

Abstract
OTHER THEMES PUBLISHED IN THIS IMMUNOLOGY IN THE CLINIC REVIEW SERIES
Metabolic diseases, host responses, cancer, autoinflammatory diseases, allergy.
Type 1 diabetes (T1D) is an autoimmune disease arising as a consequence of a misdirected T cell response to the pancreatic beta cell. In recent years, there has been a growing interest in the innate immune system as a regulator of disease development. Genome-wide association studies have identified diabetes-associated polymorphisms in genes encoding proteins with functions related to the innate immune response. Moreover, enteroviruses, known to activate a strong innate immune response, have been implicated in the disease pathogenesis. In this review, we discuss the innate immune response elicited by enteroviruses and how this response may regulate T1D development.
Keywords: enterovirus, innate immunity, interferon, RIG-I-like receptors, Type 1 diabetes
Introduction
Type 1 diabetes (T1D) is a chronic metabolic disease resulting from the loss of functional pancreatic beta cells. Although it is not entirely clear how beta cells are damaged and destroyed, numerous observations have implicated autoreactive T cells in this process. Both genetic and environmental factors regulate disease susceptibility. While it is clear that the major histocompatibility complex (MHC) genes on chromosome 6 provide a strong genetic susceptibility, recent genome-wide association studies have identified additional T1D risk loci containing candidate genes with functions important for the innate immune system. This is of interest because infections with enteroviruses, a group of viruses known to activate a strong innate immune response, have been implicated in T1D [1],[2]. Here we review current knowledge on the early innate immune response to enterovirus infections. We also discuss how the T1D-associated polymorphisms in genes related to innate immunity may affect the response to infection and thereby possibly susceptibility to T1D development.
Viral recognition and the innate immune response to viral infection
The innate immune system
The innate immune system consists of anatomical barriers, secreted molecules and several different types of immune cells such as eosinophils, mono- and polynuclear phagocytes and natural killer (NK) cells. It provides the first line of defence against microbes and stimulates the subsequent activation of the more specific adaptive immune response. During an acute viral infection, efficient recognition of the infecting virus is imperative for a robust immune response to evolve [3],[4].
Viral recognition via pattern recognition receptors (PRRs)
Detection of invading pathogens occurs via pattern recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) (examples of PRRs discovered in humans are listed in Table 1) [3],[5]–[7]. Viruses are detected mainly via two receptor families, the Toll-like receptors (TLRs) and the retinioic acid inducible gene 1 (RIG-I) like receptors (RLRs) [8]. Of the 10 TLRs described in humans (13 in mice) TLR-3, -7, -8 and -9 have been identified as sensors of viral RNA or DNA. However, there are studies suggesting a role for additional TLRs in viral recognition, such as TLR-2, -4 and -6 [3],[9].
Table 1.
| Receptor | Cellular localization | Ligands | Main adaptor protein used for signalling |
|---|---|---|---|
| TLR-1 | Cell surface | Triacyl lipopeptides | MyD88 |
| TLR-2 | Cell surface | Peptidoglycan, dsDNA viruses, haemagglutinin | MyD88 |
| TLR-3 | Endosome | ssRNA viruses, dsRNA viruses | TRIF |
| TLR-4 | Cell surface and endosome | Lipopolysaccharide | MyD88 and TRIF |
| TLR-5 | Cell surface | Flagellin from flagellated bacteria | MyD88 |
| TLR-6 | Cell surface | Diacyl lipopeptides from mycoplasma, lipoteic acid | MyD88 |
| TLR-7 | Endolysosome | ssRNA viruses | MyD88 |
| TLR-8 | Endolysosome | ssRNA viruses | MyD88 |
| TLR-9 | Endolysosome | CpG motifs, DNA viruses | MyD88 |
| RIG-I | Cytoplasm | Short dsRNA with triphosphate or monophophate at 5′ end, several negative sense ssRNA viruses | IPS-1/MAVS/Cardif/VISA |
| MDA5 | Cytoplasm | Long dsRNA, several positive sense ssRNA viruses | IPS-1/MAVS/Cardif/VISA |
| DDX1/DDX21/DDX36 | Cytoplasm | dsRNA | TRIF |
TLR: Toll-like receptor; RIG-1: retinoic acid-inducible gene 1 protein; MDA: melanoma differentiation-associated protein; DDX: human dead-box protein; MyD88: myeloid differentiation protein 88; TRIF: TIR-domain-containing adapter-inducing interferon-β; IPS-1: interferon-β promoter stimulator 1; MAVS: mitochondrial anti-viral-signalling protein; VISA: virus-induced signalling adapter.
TLR-3, -7, -8 and -9 are expressed in intracellular vesicles (e.g. endosomes, endoplasmatic reticulum, lysosomes and endolysosomes), whereas TLR-1, -2, -4, -5 and -6 are expressed primarily on the cell surface [5],[9]. The RLR family consists of the DExD/H helicases melanoma differentiation-associated factor 5 [MDA5, also denoted interferon (IFN)-induced helicase 1, IFIH1], RIG-I and laboratory of genetics and physiology 2 (LGP2), which are expressed in the cytoplasm. MDA5 and RIG-I recognize viral RNA, whereas LGP2 may act as a regulator of RIG-I and MDA5 function [3],[6]. Recently, three additional members of the DExD/H helicase family – DDX1, DDX21 and DHX36 – were shown to contribute to the recognition of double-stranded (ds)RNA in dendritic cells (DCs) [7]. The continuous identification of new receptors recognizing viral genomes indicates that additional PRRs are yet to be discovered.
