

Abstract
OTHER THEMES PUBLISHED IN THIS IMMUNOLOGY IN THE CLINIC REVIEW SERIES
Metabolic diseases, host responses, cancer, autoinflammatory diseases, allergy.
Type 1 diabetes results from an interaction between genetic and environmental factors. Coxsackieviruses B (CV-B) are major environmental candidates, as suggested by epidemiological and experimental studies. The mechanisms leading to the disease involve interactions between the virus, host target tissue (pancreas) and the immune system. The infection of target cells with viruses can be prevented by antibodies. Conversely, the infection can be enhanced by antibodies. The antibody-dependent enhancement (ADE) of infection has been described with various viruses, especially Picornaviruses. In mice infected with CV-B3 this phenomenon resulted in an extended inflammatory reaction and myocarditis. In the human system non-neutralizing antibodies can increase the infection of monocytes with CV-B4 and stimulate the production of interferon (IFN)-α by these cells in vitro. CV-B4/immunoglobulin (Ig)G immune complexes interacted with a specific viral receptor [Coxsackievirus and adenovirus receptor (CAR)] and with IgG Fc fraction receptors (FcγRII and FcγRIII) at the surface of monocytes. The virus–antibody complexes are internalized (CAR) and receptors for the Fc of IgG (FcγRII and FcγRIII). Such antibodies have been detected in patients with type 1 diabetes and they could be responsible for the presence of enteroviral RNA and IFN-α in peripheral blood mononuclear cells (PBMC) of these individuals. The target of enhancing antibodies has been identified as the VP4 protein, which allowed the detection of these antibodies by enzyme-linked immunosorbent assay (ELISA). It cannot be excluded that antibodies enhancing the infection with CV-B may play a role in the pathogenesis of type 1 diabetes, induced or aggravated by these viruses. They can cause a viral escape from the immune response and may participate in the spreading of viruses to β cells. Whether enhancing antibodies raised against VP4 can play a role in iterative homologous and/or heterologous CV-B infections and in the persistence of viruses within the host deserves further study.
Keywords: Coxsackievirus-B4, enterovirus, interferon alpha, monocytes, type 1 diabetes, patients
Antibodies can be foes
The rapidly increased incidence of T1 diabetes (T1D), especially in young individuals, is challenging [1]. Epidemiological studies in neighbouring populations with similar genetic profiles defend the hypothesis of an interaction between genetic and environmental factors in the pathogenesis of type 1 diabetes [2]. In addition to nutriments, drugs and toxin, viruses are environmental factors that are supposed to play a role in the development of the disease.
Coxsackievirus-B (CV-B), belonging to the human enterovirus B specie in the enterovirus genus of the Picornaviridae family, are major candidates as suggested by epidemiological and experimental studies [3]–[9].
The relationship between CV-B and type 1 diabetes probably involves an interplay between viruses, pancreatic β cells, the innate and adaptive immune systems and host genes that regulate the immune response to virus infections [6],[10],[11]. The host immune response to viral infections is advantageous, but may result and/or participate in pathogenic processes able to play a role in the development of autoimmune diseases [12],[13].
Inflammation, bystander activation of T cells, molecular mimicry and disturbance of thymic function are not mutually exclusive mechanisms, possibly involved in the CV-B-induced or aggravated development of T1D [14]. A protective role of antibodies, able to prevent viruses from infecting target cells, is well known; however, antibodies against various viruses can increase the infection of target cells [15].
This phenomenon, which is called antibody-dependent enhancement (ADE) of infection, has been observed in the infection with viruses belonging to the Picornaviridae family. Antibodies can increase the infection of animal peripheral blood mononuclear cells (PMNC) and macrophages (and an animal macrophage-like cell line) with foot and mouth disease virus (FMDV), belonging to the Aphtovirus genus of this family [16]. In mice, the ADE of CV-B infection has been investigated in vitro and in vivo. Murine anti-CV-B2 antibodies increase the infection of mouse macrophages with CV-B3 in vitro and these antibodies, inoculated with CV-B3 into mice, increased the level of virus in blood and organs (heart, pancreas, spleen) that was associated with tissue damage in exocrine pancreas and heart [17].
Mice without anti-CV-B3 antibodies, inoculated with CV-B3, had moderate heart damage and inflammatory reaction and low viral titres in heart tissue. Mice with high levels of antibodies were totally protected. In contrast, low levels of antibodies (following a first infection with a non-myocarditic CV-B3) resulted in the animals inoculated with CV-B3 in an extended inflammatory reaction and myocarditis [18].
