of NF-κB, a major prosurvival component involved in TNF-α signalling. We also found that the suppressive effect of cordycepin was mediated, at least in part, by the induction of the eIF2α–mTORC1 pathway. Cordycepin is known to inhibit ER stress-induced apoptosis in NRK-52E cells [13]. Our current results, together with the previous report, disclosed bidirectional regulation of apoptosis by cordycepin under distinct stress conditions.
In this report, we show that cordycepin induced phosphorylation of eIF2α. Currently, it is unclear how cordycepin activates eIF2α. One possibility is its suppressive effect on growth arrest and DNA damage gene 34 (GADD34) involved in dephosphorylation of eIF2α[23]. As we showed previously, GADD34 was expressed constitutively in unstimulated NRK-52E cells, and was suppressed by cordycepin treatment [13]. The down-regulation of basal GADD34 may be a possible mechanism underlying phosphorylation of eIF2α by cordycepin.
In the present study, we found that the pro-apoptotic effect of cordycepin was dependent on adenosine transporter and A3 adenosine receptor. Similarly, we found that both adenosine transporter and A3 adenosine receptor were involved in the suppression of NF-κB. These results are similar to our previous finding that the suppressive effect of cordycepin on ER stress-induced apoptosis was through adenosine transporter-dependent and -independent mechanisms, the latter of which required the A3 adenosine receptor [13]. Regarding the activation of eIF2α by cordycepin, however, the intracellular action via the transporter and the extracellular action via the receptor may play distinct roles. We demonstrated recently that the adenosine transporter, but not the A3 receptor, contributed to early phosphorylation of eIF2α by cordycepin. In contrast, the A3 receptor was involved in the maintenance of phosphorylated eIF2α in the later phase [13]. The contribution of adenosine transporter and adenosine receptor may be different, depending on the phase of NF-κB activation.
Currently, it is unclear how eIF2α causes activation of mTORC1. It is known that Akt up-regulates activity of mTORC1 [24]. One possible explanation might be that eIF2α causes activation of mTORC1 via Akt. However, our preliminary results did not show any positive effects of salubrinal on Akt phosphorylation (our unpublished data), excluding this possibility. Another possibility is that eIF2α may cause suppression of AMP-activated protein kinase (AMPK), a negative regulator of mTORC1 [25]. However, we found that (1) TNF-α did not induce activation of AMPK (our unpublished data) and (2) phosphorylation of AMPKα rather suppressed activation of NF-κB in cytokine-treated NRK-52E cells [26], also excluding this possibility. Further investigation will be required to elucidate mechanisms underlying the link between eIF2α and mTORC1.
In this report, we show that eIF2α-mediated activation of mTORC1 contributed to the suppression of NF-κB. How does mTORC1 down-regulate TNF-α-induced NF-κB activation? One possibility is its negative effect on Akt. In TNF-α signalling, Akt contributes to the activation of NF-κB [27]. Previous reports have suggested that phosphorylation of mTORC1 by Akt, in turn, regulates the Akt pathway negatively [28],[29]. Indeed, our preliminary results showed that basal and inducible phosphorylation of Akt was depressed by the treatment with salubrinal in TNF-α-exposed cells (our unpublished data).
We identified that mTORC1 was involved in the suppression of NF-κB by cordycepin. However, inhibition of mTORC1 by rapamycin reversed only partially the suppressive effects of cordycepin and salubrinal (Fig. 5c,d), suggesting that cordycepin may also suppress NF-κB signalling in another way, i.e. independently of mTORC1. It is known that cordycepin inhibits transcription via blockade of mRNA polyadenylation, presumably by causing chain termination following its incorporation as cordycepin triphosphate [30]. Although, in our experimental condition, cordycepin did not suppress luciferase activity driven by viral promoters and housekeeping gene promoters, we cannot exclude the possibility that inhibition of transcription is part of the suppressive effect of cordycepin on NF-κB.
Recently, we reported that cordycepin attenuated ER stress-triggered renal tubular injury in vitro and in vivo[13]. In the present report, we also show that cordycepin inhibited activation of NF-κB by TNF-α in renal tubular cells, suggesting that cordycepin is useful for the treatment of inflammatory kidney diseases. However, therapeutic utility of cordycepin on kidney diseases might have a ‘Janus face’. Although it is renoprotective in ER stress-related diseases, it could be renotoxic in some pathologies where TNF-α plays a critical role. For example, some reports have suggested that TNF-α was causative of renal tubular apoptosis in urinary tract obstruction and ischaemia–reperfusion injury [31],[32]. If so, in these pathological situations, treatment with cordycepin might exacerbate renal diseases via enhancement of apoptosis.
