

Abstract
Myostatin is a potent inhibitor of muscle development. Genetic deletion of myostatin in mice results in muscle mass increase, with muscles often weighing three times their normal values. Contracting muscle transfers tension to skeletal elements through an elaborate connective tissue network. Therefore, the connective tissue of skeletal muscle is an integral component of the contractile apparatus. Here we examine the connective tissue architecture in myostatin null muscle. We show that the hypertrophic muscle has decreased connective tissue content compared with wild-type muscle. Secondly, we show that the hypertrophic muscle fails to show the normal increase in muscle connective tissue content during ageing. Therefore, genetic deletion of myostatin results in an increase in contractile elements but a decrease in connective tissue content. We propose a model based on the contractile profile of muscle fibres that reconciles this apparent incompatible tissue composition phenotype.
Keywords: connective tissue, epimysium, muscle, myostatin, perimysium
Introduction
Skeletal muscle is a highly adaptable tissue. The overall size of skeletal muscle is readily increased through the application of mechanical load. Conversely, mass can be decreased through inadequate energy supply and is a feature of the ageing process. Changes in muscle mass are accompanied by alterations in the metabolic, biochemical and molecular profile of the tissue (Matsakas & Patel, 2009a,b). The benefits to human health of regimes that maintain muscle mass extend beyond those associated with locomotion. One relevant example is the recent study that shows the incidence of diabetes is related to decreased muscle mass (Srikanthan & Karlamangla, 2011).
A huge amount of research has been undertaken to identify the key molecular components that regulate muscle growth. A large body of literature has shown that insulin-like growth factor (IGF-I and IGF-II)-mediated signalling, working through the type I IGF receptor (IGF-IR), promotes skeletal muscle fibre growth and hypertrophy by activating pathways that simultaneously increase protein synthesis and decrease proteolysis (Glass, 2003). These pathways are physiologically important as production of IGF-I is stimulated by exercise (Nindl & Pierce, 2010). Another potent regulator of muscle development is myostatin; an evolutionarily conserved member of the transforming growth factor (TGF)-β super-family of signalling proteins. In contrast to IGF-I, myostatin functions as an inhibitor of skeletal muscle development. Loss of function mutations in the myostatin gene have been described in numerous mammalian species, all of which develop a hypermuscular phenotype through varying degrees of muscle fibre hypertrophy and fibre hyperplasia (Mcpherron & Lee, 1997; Mcpherron et al. 1997; Schuelke et al. 2004). In addition, myostatin controls the metabolic profile of skeletal muscle by promoting an oxidative phenotype. Deletion of myostatin results in the development of muscle with a glycolytic phenotype (Mcpherron et al. 1997).
Most studies investigating the myostatin have focused on the muscle fibre component of skeletal muscle. However, skeletal muscle is an organ system composed not only of myofibres but also of nerves, sensory cells, blood vessels and connective tissue. In order to generate a working organ, these differing tissues must develop in a coordinated manner. The connective tissue plays a pivotal role in the transfer of contraction tension generated by the myofibre to the skeletal elements. Connective tissue throughout the body fails to develop to its normal levels in mice and cattle that harbour a mutation in their myostatin genes (Bailey et al. 1982; Mcpherron et al. 1997), and Mendias et al. (2006) reported a reduction in muscle collagen (through the measurement of hydroxyproline) in the skeletal muscle of myostatin null mice. These findings have been complimented by studies showing that myostatin directly promotes muscle fibroblast and tendon fibroblast proliferation (Mendias et al. 2006; Zhu et al. 2007). Furthermore, a number of elegant studies have shown that deletion of myostatin accelerates muscle regeneration and inhibits fibrosis in acute and chronic injury models (Wagner et al. 2002; Mccroskery et al. 2005; Zhu et al. 2007). In this study we focused on the relationship between myofibre development and its connective tissue in the myostatin null mice. We show that in myostatin null mice both the perimysial and endomysial compartments are reduced in muscles through a decrease in the amount of laminin, collagen I, III and IV. We propose a model that suggests that the changes in the connective tissue composition in the myostatin null mice are adapted to the contractile phenotype of the muscle.