It cannot be discounted that the ADE of CV-B infection may play a role in the CV-B-induced pathogenesis of type 1 diabetes, as presented in the next sections.
Antibody-dependent enhancement of CV-B infection in the human system
Antibodies, Coxsackievirus B and interferon (IFN)-α
The circulation of IFN-α, together with the presence of IFN mRNA and CV-B RNA in peripheral blood of patients with type 1 diabetes [9],[19],[20], prompted our group to investigate the mechanisms of CV-B-induced production of IFN-α by PBMC, and thereafter we discovered the ADE of the CV-B4-induced production of IFN-α by PBMC [21]. It was observed that immunoglobulin (Ig)G, devoid of neutralizing antibodies present in serum/plasma of T1D patients and controls, interacting with the virus and FcγRII and FcγRIII, increased the CV-B4-induced synthesis of IFN-α by human PBMC in vitro, and that CV-B4/IgG immune complexes bound Coxsackievirus and adenovirus receptor (CAR) and IgG Fc fraction receptors (FcγRII and FcγRIII) at the surface of monocytes [21]. In the presence of CV-B4, PBMC of patients with type 1 diabetes but not those of healthy controls produced high amounts of IFN-α, which was due to anti-CV-B4 IgG bound to the surface of PBMC through FcγRI and FcγRIII. When plasma samples from patients with T1D were preincubated with CV-B4, their enhancing activity was higher than those from healthy controls [22].
Antibodies and Coxsackievirus B infection
It was demonstrated that the cells producing IFN-α in response to CV-B4/IgG complexes were CD14+ monocytes within monocyte-enriched PBMC [21]. This observation prompted us to speculate that the synthesis of IFN-α in these conditions was due to the infection of monocytes with CV-B4. Indeed, non-neutralizing anti-CV-B4 IgG increased the in-vitro infection of monocytes from peripheral blood with CV-B4, as suggested by the double indirect immunofluorescence staining of PBMC with CD14 antibodies and VP1 viral capsid antibodies [23]. Productive viral replication was obtained in monocytes infected with CV-B4 preincubated with plasma, as demonstrated by the presence of intracellular plus-sense and minus-sense CV-B RNA strands detected by reverse transcription–polymerase chain reaction (RT–PCR) and by the release of infectious particles in culture supernatant fluids [23]. The role of CAR, FcγRII and FcγRIII in infection with CV-B4 mixed with plasma, and the role of viral RNA entry in PBMC irrespective of viral RNA replication, as the mechanism of CV-B4-induced synthesis of IFN-α in these cells, was demonstrated (see Fig. 1) [23].
Fig. 1.
Antibodies enhance the infection with Coxsackievirus-B4 (CV-B4). Antibodies (IgG) bind CV-B4 and the complex interacts with the Coxsackievirus and adenovirus receptor (CAR) and with immunoglobulin (Ig)G Fc fraction receptors (FcγRII and FcγRIII) at the surface of monocytes. The virus–antibody complexes are internalized within the cell and viral RNA is uncoated. Entry of viral RNA stimulates the synthesis of interferon (IFN)-α. Viral RNA and proteins are produced, assembled and viral particles are released. The target of enhancing antibodies is a region of VP4.
The target protein of antibodies increasing both the CV-B3- and CV-B4-induced IFN-α production by PBMCs and CV-B4 infection of PBMC was identified as capsid protein VP4 [24],[25]. The target epitope(s) of antibodies increasing the CV-B3- or CV-B4-induced IFN-α production by PBMCs is (are) located between amino acids 11 and 30 on protein VP4, which is made of 69 amino acids [26].
The ability of facilitating antibodies from human plasma to bind VP4 is intriguing because, in contrast with VP1 VP2 and VP3, which are the three other viral capsid proteins, VP4 is buried in the capsid according to X-ray crystallography studies performed with viral particles at −196°C [27]. However, at physiological temperatures the viral conformation is different, as antibodies in plasma can bind CV-B through VP4 at 37°C. It can be explained by distorting the viral particle at 37°C, consequently enabling exposure of VP4 on the capsid surface, as described previously for poliovirus [28]. Whether a part of VP4 recognized by enhancing antibodies is exhibited continuously by the virion or is exposed following discontinuous conformational changes is an open question.