Engagement of the cognant PRR ligands activates downstream signalling cascades, which lead to the production of proinflammatory cytokines such as type I interferons (IFNs) and interleukins. The PRRs utilize specific adaptor proteins that converge in the activation of three families of transcription factors, nuclear factor kappa B (NF-κB), IFN regulatory factors (IRFs) and ATF-2/cJun. TLRs signal primarily via engagement of TIR domain-containing adaptor proteins such as myeloid differentiation protein 88 (MyD88), TIR-domain-containing adapter-inducing IFN-β (TRIF, also denoted TICAM1), Toll-interleukin (IL)-1 receptor (TIR) domain-containing adapter protein (TIRAP, also denoted Mal) and translocating train-associating membrane (TRAM), while the RLRs activate the IFN-β promoter stimulator-1 (IPS-1, also denoted MAVS, Cardif or VISA) located in the mitochondrial membrane (Table 1) [3],[5]–[7].
The role of IFNs in the early immune response to an acute viral infection
The interaction between viral sensors and viral nucleic acids induces the production of type I IFNs. Although DCs and macrophages appear to be the main producers of these cytokines, several studies have shown that non-haematopoietic cells also can produce type I IFNs during virus infections [10]. The strong anti-viral activities of the IFNs are attributed to their ability to induce the transcription of hundreds of genes, many with distinct anti-viral activities such as protein kinase R (PKR) and 2′5′-oligoadenylate synthase (2′5′OAS). This results in the activation of a so-called anti-viral state in responding cells that aims to block viral replication and prevent uninfected cells from becoming infected [4]. The IFNs also induce or up-regulate the expression of PRRs and related signalling molecules [e.g. MDA5, RIG-I and signal transducers and activators of transcription (STATs)], allowing cells to recognize and respond more efficiently to infecting viruses [4],[11]. In addition to the direct anti-viral actions, IFNs modulate various cellular responses of the innate immune system including cytotoxicity of NK cells and maturation of antigen-presenting cells (APCs), thereby contributing to the killing of virus-infected cells and the shaping of the adaptive immune response [9],[12].
Innate immune response to enteroviruses
Enteroviruses are single-stranded RNA viruses belonging to the picornavirus family. Examples of enteroviruses are Coxsackie-, polio- and human rhinoviruses. The enteroviruses implicated mainly in T1D are the Coxsackie type B viruses (CVBs) [13]–[15]. An intact innate immune response is critical for host survival during enterovirus infection. The rapid induction of IFNs is important, as mice unresponsive to type I IFNs or lacking IFN-β have an increased and early mortality after, for example, CVB infection [16]–[18]. Studies in mice lacking selected genes with known anti-viral activity [e.g. inducible nitric oxide synthase (iNOS), PKR and RNaseL] suggest that the early protection provided by the IFNs is dependent upon the induced expression of proteins involved in anti-viral defence [19]–[21]. Also, the pancreatic beta cell relies heavily on IFNs, as pancreatic beta cells that cannot respond to these cytokines succumb to enterovirus infection resulting in T1D in the host [17],[22].
Recognition of enterovirus
From studies in mice and in-vitro experiments using human cells we are beginning to gain some understanding into how the host senses enteroviruses such as CVBs. In-vitro experiments have indicated that CVBs can be recognized by TLR-4, -7 and -8 [23]–[25]. TLR-7 and -8 are important for human cardiac cells as well as plasmacytoid DCs to induce an inflammatory response upon CVB infection [23]. Recognition of CVB4 and subsequent cytokine secretion was shown to be dependent upon TLR-4 in pancreatic cell lines [24]. However, several findings indicate that signalling via TLR-4, -7 and -8 contribute to immunopathology rather than protection of the host. Mice deficient in MyD88, an adaptor protein in the signalling cascades induced by, for example, TLR-4, -7 and -8, did not demonstrate increased pathology and mortality after infection with CVB. Instead, the animals had a better survival and less infiltrating lymphocytes in the heart and pancreas compared to wild-type mice [26]. A separate study showed that CVB3 infection resulted in lower viral replication and pathology in the heart of TLR-4 knock-out mice compared to wild-type littermates [27].
More important roles in protecting the host seem to be played by TLR-3 and MDA5. Both tlr3−/− and mda5/ifih1−/− animals showed heightened mortality after infection with CVB [28]–[31]. In-vivo and in-vitro studies demonstrated high levels of CVB3 replication and an impaired inflammatory response in the absence of TLR-3 [29],[30]. Mice lacking TRIF, an adaptor protein of TLR-3 and -4 signalling, showed higher viral titres in sera and increased mortality upon CVB infection [29],[32]. However, treatment of trif−/− mice with IFN-β improved survival significantly [32]. Mice lacking MDA5 suffered from increased tissue damage and heightened mortality after infection with CVB, and this was paralleled with a reduced capacity to induce IFNs [28],[31], further stressing the importance for IFNs in protection against enterovirus.
Collectively, studies on TLRs and RLRs in CVB infection highlight the possible double-edged outcome of viral recognition. On one hand, TLR-3 and MDA5 provide protection against viral-induced damage. On the other hand, MyD88-dependent TLR signals contribute to tissue damage and impaired host survival. Studies using MDA5- or TLR-3-deficient animals also suggest that one signalling pathway alone is not sufficient to induce a proper immune response and that the actions of several receptors are required.
The possible role of the innate immune response to enteroviruses in regulating T1D
How does an enterovirus infection contribute to T1D development?
Numerous observations, ranging from clinical case reports to results emanating from large epidemiological studies, indicate that enterovirus infections play a role in T1D. The virus has been isolated from patients at disease onset. It has been found more frequently in blood or serum from T1D patients compared to control individuals, the appearance of autoantibodies coincide with enterovirus infections in some individuals, and the virus has been found more often in the gut and pancreatic islets of T1D patients compared to control individuals [13]–[15],[33]. There are, however, studies that have failed to confirm these observations, opening the possibility that enterovirus infections contributes to some, but not all, cases of T1D. Studies on pancreata from humans with T1D have indeed demonstrated different histopathological patterns, indicating the existence of different disease mechanisms leading to the same outcome, clinical T1D [34],[35].