Several previous reports have suggested pro-apoptotic effects of cordycepin on malignant cells, whereas molecular mechanisms involved are largely unknown. Chen et al. reported that transcriptional expression of MET, a survival gene for multiple myeloma, was suppressed selectively in cordycepin-treated myeloma cells without affecting levels of other anti-apoptotic proteins including Bcl-2, XIAP, Mcl-1 and survivin. They also showed that stabilization of p53 was not involved in the pro-apoptotic effect of cordycepin [11]. Our current results indicate a novel mechanism by which cordycepin exerts its pro-apoptotic effect, especially under pathological conditions where NF-κB plays a significant role. For example, NF-κB has tumour-promoting effects in many types of cancers [33]. The mechanism described here may explain the anti-tumour effect of cordycepin in vivo, which has been reported by some investigators [5],[34].
Acknowledgments
We thank Dr Kazuo Umezawa (Keio University) for the gift of DHMEQ, and Dr David Piwnica-Worms for kindly providing us with pCMV-Fluc and pIκBα-Fluc. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (no. 20390235) to M. Kitamura.
Disclosure
None.
References
- 1.Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–7. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 2.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–3. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 3.Wajant H, Henkler F, Scheurich P. The TNF-receptor-associated factor family: scaffold molecules for cytokine receptors, kinases and their regulators. Cell Signal. 2001;13:389–400. doi: 10.1016/s0898-6568(01)00160-7. [DOI] [PubMed] [Google Scholar]
- 4.Cho HJ, Cho JY, Rhee MH, Kim HS, Lee HS, Park HJ. Inhibitory effects of cordycepin (3′-deoxyadenosine), a component of Cordyceps militaris, on human platelet aggregation induced by thapsigargin. J Microbiol Biotechnol. 2007;17:1134–8. [PubMed] [Google Scholar]
- 5.Nakamura K, Konoha K, Yoshikawa N, et al. Effect of cordycepin (3'-deoxyadenosine) on hematogenic lung metastatic model mice. In Vivo. 2005;19:137–41. [PubMed] [Google Scholar]
- 6.Koc Y, Urbano AG, Sweeney EB, McCaffrey R. Induction of apoptosis by cordycepin in ADA-inhibited TdT-positive leukemia cells. Leukemia. 1996;10:1019–24. [PubMed] [Google Scholar]
- 7.De Julian-Ortiz JV, Galvez J, Munoz-Collado C, Garcia-Domenech R, Gimeno-Cardona C. Virtual combinatorial syntheses and computational screening of new potential anti-herpes compounds. J Med Chem. 1999;42:3308–14. doi: 10.1021/jm981132u. [DOI] [PubMed] [Google Scholar]
- 8.Sugar AM, McCaffrey RP. Antifungal activity of 3'-deoxyadenosine (cordycepin) Antimicrob Agents Chemother. 1998;42:1424–7. doi: 10.1128/aac.42.6.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim HG, Shrestha B, Lim SY, et al. Cordycepin inhibits lipopolysaccharide-induced inflammation by the suppression of NF-κB through Akt and p38 inhibition in RAW264.7 macrophage cells. Eur J Pharmacol. 2006;545:192–9. doi: 10.1016/j.ejphar.2006.06.047. [DOI] [PubMed] [Google Scholar]
- 10.Wu WC, Hsiao JR, Lian YY, Lin CY, Huang BM. The apoptotic effect of cordycepin on human OEC-M1 oral cancer cell line. Cancer Chemother Pharmacol. 2007;60:103–11. doi: 10.1007/s00280-006-0354-y. [DOI] [PubMed] [Google Scholar]
- 11.Chen LS, Stellrecht CM, Gandhi V. RNA-directed agent, cordycepin, induces cell death in multiple myeloma cells. Br J Haematol. 2008;140:682–91. doi: 10.1111/j.1365-2141.2007.06955.x. [DOI] [PubMed] [Google Scholar]
- 12.Shi P, Huang Z, Tan X, Chen G. Proteomic detection of changes in protein expression induced by cordycepin in human hepatocellular carcinoma BEL-7402 cells. Methods Find Exp Clin Pharmacol. 2008;30:347–53. doi: 10.1358/mf.2008.30.5.1186085. [DOI] [PubMed] [Google Scholar]
- 13.Kitamura M, Kato H, Saito Y, et al. Aberrant, differential and bidirectional regulation of the unfolded protein response towards cell survival by 3′-deoxyadenosine. Cell Death Differ. 2011;18:1876–88. doi: 10.1038/cdd.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ariga A, Namekawa J, Matsumoto N, Inoue J, Umezawa K. Inhibition of tumor necrosis factor-α-induced nuclear translocation and activation of NF-κB by dehydroxymethylepoxyquinomicin. J Biol Chem. 2002;277:24625–30. doi: 10.1074/jbc.M112063200. [DOI] [PubMed] [Google Scholar]