It was observed that there was no cross-reaction between enhancing antibodies directed towards the VP4 protein of CV-B4 and those directed towards the VP4 protein of CV-B3 [24]. Bioinformatic analysis indicates that the 16–24 region of CV-B4 VP4 is distinct from that of CV-B3 VP4. The 16–24 region of VP4 could be the target of enhancing antibodies and the amino acid pattern of this area would participate in the interserotype specificity. This region could bear the intraserotype specificity of CV-B; approximately 64% of homology was displayed by the in silico approach between the 16–24 region of the VP4 of one CV-B4, taken as reference, and those of other CV-B4 VP4 sequences available from the GenBank database. Homology between the 16–24 regions of VP4 in various CV-B3 sequences was also observed (68·5%), but CV-B3 VP4 had homologies with those of CV-B1 and CV-B5 (43·6 and 21·3%, respectively), suggesting that a cross-reaction between CV-B3, CV-B1 and CV-B5 cannot be excluded with regard to the binding of enhancing antibodies [29].
Conclusion and perspective: the antibody-dependent enhancement of CV-B infection in the pathogenesis of T1D
In vivo, the ADE of infection can cause a viral escape from the immune response and enables viruses to spread along the whole organism and to persist for longer in increased quantity. This phenomenon is responsible for the exacerbation of diseases such as haemorrhagic dengue or respiratory syncitial virus (RSV) bronchiolitis, and is also suggested to be involved in the evolution of acquired immune deficiency virus (AIDS). Moreover, studies of dengue virus (DV) suggest that ADE of DV infection is related to a shift from a T helper type 1 (Th1) type immune response to a Th2 type response [15]. The pathogenic effects of ADE in various viral infections suggest that the hypothesis of a role of the ADE of CV-B infection in human beings, especially in the context of the pathogenesis of T1D, cannot be discounted (see Fig. 2).
Fig. 2.
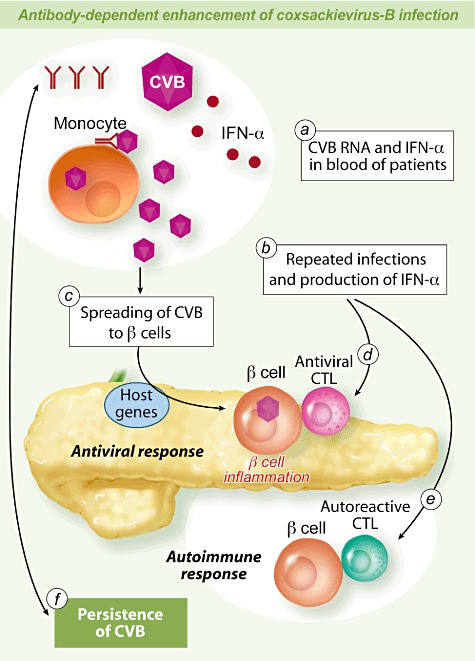
Antibody-dependent enhancement of Coxsackievirus-B (CV-B4) infection and pathogenesis of type 1 diabetes (T1D). Antibodies can enhance the infection of monocytes with CV-B in vitro. Enhancing antibodies and CV-B have been detected in patients with T1D. The presence of CV-B and interferon (IFN)-α in peripheral blood mononuclear cells (PBMC) of patients with T1D can be a result of the antibody-dependent enhancement (ADE) of CV-B infection (a). ADE can play a role in homologous or heterologous repeated infections with CV-B and iterative production of IFN-α that may be associated with the development of autoimmunity (b). Antibodies enhancing the infection of monocytes with CV-B can participate in the spreading of viruses to β cells and can be responsible for a higher viral load which can increase the damage to β cells (c). The infection of β cells with CV-B results in inflammation and innate immune response which is influenced by host genes, followed by a specific immune response with anti-viral cytotoxic T lymphocytes (CTL) that disrupt and clear virus-infected cells contributing to β cell antigen release (d). Innate and adaptive immune response to CV-B can result in the activation of autoreactive CTL directed towards β cells (e). The ADE of CV-B infection can result in the persistence of CV-B in pancreas and/or other organs and, conversely, the persistence of viruses can participate in maintaining the production of enhancing antibodies (f).