How do enterovirus infections contribute to diabetes development? There is currently no answer to this question. Numerous mechanisms have been proposed, and some have been backed up by experimental studies. The mechanisms that have received most attention are direct infection of target cells and bystander activation of autoreactive T cells, and it is possible that these mechanisms act simultaneously [36],[37].
The discovery of enterovirus RNA and/or enterovirus proteins in pancreatic islets of T1D patients [38]–[41], and in-vitro studies demonstrating that infection of human and rodent islets leads to functional impairments and widespread beta cell death (e.g. [17],[20],[42]–[45]) support the idea that direct infection of the islets contribute to disease development. Although many enteroviruses such as CVBs have a lytic effect when studied in cell culture, the observations in human T1D suggest that the virus is establishing a persistent infection of the islet cells. That enteroviruses can cause persistent infections is well documented (e.g. [46]–[48]). Other picornaviruses such as Theilers murine encephalomyelitis virus can persist as a ‘smouldering’ infection despite the activation of a strong immune response by the host (reviewed in [49]). However, if and how a persistent infection of beta cells contributes to T1D development remains to be clarified.
A virus infection often results in the production of proinflammatory cytokines (e.g. IFNs) and the activation of APCs such as DCs. If this coincides with tissue damage, endogenous antigens might be taken up and presented by the APC. In an individual with a genetic predisposition, this may result in the aberrant activation of self-reactive T cells, so-called ‘bystander’ activation. Support for this mechanism in enterovirus-induced T1D comes from studies in animal models. A CVB4 infection results in the activation of beta cell-specific T cells and T1D in non-obese diabetic (NOD) mice carrying a T cell receptor transgene specific for a MHC class II-restricted beta cell antigen (BDC2·5 mice). Diabetes development in this model was dependent upon a pre-existing insulitis [50], suggesting that the virus accelerated an already ongoing diabetogenic process rather than initiating it. Interestingly, recent data from the Diabetes and Autoimmunity Study in the Young (DAISY) suggests that progression from islet autoimmunity to clinical disease may increase after an enterovirus infection [12], and is thus in agreement with the animal model. Yet other mechanisms by which enteroviruses may trigger T1D have been suggested [36], but for these experimental evidence is still mainly lacking.
How does the innate immune response to enteroviruses regulate T1D development?
As mentioned above, studies in experimental models have demonstrated that the innate immune response is crucial for host survival during enterovirus infection. A weak innate immune response, for example by the inability to mount an efficient IFN response, is associated with unrestricted viral replication and increased viral spread. Tissue damage may ensue as a direct effect of the infecting virus, and it is possible that the inefficient immune response favours viral persistence. A strong innate immune response is important for efficient restriction of viral replication and spread. However, immunopathologies may appear in the wake of a very potent immune response (Fig. 1). This is exemplified by lower mortality and less cardiac and pancreatic inflammation in mice lacking MyD88 [26].
Fig. 1.
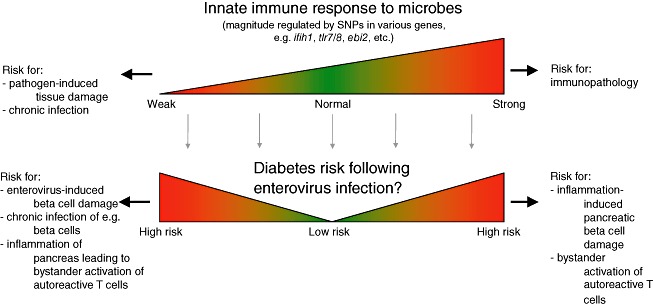
An imbalanced innate immune response may have adverse consequences for the outcome of enterovirus infections and thereby contribute to Type 1 diabetes (T1D) development. During an acute viral infection, the innate immune system plays an important role by limiting early viral replication and spread, thereby allowing the host to mount an adaptive immune response. Although a strong innate immune response may be efficient in limiting viral dissemination and damage, it may also contribute to unwanted tissue damage. A weak response may favour viral replication and spread and thereby also tissue damage. Numerous gene loci associated with risk for T1D development contain gene candidates involved in the innate immune response. While it remains unclear how these different gene versions are involved in the disease process, some are likely to affect the response to an enterovirus infection.
Without knowledge of how enterovirus infections are involved in the aetiopathogenesis of T1D, only speculations can be made regarding how the innate immune response to infection may affect disease development. Assuming that enteroviruses need to access the pancreas and the beta cell in order to promote T1D, then it can be expected that a strong innate immune response to the virus will be protective. By already mounting an efficient response in the intestine, the port of entry for most enteroviruses, the host is likely to prevent systemic spread. A robust IFN response will lower permissiveness of parenchymal cells to infection and lower the risk for virally instigated damage [17],[51]. In this scenario a weak response would be counterproductive, as it may allow the virus to replicate and spread systemically. The risk for beta cells to become infected would increase and the virus may have a better chance to establish persistent infection. In this respect, it is of interest to note that enteroviruses have been found more frequently in duodenum samples and pancreatic islets from T1D patients than in healthy controls [38],[40],[41],[52]. At this stage it is, however, unknown whether T1D patients are infected more frequently by enteroviruses or increasingly permissive to the establishment of persistent enterovirus infections compared to healthy individuals.