Materials and methods
Animal maintenance
All procedures were approved by local ethical review and covered by UK Home Office licenses. Healthy male C57/BL6 mstn+/+ and myostatin null mice (mstn−/−) were bred and maintained in the biological resource unit of University of Reading. They were housed under standard environmental conditions (20–22 °C, 12–12 h light–dark cycle), and were provided with food and tap water ad libitum. mstn−/− founder mice were obtained from Dr Lee at Johns Hopkins University School of Medicine, USA. Three animals for each genotype and age group (4 and 19 months) were used in the study. Animals were killed by cervical dislocation.
Antibodies
Mouse monoclonal antibodies against collagen I and III (Abcam, Cambridge, UK) were used at concentrations of 1 : 3000 or 1 : 1000, respectively, for Western blotting or 1 : 500 for immunohistology. Rabbit polyclonal antibody against collagen IV and matrix metalloproteinase (MMP)-9 (Abcam) were used at a concentration of 1 : 4000 for Western blotting or 1 : 700 for immunohistology. A monoclonal antibody against alpha tubulin (Epitomics, USA) was used at 1 : 2000 as a loading control for Western blots. Alexafluor-conjugated secondary antibodies were used at 1 : 200 for immunohistology.
Skeletal muscle cryo-scanning electron microscopy (SEM) for extracellular matrix
Following dissection, skeletal muscles of the fore and hind limbs were fixed in 2.5% glutaraldehyde in 0.1 m phosphate buffer (pH 7.3) for 3 days. The samples were then immersed in 10% NaOH for 5 days to dissolve contractile proteins, during which time the muscles became transparent. Thereafter, the samples were rinsed in distilled water for 5 days at 4 °C. This was followed by coating the samples with 1% tannic acid aqueous solution for 3 h in order to detect the fine connective tissue network. The samples were washed again with distilled water and left overnight at 4 °C in order to remove the excess tannic acid. Subsequently, the samples were fixed again in 1% osmium tetroxide for 1 h. Samples were then dehydrated in ascending concentrations of ethanol (70–100%) at 15-min intervals. Thereafter, the muscle sample was fixed onto a microscope stub in a vertical position using a mixture of charcoal and Tissue Tech. Samples were then immersed in liquid nitrogen before being transferred to a high-vacuum cryo-chamber to be fractured. The surface was coated with gold and the sample was introduced into the observation unit. SEM was performed using FEI, Quanta, 600 F (Holland). Fractured cross-sections were scanned at multiple magnifications.
Immunohistochemical staining
Immunohistology was performed as previously described, with the addition of an antigen retrieval step (Matsakas et al. 2009). Briefly, samples were incubated in citrate buffer (10 mm, 0.05% Tween 20, pH 6.0) preheated to boiling point for 15 min. All primary antibodies were pre-blocked in wash buffer 30 min prior to use and incubated overnight at 4 °C with the muscle sections. Primary antibodies were visualised using Alexafluor 488 and 633 secondary antibodies. All secondary antibodies were pre-blocked in wash buffer and kept in the dark for a minimum of 30 min prior to use. The samples were incubated with the secondary antibody in the dark at room temperature for 45–60 min. Slides were mounted in fluorescent medium (Calbiochem, Middlesex, UK). Myonuclei were visualised using 4′, 6-diamidino-2-phenylindole (DAPI).
Western blotting
Frozen muscles were wrapped in aluminium foil and pulverised using mortar and pestle. Thereafter, the muscle was mixed with 250 μL of lysis buffer [50 mm HCl (pH 8.0), 200 mm NaCl, 50 mm NaF, 1 mm sodium orthovanadate, 0.3% IGEPAL, and protease inhibitors: pepstatin A (1 μg mL−1), aprotinin (10 μg mL−1), leupeptin (10 μg mL−1) and phenylmethylsulphonyl fluoride (1 mm)]. Muscle was further pulverised using a manual homogeniser, and then centrifuged at 13 000 g for 10 min to remove insoluble components. Protein content in the soluble fraction was determined using the Bradford assay (Sigma UK). Soluble protein (20 μg) was resolved by 8% sodium dodecyl sulphate–polyacrylamide gel electrophoresis under non-reducing conditions before transferring to a polyvinylidene fluoride membrane. The membrane was blocked using 5% (w/v) bovine serum albumin in TBST (20 mm Tris, 140 mm NaCl and 0.1% Tween20 pH 7.6) for 1 h prior to probing with the primary antibody (diluted in blocking buffer) for 12 h. Subsequently, the membrane was washed three times with TBST and was then incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. This was followed by TBST wash to remove any unbound secondary antibody. Immunoreactivity was visualised using enhanced chemiluminescence. Multiple exposures (3–30 s) were taken to obtain sub-saturation band intensities. The X-ray films were scanned using a Bio-Rad Gs-710 imaging densitometer. Band intensities from scanned images of Western blots were quantified using Quanti-Scan software from BioSoft (version Windows 3.x, Cambridge, UK). Band intensity was normalised to alpha tubulin abundance.