The possible role of ADE of CV-B infection in vivo must be considered. Indeed, production of IFN-α by PBMC associated with the presence of CV-B that is observed in the blood of patients with T1D can be due to enhancing antibodies, as in diabetic patients the CV-B4-induced in-vitro production of IFN-α by their PBMC depended upon enhancing anti-CV-B4 IgG contained in their plasma or bound at the surface of their mononuclear cells [22]. Enteroviral RNA sequences presenting homologies with CV-B (CV-B2, CV-B3 or CV-B4) found by RT–PCR in circulating blood of patients with type 1 diabetes may be the result of an increased infection of PBMC through an antibody-dependent mechanism [23].
Most human beings have been infected by at least one of the six CV-B serotypes during their lifetime. Whether enhancing antibodies raised against VP4 can play a role in homologous and/or heterologous CV-B (HEV-Bs) infections deserves further study.
These antibodies and repeated infection with CV-B can result in the iterative production of IFN-α, which is not blameless, as abnormal activation of IFN-α is associated with the development of autoimmune reactions and may play a part in autoimmunity towards β cells in genetically predisposed individuals [30].
Another issue in which antibodies may intervene is the spreading of CV-B to pancreas islet β cells through enhancement of the infection with CV-B of monocytes acting as a Trojan horse. The infection of β cells by CV-B through this route can play a role in the induction of an inflammatory response that is thought to be the initial disturbance of β cells in the natural history of T1D [31],[32].
The ADE of CV-B infection and the spreading of viruses can be responsible for an increased quantity of viral particles in the host, the role of which in the CV-B-induced or aggravated pathogenesis of T1D could be critical, as suggested by studies in animal models [33]. It remains to determine whether pre-existing anti-CV-B antibodies exacerbate CV-B infection and result in damage in b-cells in humans beings like antibodies in CV-B3-induced heart tissue lesions in mice [17],[18].
The persistence of CV-B has been suggested to be one of the mechanisms by which these viruses could induce β cell destruction through prolonged activation of the immune system, resulting in the induction of an anti-viral and autoimmune response [34]. The possible role of enhancing antibodies in the persistence of CV-B into the host and, conversely, the role of a persistent CV-B infection in the production of antibodies able to enhance reinfections with CV-B, should not be discarded.
In addition to a possible role of the ADE of CV-B infection in the pathogenesis of virus-induced T1D, the existence of enhancing antibodies should prompt us to take precautions when designing vaccines. Antigens inducing facilitating antibodies able to worsen a subsequent infection can compromise vaccine safety, as has occurred in children vaccinated with formalin-inactivated anti-RSV preparation which resulted in an increased RSV replication in macrophages associated with severe pulmonary pathology [35],[36].
Acknowledgments
The studies performed by the authors or in progress have been or are supported by EU FP6 Integrated Project EURO-THYMAIDE (contract LSHB-CT-2003-503410), EU FP7 PEVNET Project (FP7-HEALTH-2010-single-stage no. 261441), a grant from Nord-Pas-de-Calais Région (ArCir convention 2004/018), CHRU Lille, the ministère de l'Education nationale de la recherche et de la technologie, Université de Lille 2, France and the comité mixte de coopération universitaire franco-tunisien (CMCU 2008N808/G0808). Didier Hober was Fondation pour la Recherche Médicale 2008 prize winner, and is a member of the Virus in Diabetes International Study group (VIDIS group).
Disclosure
None.