An alternative scenario is that the innate immune response to infection is triggering the activation of self-reactive T cells. The stronger the response, the greater the risk that T cells become activated. Several autoimmune diseases are associated with the excess production of IFNs [53]. At first thought, it is tempting to propose that a strong innate immune response to the virus would automatically increase the risk for bystander activation of autoreactive T cells. This may, however, not be the case, as a strong response is more likely to prevent the virus from infecting its host productively and thereby also hinder the virus to access the pancreas. In contrast, a weak response increases the risk for systemic spread and the induction of an innate immune response in tissues targeted by the virus. Thereby, infection of an individual with a weak initial response to the virus may result in higher systemic levels of proinflammatory cytokines and an increased expression of anti-viral defence genes than in an individual with a potent initial response. This scenario is supported by studies demonstrating that ifih1/mda5−/− mice in comparison to wild-type mice have an impaired ability to respond to the virus express higher levels of IFN-β, 2′5′OAS and CXCL10 mRNA in pancreas and liver after infection with CVB3 [28]. In short, a weak initial response to the virus may contribute later to the activation of autoreactive T cells.
T1D-associated polymorphisms in genes important for the innate immune response – could they alter the immune response to enteroviruses?
Genetic susceptibility to T1D is regulated primarily by the HLA gene locus. Genome-wide association studies have identified additional loci containing regions associated with altered risk for T1D development. Some contain candidate genes with a role in the innate immune response, and it is possible that the T1D-associated non-synonymous single nucleotide polymorphisms (SNPs) alter the biological functions of the translated proteins. Considering the prominent role for the innate immune response during enterovirus infection, it is possible that some of these proteins contribute to the host defence against these viruses. Thus, learning more about the functions of the proteins these genes encode and how the different versions affect the host response to enterovirus infections may provide additional clues on the aetiopathogenesis of T1D. Below we discuss some of the T1D-associated candidate genes and how altered functions of those may affect the host response to infection.
MDA5/IFIH1
In 2006 Todd and colleagues discovered a novel T1D susceptibility locus, in which the main candidate gene was identified as the mda5/ifih1 gene [54]. Soon thereafter it became clear that the mda5/ifih1 gene contains several SNPs associated with altered risk for T1D development [55], and the associations have been confirmed in other patient cohorts (e.g. [56],[57]). Moreover, genetic variants of mda5/ifih1 have been associated with other autoimmune conditions including psoriasis [58], autoimmune thyroid disease [59] and systemic lupus erythematosis (SLE) [60],[61].
The ifih1/mda5 gene encodes the RLR family member MDA5, known to recognize dsRNA generated during the replication of certain picornaviruses (e.g. enteroviruses), as well as synthetic dsRNA (poly I : C). MDA5 is important for the induction of type I IFN production by members of the picornavirus family but also other types of viruses, such as West Nile, Dengue and measles virus [10]. This viral sensor is expressed at low levels in numerous cell types, including human pancreatic islets, and its expression is induced by, for example, IFNs [11]. Using mda5/ifih1−/− mice, we and others have demonstrated that MDA5 is crucial for host survival during CVB3 infection and that it plays an important role in regulating viral replication [28],[31]. These observations strongly implicate MDA5 in the host response to enterovirus infections.
How the different T1D-associated SNPs affect the function of the MDA5 protein remains to be established in detail. In an experimental system based on over-expression of different MDA5 mutants in mda5/ifih1−/− cells it was found that some, but not all, of the the SNPs associated with lower risk for T1D development led to defective binding of dsRNA (poly I : C) and/or reduced induction of IFN-β mRNA expression following poly I : C stimulation [62]. Consistent with predictions made based upon the MDA5 protein structure, the common coding-change variant rs1990760 (A946T) did not demonstrate altered functions in this assay. Protective mda5/ifih1 rs1990760 haplotypes have, however, been associated with reduced MDA5 expression levels and/or function following stimulation with IFN-β or poly I : C [56],[57],[63], although some studies have not been able to confirm this [64]. Collectively, most of these observations suggest that lower expression and/or functional activity of MDA5 is protective in T1D. A contrasting observation regarding the rs1990760 (A946T) SNP was, however, made in patients with SLE; patients who had anti-dsDNA antibodies and were homozygous for the rs1990760 T (946T) SNP, the genotype associated with increased risk for autoimmune diseases, demonstrated lower serum IFN-α levels than those being homozygous for the protective allele, rs1990760 A (A946) [65]. This study highlights the complexity of studies on how gene variants affect the immunological response and biological outcomes. Deficiencies in one immune function may be compensated by other mechanisms, and results from in-vitro studies may not translate readily to the in-vivo conditions.
To date, no studies have shown how the T1D-associated genetic variants of mda5/ifih1 affect the host response to enteroviruses. Such studies are more difficult to perform and interpret, as the viruses have many means to modulate intracellular signalling pathways and cytokine production (e.g. [66]). None the less, it may be speculated that a gain-of-function mechanism is beneficial for viral clearance but may contribute in parallel to the activation of self-reactive T cells by bystander mechanisms. A loss-of-function mechanism may, as demonstrated in mda5/ifih1−/− mice [28],[31], instead increase the risk for systemic viral spread, tissue pathology and possibly the establishment of persistent infections. Increased knowledge on how the different polymorphisms in mda5/ifih1 affect the host immune response to enterovirus infections may uncover mechanisms by which these viruses contribute to T1D development.
DHCR7 and CYP2R1, genes involved in vitamin D metabolism
Vitamin D is known historically for its importance in calcium homeostasis and bone mineralization. More recently, it has been recognized as a modulator of both the innate and the adaptive immune system [67],[68]. Vitamin D is obtained via sunlight exposure to the skin, diet and supplements. Following sun exposure, the biologically active form of vitamin D, 1,25-dihydroxyvitamin D3, is produced from 7-dehydrocholesterol. This process involves several enzymatic reactions conducted by specific enzymes, including 7-dehydrocholesterol reductase (DHCR7) that catalyzes the conversion of 7-dehydrocholesterol to cholesterol, and CYP2R1 that hydroxylates 25-dihydroxyvitamin D3 to the active form, 1,25-dihydroxyvitamin D3. Circulating vitamin D levels are regulated not only by the amount of sun exposure and vitamin D content in the diet, but also polymorphisms in genes involved in vitamin D production such as DHCR7 and CYP2R1 [69].