- 15.Gross S, Piwnica-Worms D. Monitoring proteasome activity in cellulo and in living animals by bioluminescent imaging: technical considerations for design and use of genetically encoded reporters. Methods Enzymol. 2005;399:512–30. doi: 10.1016/S0076-6879(05)99035-6. [DOI] [PubMed] [Google Scholar]
- 16.Kitamura M, Suto T, Yokoo T, Shimizu F, Fine LG. Transforming growth factor-β1 is the predominant paracrine inhibitor of macrophage cytokine synthesis produced by glomerular mesangial cells. J Immunol. 1996;156:2964–71. [PubMed] [Google Scholar]
- 17.Rollins BJ, Morrison ED, Stiles CD. Cloning and expression of JE, a gene inducible by platelet-derived growth factor and whose product has cytokine-like properties. Proc Natl Acad Sci USA. 1988;85:3738–42. doi: 10.1073/pnas.85.11.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamura K, Yoshikawa N, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M. Antitumor effect of cordycepin (3′-deoxyadenosine) on mouse melanoma and lung carcinoma cells involves adenosine A3 receptor stimulation. Anticancer Res. 2006;26:43–7. [PubMed] [Google Scholar]
- 19.Jiang HY, Wek SA, McGrath BC, et al. Phosphorylation of the α subunit of eukaryotic initiation factor 2 is required for activation of NF-κB in response to diverse cellular stresses. Mol Cell Biol. 2003;23:5651–63. doi: 10.1128/MCB.23.16.5651-5663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng J, Lu PD, Zhang Y, et al. Translational repression mediates activation of NF-κB by phosphorylated translation initiation factor 2. Mol Cell Biol. 2004;24:10161–8. doi: 10.1128/MCB.24.23.10161-10168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyce M, Bryant KF, Jousse C, et al. A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science. 2005;307:935–9. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 22.Nakajima S, Hiramatsu N, Hayakawa K, et al. Selective abrogation of BiP/GRP78 blunts activation of NF-κB through the ATF6 branch of the UPR: involvement of C/EBPβ and mTOR-dependent dephosphorylation of Akt. Mol Cell Biol. 2011;31:1710–18. doi: 10.1128/MCB.00939-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J Cell Biol. 2001;153:1011–22. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 25.Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009;196:65–80. doi: 10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chi Y, Li K, Yan Q, et al. Nonsteroidal anti-inflammatory drug flufenamic acid is a potent activator of AMPK. J Pharmacol Exp Ther. 2011;339:257–66. doi: 10.1124/jpet.111.183020. [DOI] [PubMed] [Google Scholar]
- 27.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-κB activation by tumour necrosis factor requires the Akt serine–threonine kinase. Nature. 1999;401:82–5. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 28.Tremblay F, Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem. 2001;276:38052–60. doi: 10.1074/jbc.M106703200. [DOI] [PubMed] [Google Scholar]
- 29.Park S, Zhao D, Hatanpaa KJ, et al. RIP1 activates PI3K-Akt via a dual mechanism involving NF-κB-mediated inhibition of the mTOR-S6K-IRS1 negative feedback loop and down-regulation of PTEN. Cancer Res. 2009;69:4107–11. doi: 10.1158/0008-5472.CAN-09-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Müller WE, Seibert G, Beyer R, Breter HJ, Maidhof A, Zahn RK. Effect of cordycepin on nucleic acid metabolism in L5178Y cells and on nucleic acid-synthesizing enzyme systems. Cancer Res. 1977;37:3824–33. [PubMed] [Google Scholar]
- 31.Misseri R, Meldrum DR, Dinarello CA, et al. TNF-α mediates obstruction-induced renal tubular cell apoptosis and proapoptotic signaling. Am J Physiol Renal Physiol. 2005;288:F406–11. doi: 10.1152/ajprenal.00099.2004. [DOI] [PubMed] [Google Scholar]
- 32.Choi DE, Jeong JY, Lim BJ, Na KR, Shin YT, Lee KW. Pretreatment with the tumor necrosis factor-α blocker etanercept attenuated ischemia–reperfusion renal injury. Transplant Proc. 2009;41:3590–6. doi: 10.1016/j.transproceed.2009.05.042. [DOI] [PubMed] [Google Scholar]
- 33.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol. 2011;12:715–23. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 34.Yoshikawa N, Nakamura K, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M. Antitumour activity of cordycepin in mice. Clin Exp Pharmacol Physiol. 2004;31(Suppl. 2):S51–3. doi: 10.1111/j.1440-1681.2004.04108.x. [DOI] [PubMed] [Google Scholar]