References
- 1.Vehik K, Dabelea D. The changing epidemiology of type 1 diabetes: why is it going through the roof? Diabetes Metab Res Rev. 2011;27:3–13. doi: 10.1002/dmrr.1141. [DOI] [PubMed] [Google Scholar]
- 2.Kondrashova A, Reunanen A, Romanov A, et al. A six-fold gradient in the incidence of type 1 diabetes at the eastern border of Finland. Ann Med. 2005;37:67–72. doi: 10.1080/07853890410018952. [DOI] [PubMed] [Google Scholar]
- 3.Jaïdane H, Hober D. Role of coxsackievirus B4 in the pathogenesis of type 1 diabetes. Diabetes Metab. 2008;34:537–48. doi: 10.1016/j.diabet.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 4.Jaïdane H, Sane F, Gharbi J, Aouni M, Romond MB, Hober D. Coxsackievirus B4 and type 1 diabetes pathogenesis: contribution of animal models. Diabetes Metab Res Rev. 2009;25:591–603. doi: 10.1002/dmrr.995. [DOI] [PubMed] [Google Scholar]
- 5.Jaïdane H, Sauter P, Sane F, Goffard A, Gharbi J, Hober D. Enteroviruses and type 1 diabetes: towards a better understanding of the relationship. Rev Med Virol. 2010;20:265–80. doi: 10.1002/rmv.647. [DOI] [PubMed] [Google Scholar]
- 6.Hober D, Sane F. Enteroviruses and type 1 diabetes. BMJ. 2011;342:c7072. doi: 10.1136/bmj.c7072. [DOI] [PubMed] [Google Scholar]
- 7.Yeung WC, Rawlinson WD, Craig ME. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ. 2011;342:d35. doi: 10.1136/bmj.d35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stene LC, Rewers M. Immunology in the clinic review series; focus on type 1 diabetes and viruses: the enterovirus link to type 1 diabetes: critical review of human studies. Clin Exp Immunol. 2012;168:12–23. doi: 10.1111/j.1365-2249.2011.04555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lind K, Hühn MH, Flodström-Tullberg M. Immunology in the clinic review series; focus on type 1 diabetes and viruses: the innate immune response to enteroviruses and its possible role in regulating type 1 diabetes. Clin Exp Immunol. 2012;168:30–8. doi: 10.1111/j.1365-2249.2011.04557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hober D, Sane F. Enteroviral pathogenesis of type 1 diabetes. Discov Med. 2010;10:151–60. [PubMed] [Google Scholar]
- 11.Hober D, Sauter P. Pathogenesis of type 1 diabetes mellitus: interplay between enterovirus and host. Nat Rev Endocrinol. 2010;6:279–89. doi: 10.1038/nrendo.2010.27. [DOI] [PubMed] [Google Scholar]
- 12.Fujinami RS, von Herrath MG, Christen U, Whitton JL. Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev. 2006;19:80–94. doi: 10.1128/CMR.19.1.80-94.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tauriainen S, Oikarinen S, Oikarinen M, Hyoty H. Enteroviruses in the pathogenesis of type 1 diabetes. Semin Immunopathol. 2011;33:45–55. doi: 10.1007/s00281-010-0207-y. [DOI] [PubMed] [Google Scholar]
- 14.Sane F, Moumna I, Hober D. Group B coxsackieviruses and autoimmunity: focus on Type 1 diabetes. Exp Rev Clin Immunol. 2011;7:357–66. doi: 10.1586/eci.11.11. [DOI] [PubMed] [Google Scholar]
- 15.Sauter P, Hober D. Mechanisms and results of the antibody-dependent enhancement of viral infections and role in the pathogenesis of coxsackievirus B-induced diseases. Microbes Infect. 2009;11:443–51. doi: 10.1016/j.micinf.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Baxt B, Mason PW. Foot-and-mouth disease virus undergoes restricted replication in macrophage cell cultures following Fc receptor-mediated adsorption. Virology. 1995;207:503–9. doi: 10.1006/viro.1995.1110. [DOI] [PubMed] [Google Scholar]
- 17.Girn J, Kavoosi M, Chantler J. Enhancement of coxsackievirus B3 infection by antibody to a different coxsackievirus strain. J Gen Virol. 2002;83:351–8. doi: 10.1099/0022-1317-83-2-351. [DOI] [PubMed] [Google Scholar]
- 18.Kishimoto C, Kurokawa M, Ochiai H. Antibody-mediated immune enhancement in coxsackievirus B3 myocarditis. J Mol Cell Cardiol. 2002;34:1227–38. doi: 10.1006/jmcc.2002.2087. [DOI] [PubMed] [Google Scholar]
- 19.Chehadeh W, Weill J, Vantyghem MC, et al. Increased level of interferon-alpha in blood of patients with insulin-dependent diabetes mellitus: relationship with coxsackievirus B infection. J Infect Dis. 2000;181:1929–39. doi: 10.1086/315516. [DOI] [PubMed] [Google Scholar]