Vitamin D signals by the binding of 1,25-dihydroxyvitamin D3 to the intracellular vitamin D receptor (VDR) which, in turn, forms homodimers or VDR/retinoid X receptor (RXR) heterodimers. This complex translocates to the nucleus, where it binds to vitamin D response elements (VDREs). VDREs have been identified in genes known to participate in processes such as cell proliferation, differentiation and immunomodulation, and also appear to be represented near genes associated with autoimmune diseases such as T1D, rheumatoid arthritis and Crohn's disease [68],[70].
Numerous epidemiological studies have suggested that vitamin D deficiency plays a role in T1D (reviewed in [68]). The disease has a seasonal onset, with an increased number of patients diagnosed during the winter months than the summer months. Newly diagnosed T1D patients have lower levels of 25-dihydroxyvitamin D3 than healthy controls. Moreover, polymorphisms in the genes encoding DHCR7 and CYP2R1 genes have been associated with T1D risk [71]–[73]. Initial studies using small numbers of patients indicated that certain SNPs in the VDR gene were also associated with increased risk for T1D development, but this was not confirmed in a study using a larger patient cohort (reviewed in [68]). A recent study showed that serum levels of the vitamin D-binding protein (VDBP), a protein that binds and transports vitamin D in the circulation, were significantly lower in T1D patients compared to controls [74]. The biological consequences of low VDBP, however, remain to be established. Moreover, how low vitamin D levels contribute to T1D development is not known, but it has been speculated that they may be related to its immunomodulatory effects.
Recent studies have brought attention to the anti-viral activities of vitamin D [67]. Vitamin D deficiency appears to increase the risk for viruses infecting the respiratory tract. SNPs in the gene encoding the VDR are associated with severe outcomes of respiratory syncytial virus (RSV) infections. The exact mechanisms by which vitamin D exerts its anti-viral activity have not been identified fully. Some of its anti-microbial effects have been ascribed to the induced expression of anti-microbial peptides LL37 (cathelicidin) and human beta defensin 2 [67], and a recent study demonstrated that vitamin D alone triggered the expression of IFN-β and MxA (MxA has anti-viral functions) in hepatoma cells [75]. At present it is also unknown whether vitamin D status affects the outcome of enterovirus infections. However, CVB3-induced cardiac fibrosis was reduced in mice treated with the vitamin D analogue ZK191784 [75]. Protection from fibrosis was accompanied by a reduced expression of the signal-transducing molecules phospho-extracellular-regulated kinase (pERK) and phospho-protein kinase B (pAkt), as well as fibrosis-inducing proteins, in heart tissue. Viral loads in hearts on day 8 post-infection were, however, not different between controls and animals treated with ZK191784, indicating that treatment with the analogue led mainly to the prevention of immunopathology. Based on the hitherto documented anti-viral effects of vitamin D in other virus infections, and the epidemiological observations linking vitamin D deficiency to heightened susceptibility to viral infections, it may be reasonable to hypothesize that suboptimal vitamin D levels increase risk for enterovirus infections. The gene polymorphisms associated both with low production of the active form of vitamin D and heightened risk for T1D may therefore increase permissiveness to infection.
TLR-7 and -8
Additional T1D susceptibility genes or candidate genes that may have a function in the host response to enterovirus infections are the genes encoding TLR-7 and -8. As mentioned above, TLR-7 and -8 are important for the host recognition of these viruses [23],[25]. An SNP located 30 kb centromeric of the candidate genes TLR-7 and -8 is associated with increased risk for the disease [76]. It is currently unknown whether this SNP or variants in linkage disequilibrium with it affect the expression of TLR-7 or -8; however, such change could potentially alter the magnitude of the host response to enterovirus infection.
EBI2 (GPR183)
The Epstein–Barr virus-induced gene 2 (EBI2, also denoted GPR183) negatively regulates an IRF-7-driven inflammatory network enriched for genes involved in the anti-viral response including the transcription factor IRF7 itself. A T1D-associated SNP was identified recently within the EBI2 gene (rs9585056, [77]). The minor allele confers increased risk for T1D development and appears to lower EBI2 gene expression, thereby increasing the expression of IRF7-driven genes. As IRF7 regulates many genes, including type I IFNs, it is possible that individuals harbouring this allele have a stronger immune response to enterovirus infection. This could, on one hand, provide a better protection from the virus, but on the other hand also increase the risk for a bystander activation of autoreactive T cells if the virus gains access to the pancreas.
Concluding remarks
Via animal models and in-vitro studies using human cells and tissues we have come closer to understanding the intricate immune response to enterovirus infection. It is evident that imbalanced responses may result in immunopathology when the proinflammatory response is not controlled, and inefficient virus clearance leading to virally instigated damage when the response is too weak. SNPs associated with altered risk for T1D development have been found in genes presumably playing important roles in the innate immune response to enteroviruses. While we are beginning to understand partly how these gene variants affect gene functions, studies on host gene–microbe interactions are still in their infancy. The increasing complexity of the accumulating data highlights the need for future studies in this area of research. Although it will be a challenge to understand the biological complexity, such knowledge may enable us to decipher pathways involved in the regulation of T1D.
Acknowledgments
K. L. and M. F.-T. are supported by the Karolinska Institutet, Stockholm, Sweden. Studies on enteroviruses in the M. F.-T. laboratory are supported by the Swedish MRC, the Swedish Diabetes Association Research Foundation, the Swedish Child Diabetes Foundation, the European Union (Collaborative project PEVNET) and the European Foundation for the Study of Diabetes. M.H. is supported by Oxford University, Oxford, U.K.
Disclosure
The authors have no conflicts of interest to declare.
References
- 1.Coppieters KT, Wiberg A, Tracy SM, von Herrath MG. Immunology in the clinic review series; focus on type 1 diabetes and viruses: the role of viruses in type 1 diabetes: a difficult dilemma. Clin Exp Immunol. 2012;168:5–11. doi: 10.1111/j.1365-2249.2011.04554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stene LC, Rewers M. Immunology in the clinic review series; focus on type 1 diabetes and viruses: the enterovirus link to type 1 diabetes: critical review of human studies. Clin Exp Immunol. 2012;168:12–23. doi: 10.1111/j.1365-2249.2011.04555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 4.Stetson DB, Medzhitov R. Antiviral defense: interferons and beyond. J Exp Med. 2006;203:1837–41. doi: 10.1084/jem.20061377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–15. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 6.Jeong E, Lee JY. Intrinsic and extrinsic regulation of innate immune receptors. Yonsei Med J. 2011;52:379–92. doi: 10.3349/ymj.2011.52.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Z, Kim T, Bao M, et al. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity. 2011;34:866–78. doi: 10.1016/j.immuni.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grieco F, Sebastiani G, Spagnuolo I, Patti A, Dotta F. Immunology in the clinic review series; focus on type 1 diabetes and viruses: how viral infections modulate beta cell function. Clin Exp Immunol. 2012;168:24–9. doi: 10.1111/j.1365-2249.2011.04556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–50. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 10.Swiecki M, McCartney SA, Wang Y, Colonna M. TLR7/9 versus TLR3/MDA5 signaling during virus infections and diabetes. J Leukoc Biol. 2011;90:691–701. doi: 10.1189/jlb.0311166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hultcrantz M, Hühn M, Wolf HM, et al. Interferons induce an antiviral state in human pancreatic islets. Virology. 2007;367:92–101. doi: 10.1016/j.virol.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 12.Stene LC, Oikarinen S, Hyoty H, et al. Enterovirus infection and progression from islet autoimmunity to type 1 diabetes: the Diabetes and Autoimmunity Study in the Young (DAISY) Diabetes. 2010;59:3174–80. doi: 10.2337/db10-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roivainen M, Klingel K. Virus infections and type 1 diabetes risk. Curr Diab Rep. 2010;10:350–6. doi: 10.1007/s11892-010-0139-x. [DOI] [PubMed] [Google Scholar]
- 14.Tauriainen S, Oikarinen S, Oikarinen M, Hyoty H. Enteroviruses in the pathogenesis of type 1 diabetes. Semin Immunopathol. 2011;33:45–55. doi: 10.1007/s00281-010-0207-y. [DOI] [PubMed] [Google Scholar]
- 15.Yeung WC, Rawlinson WD, Craig ME. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ (Clin Res Ed) 2011;342:d35. doi: 10.1136/bmj.d35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deonarain R, Cerullo D, Fuse K, Liu PP, Fish EN. Protective role for interferon-beta in coxsackievirus B3 infection. Circulation. 2004;110:3540–3. doi: 10.1161/01.CIR.0000136824.73458.20. [DOI] [PubMed] [Google Scholar]
- 17.Flodstrom M, Maday A, Balakrishna D, Cleary MM, Yoshimura A, Sarvetnick N. Target cell defense prevents the development of diabetes after viral infection. Nat Immunol. 2002;3:373–82. doi: 10.1038/ni771. [DOI] [PubMed] [Google Scholar]
- 18.Wessely R, Klingel K, Knowlton KU, Kandolf R. Cardioselective infection with coxsackievirus B3 requires intact type I interferon signaling: implications for mortality and early viral replication. Circulation. 2001;103:756–61. doi: 10.1161/01.cir.103.5.756. [DOI] [PubMed] [Google Scholar]
- 19.Flodstrom M, Horwitz MS, Maday A, Balakrishna D, Rodriguez E, Sarvetnick N. A critical role for inducible nitric oxide synthase in host survival following coxsackievirus B4 infection. Virology. 2001;281:205–15. doi: 10.1006/viro.2000.0801. [DOI] [PubMed] [Google Scholar]
- 20.Flodstrom-Tullberg M, Hultcrantz M, Stotland A, et al. RNase L and double-stranded RNA-dependent protein kinase exert complementary roles in islet cell defense during coxsackievirus infection. J Immunol. 2005;174:1171–7. doi: 10.4049/jimmunol.174.3.1171. [DOI] [PubMed] [Google Scholar]
- 21.Zaragoza C, Ocampo CJ, Saura M, et al. Inducible nitric oxide synthase protection against coxsackievirus pancreatitis. J Immunol. 1999;163:5497–504. [PubMed] [Google Scholar]
- 22.Flodstrom M, Tsai D, Fine C, Maday A, Sarvetnick N. Diabetogenic potential of human pathogens uncovered in experimentally permissive beta-cells. Diabetes. 2003;52:2025–34. doi: 10.2337/diabetes.52.8.2025. [DOI] [PubMed] [Google Scholar]
- 23.Triantafilou K, Orthopoulos G, Vakakis E, et al. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell Microbiol. 2005;7:1117–26. doi: 10.1111/j.1462-5822.2005.00537.x. [DOI] [PubMed] [Google Scholar]
- 24.Triantafilou K, Triantafilou M. Coxsackievirus B4-induced cytokine production in pancreatic cells is mediated through toll-like receptor 4. J Virol. 2004;78:11313–20. doi: 10.1128/JVI.78.20.11313-11320.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang JP, Asher DR, Chan M, Kurt-Jones EA, Finberg RW. Cutting edge: antibody-mediated TLR7-dependent recognition of viral RNA. J Immunol. 2007;178:3363–7. doi: 10.4049/jimmunol.178.6.3363. [DOI] [PubMed] [Google Scholar]
- 26.Fuse K, Chan G, Liu Y, et al. Myeloid differentiation factor-88 plays a crucial role in the pathogenesis of Coxsackievirus B3-induced myocarditis and influences type I interferon production. Circulation. 2005;112:2276–85. doi: 10.1161/CIRCULATIONAHA.105.536433. [DOI] [PubMed] [Google Scholar]