- 20.Coppieters KT, Wiberg A, Tracy SM, von Herrath MG. Immunology in the clinic review series: focus on type 1 diabetes and viruses: the role of viruses in type 1 diabetes: a difficult dilemma. Clin Exp Immunol. 2012;168:5–11. doi: 10.1111/j.1365-2249.2011.04554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chehadeh W, Bouzidi A, Alm G, Wattre P, Hober D. Human antibodies isolated from plasma by affinity chromatography increase the coxsackievirus B4-induced synthesis of interferon-alpha by human peripheral blood mononuclear cells in vitro. J Gen Virol. 2001;82:1899–907. doi: 10.1099/0022-1317-82-8-1899. [DOI] [PubMed] [Google Scholar]
- 22.Hober D, Chehadeh W, Weill J, et al. Circulating and cell-bound antibodies increase coxsackievirus B4-induced production of IFN-alpha by peripheral blood mononuclear cells from patients with type 1 diabetes. J Gen Virol. 2002;83:2169–76. doi: 10.1099/0022-1317-83-9-2169. [DOI] [PubMed] [Google Scholar]
- 23.Hober D, Chehadeh W, Bouzidi A, Wattre P. Antibody-dependent enhancement of coxsackievirus B4 infectivity of human peripheral blood mononuclear cells results in increased interferon-alpha synthesis. J Infect Dis. 2001;184:1098–108. doi: 10.1086/323801. [DOI] [PubMed] [Google Scholar]
- 24.Chehadeh W, Lobert PE, Sauter P, et al. Viral protein VP4 is a target of human antibodies enhancing coxsackievirus B4- and B3-induced synthesis of alpha interferon. J Virol. 2005;79:13882–91. doi: 10.1128/JVI.79.22.13882-13891.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sauter P, Lobert PE, Lucas B, et al. Role of the capsid protein VP4 in the plasma-dependent enhancement of the Coxsackievirus B4E2-infection of human peripheral blood cells. Virus Res. 2007;125:183–90. doi: 10.1016/j.virusres.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 26.Sauter P, Chehadeh W, Lobert PE, et al. A part of the VP4 capsid protein exhibited by coxsackievirus B4 E2 is the target of antibodies contained in plasma from patients with type 1 diabetes. J Med Virol. 2008;80:866–78. doi: 10.1002/jmv.21171. [DOI] [PubMed] [Google Scholar]
- 27.Muckelbauer JK, Kremer M, Minor I, et al. Structure determination of coxsackievirus B3 to 3.5 A resolution. Acta Crystallogr D Biol Crystallogr. 1995;51:871–87. doi: 10.1107/S0907444995002253. [DOI] [PubMed] [Google Scholar]
- 28.Li Q, Yafal AG, Lee YM, Hogle J, Chow M. Poliovirus neutralization by antibodies to internal epitopes of VP4 and VP1 results from reversible exposure of these sequences at physiological temperature. J Virol. 1994;68:3965–70. doi: 10.1128/jvi.68.6.3965-3970.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goffard A, Sane F, Soumillon M, Hober D. Specificity of the coxsackievirus B4 VP4 capsid protein investigated in silico. Discov Med. 2011;12:153–8. [PubMed] [Google Scholar]
- 30.Stewart TA, Hultgren B, Huang X, Pitts-Meek S, Hully J, MacLachlan NJ. Induction of type 1 diabetes by interferon-α in transgenic mice. Science. 1993;260:1942–46. doi: 10.1126/science.8100367. [DOI] [PubMed] [Google Scholar]
- 31.Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–26. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- 32.Grieco F, Sebastiani G, Spagnuolo I, Patti A, Dotta F. How viral infections modulate beta-cell function. Clin Exp Immunol. 2012;168:24–9. doi: 10.1111/j.1365-2249.2011.04556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanno T, Kim K, Kono K, Drescher KM, Chapman NM, Tracy S. Group B coxsackievirus diabetogenic phenotype correlates with replication efficiency. J Virol. 2006;80:5637–43. doi: 10.1128/JVI.02361-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hyoty H. Enterovirus infections and type 1 diabetes. Ann Med. 2002;34:138–47. [PubMed] [Google Scholar]
- 35.Prince GA, Jenson AB, Hemming VG, et al. Enhancement of respiratory syncytial virus pulmonary pathology in cotton rats by prior intramuscular inoculation of formalin-inactivated virus. J Virol. 1986;57:721–28. doi: 10.1128/jvi.57.3.721-728.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gimenez HB, Keir HM, Cash P. In vitro enhancement of respiratory syncytial virus infection of U937 cells by human sera. J Gen Virol. 1989;70:89–96. doi: 10.1099/0022-1317-70-1-89. [DOI] [PubMed] [Google Scholar]