- 27.Fairweather D, Yusung S, Frisancho S, et al. IL-12 receptor beta 1 and Toll-like receptor 4 increase IL-1 beta- and IL-18-associated myocarditis and coxsackievirus replication. J Immunol. 2003;170:4731–7. doi: 10.4049/jimmunol.170.9.4731. [DOI] [PubMed] [Google Scholar]
- 28.Huhn MH, McCartney SA, Lind K, Svedin E, Colonna M, Flodstrom-Tullberg M. Melanoma differentiation-associated protein-5 (MDA-5) limits early viral replication but is not essential for the induction of type 1 interferons after Coxsackievirus infection. Virology. 2010;401:42–8. doi: 10.1016/j.virol.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 29.Negishi H, Osawa T, Ogami K, et al. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc Natl Acad Sci USA. 2008;105:20446–51. doi: 10.1073/pnas.0810372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richer MJ, Horwitz MS. Coxsackievirus infection as an environmental factor in the etiology of type 1 diabetes. Autoimmun Rev. 2009;8:611–15. doi: 10.1016/j.autrev.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 31.Wang JP, Cerny A, Asher DR, Kurt-Jones EA, Bronson RT, Finberg RW. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J Virol. 2010;84:254–60. doi: 10.1128/JVI.00631-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riad A, Westermann D, Zietsch C, et al. TRIF is a critical survival factor in viral cardiomyopathy. J Immunol. 2011;186:2561–70. doi: 10.4049/jimmunol.1002029. [DOI] [PubMed] [Google Scholar]
- 33.Stene LC, Rewers M. The enterovirus link to type 1 diabetes: critical review of human studies. Clin Exp Immunol. 2012;168:12–23. doi: 10.1111/j.1365-2249.2011.04555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richardson SJ, Willcox A, Bone AJ, Morgan NG, Foulis AK. Immunopathology of the human pancreas in type-I diabetes. Semin Immunopathol. 2011;33:9–21. doi: 10.1007/s00281-010-0205-0. [DOI] [PubMed] [Google Scholar]
- 35.Rowe PA, Campbell-Thompson ML, Schatz DA, Atkinson MA. The pancreas in human type 1 diabetes. Semin Immunopathol. 2011;33:29–43. doi: 10.1007/s00281-010-0208-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flodstrom-Tullberg M. Viral infections: their elusive role in regulating susceptibility to autoimmune disease. Microbes Infect. 2003;5:911–21. doi: 10.1016/s1286-4579(03)00161-8. [DOI] [PubMed] [Google Scholar]
- 37.Munz C, Lunemann JD, Getts MT, Miller SD. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol. 2009;9:246–58. doi: 10.1038/nri2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dotta F, Censini S, van Halteren AG, et al. Coxsackie B4 virus infection of beta-cells and NK cell insulitis in recent onset type 1 diabetic patients. Proc Natl Acad Sci USA. 2007;104:5115–20. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oikarinen M, Tauriainen S, Honkanen T, et al. Analysis of pancreas tissue in a child positive for islet cell antibodies. Diabetologia. 2008;51:1796–802. doi: 10.1007/s00125-008-1107-8. [DOI] [PubMed] [Google Scholar]
- 40.Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia. 2009;52:1143–51. doi: 10.1007/s00125-009-1276-0. [DOI] [PubMed] [Google Scholar]
- 41.Ylipaasto P, Klingel K, Lindberg AM, et al. Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet beta cells. Diabetologia. 2004;47:225–39. doi: 10.1007/s00125-003-1297-z. [DOI] [PubMed] [Google Scholar]
- 42.Roivainen M, Rasilainen S, Ylipaasto P, et al. Mechanisms of coxsackievirus-induced damage to human pancreatic beta-cells. J Clin Endocrinol Metab. 2000;85:432–40. doi: 10.1210/jcem.85.1.6306. [DOI] [PubMed] [Google Scholar]
- 43.Roivainen M, Ylipaasto P, Savolainen C, Galama J, Hovi T, Otonkoski T. Functional impairment and killing of human beta cells by enteroviruses: the capacity is shared by a wide range of serotypes, but the extent is a characteristic of individual virus strains. Diabetologia. 2002;45:693–702. doi: 10.1007/s00125-002-0805-x. [DOI] [PubMed] [Google Scholar]
- 44.Szopa TM, Gamble DR, Taylor KW. Coxsackie B4 virus induces short-term changes in the metabolic functions of mouse pancreatic islets in vitro. Cell Biochem Funct. 1986;4:181–7. doi: 10.1002/cbf.290040304. [DOI] [PubMed] [Google Scholar]
- 45.Yoon JW, Austin M, Onodera T, Notkins AL. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N Engl J Med. 1979;300:1173–9. doi: 10.1056/NEJM197905243002102. [DOI] [PubMed] [Google Scholar]
- 46.Bowles NE, Bayston TA, Zhang HY, et al. Persistence of enterovirus RNA in muscle biopsy samples suggests that some cases of chronic fatigue syndrome result from a previous, inflammatory viral myopathy. J Med. 1993;24:145–60. [PubMed] [Google Scholar]
- 47.Kim KS, Tracy S, Tapprich W, et al. 5′-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J Virol. 2005;79:7024–41. doi: 10.1128/JVI.79.11.7024-7041.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zanone MM, Favaro E, Conaldi PG, et al. Persistent infection of human microvascular endothelial cells by coxsackie B viruses induces increased expression of adhesion molecules. J Immunol. 2003;171:438–46. doi: 10.4049/jimmunol.171.1.438. [DOI] [PubMed] [Google Scholar]
- 49.Tsunoda I, Fujinami RS. Neuropathogenesis of Theiler's murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J Neuroimmune Pharmacol. 2010;5:355–69. doi: 10.1007/s11481-009-9179-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry [see comments] Nat Med. 1998;4:781–5. doi: 10.1038/nm0798-781. [DOI] [PubMed] [Google Scholar]
- 51.Ida-Hosonuma M, Iwasaki T, Yoshikawa T, et al. The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J Virol. 2005;79:4460–9. doi: 10.1128/JVI.79.7.4460-4469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oikarinen M, Tauriainen S, Honkanen T, et al. Detection of enteroviruses in the intestine of type 1 diabetic patients. Clin Exp Immunol. 2008;151:71–5. doi: 10.1111/j.1365-2249.2007.03529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crow MK. Interferon-alpha: a therapeutic target in systemic lupus erythematosus. Rheum Dis Clin North Am. 2010;36:173–86. doi: 10.1016/j.rdc.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smyth DJ, Cooper JD, Bailey R, et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet. 2006;38:617–9. doi: 10.1038/ng1800. [DOI] [PubMed] [Google Scholar]
- 55.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–9. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chistiakov DA, Voronova NV, Savost'Anov KV, Turakulov RI. Loss-of-function mutations E6 27X and I923V of IFIH1 are associated with lower poly(I : C)-induced interferon-beta production in peripheral blood mononuclear cells of type 1 diabetes patients. Hum Immunol. 2010;71:1128–34. doi: 10.1016/j.humimm.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 57.Liu S, Wang H, Jin Y, et al. IFIH1 polymorphisms are significantly associated with type 1 diabetes and IFIH1 gene expression in peripheral blood mononuclear cells. Hum Mol Genet. 2009;18:358–65. doi: 10.1093/hmg/ddn342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strange A, Capon F, Spencer CC, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42:985–90. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sutherland A, Davies J, Owen CJ, et al. Genomic polymorphism at the interferon-induced helicase (Ifih1) locus contributes to Graves' disease susceptibility. J Clin Endocrinol Metab. 2007;92:3338–41. doi: 10.1210/jc.2007-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gateva V, Sandling JK, Hom G, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41:1228–33. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harley JB, Alarcon-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–10. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shigemoto T, Kageyama M, Hirai R, Zheng J, Yoneyama M, Fujita T. Identification of loss of function mutations in human genes encoding RIG-I and MDA5: implications for resistance to type I diabetes. J Biol Chem. 2009;284:13348–54. doi: 10.1074/jbc.M809449200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Downes K, Pekalski M, Angus KL, et al. Reduced expression of IFIH1 is protective for type 1 diabetes. PLoS ONE. 2010;5:pii:e12646. doi: 10.1371/journal.pone.0012646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zouk H, Marchand L, Polychronakos C. Study of transcriptional effects in Cis at the IFIH1 locus. PLoS ONE. 2010;5:e11564. doi: 10.1371/journal.pone.0011564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinson T, Kariuki SN, Franek BS, et al. Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-alpha and serologic autoimmunity in lupus patients. J Immunol. 2011;187:1298–303. doi: 10.4049/jimmunol.1100857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mukherjee A, Morosky SA, Delorme-Axford E, et al. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011;7:e1001311. doi: 10.1371/journal.ppat.1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beard JA, Bearden A, Striker R. Vitamin D and the anti-viral state. J Clin Virol. 2011;50:194–200. doi: 10.1016/j.jcv.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wolden-Kirk H, Overbergh L, Christesen HT, Brusgaard K, Mathieu C. Vitamin D and diabetes: its importance for beta cell and immune function. Mol Cell Endocrinol. 2011;347:106–20. doi: 10.1016/j.mce.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 69.Wang TJ, Zhang F, Richards JB, et al. Common genetic determinants of vitamin D insufficiency: a genome-wide association study. Lancet. 2010;376:180–8. doi: 10.1016/S0140-6736(10)60588-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramagopalan SV, Heger A, Berlanga AJ, et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res. 2010;20:1352–60. doi: 10.1101/gr.107920.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bailey R, Cooper JD, Zeitels L, et al. Association of the vitamin D metabolism gene CYP27B1 with type 1 diabetes. Diabetes. 2007;56:2616–21. doi: 10.2337/db07-0652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cooper JD, Smyth DJ, Walker NM, et al. Inherited variation in vitamin D genes is associated with predisposition to autoimmune disease type 1 diabetes. Diabetes. 2011;60:1624–31. doi: 10.2337/db10-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lopez ER, Regulla K, Pani MA, Krause M, Usadel KH, Badenhoop K. CYP27B1 polymorphisms variants are associated with type 1 diabetes mellitus in Germans. J Steroid Biochem Mol Biol. 2004;89–90:155–7. doi: 10.1016/j.jsbmb.2004.03.095. [DOI] [PubMed] [Google Scholar]
- 74.Blanton D, Han Z, Bierschenk L, et al. Reduced serum vitamin D-binding protein levels are associated with type 1 diabetes. Diabetes. 2011;60:2566–70. doi: 10.2337/db11-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gal-Tanamy M, Bachmetov L, Ravid A, et al. Vitamin-D: an innate antiviral agent suppressing hepatitis C virus in human hepatocytes. Hepatology. 2011;54:1570–9. doi: 10.1002/hep.24575. [DOI] [PubMed] [Google Scholar]
- 76.Cooper JD, Walker NM, Smyth DJ, Downes K, Healy BC, Todd JA. Follow-up of 1715 SNPs from the Wellcome Trust Case Control Consortium genome-wide association study in type I diabetes families. Genes Immun. 2009;10(Suppl. 1):S85–94. doi: 10.1038/gene.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heinig M, Petretto E, Wallace C, et al. A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature. 2011;467:460–4. doi: 10.1038/nature09386. [DOI] [PMC free article] [PubMed] [Google Scholar]