Statistical analysis
Two-way anova was performed to examine the effect of genotype (wild type vs. myostatin null) and the effect of age (4 months old vs. 19 months old) on each parameter. P-values ≤ 0.05 were considered to be significant.
Results
Myostatin deletion decreases muscle connective tissue
Ultra-structure of connective tissue was examined by firstly eliminating all contractile proteins followed by freeze fracture and scanning using electron micrograph of muscles of the fore and hind limbs [biceps brachii m (BB) and tibialis anterior m (TA)]. The micrographs showed that the endomysium of either muscle from 4-week-old mstn+/+ showed densely packed aligned fibres (Fig. 1A,E). In contrast, in mstn−/− animals, the connective tissue fibres were less dense in both muscles of the same age (Fig. 1B,F). The differences in connective tissue density were more apparent in older animals. The endomysium from 19-month-old mstn+/+ animals showed a thick honeycomb organisation. In contrast, the connective tissue fibres from mstn−/− were less dense, and many fibres appeared to terminate prematurely rather than spanning the entire compartment (Fig. 1C,D shows TA, similar features were displayed by BB, data not shown). Our results show that myostatin deletion results in decreased density of connective tissue fibres in the perimysium and endomysium.
Fig. 1.
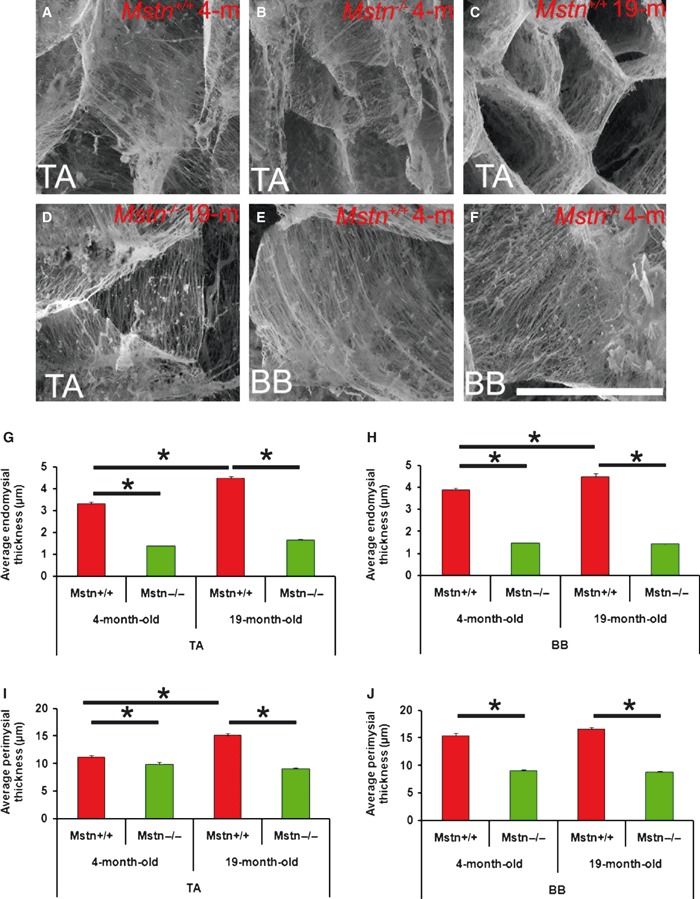
Myostatin deletion changes the morphology and thickness of the connective tissue in mouse muscles. Cryo-SEM of freeze fractured mid-belly surface of the tibialis anterior (TA) and biceps brachii (BB) muscle after solubilisation of contractile proteins. (A) Four-month-old mstn+/+ TA shows a mixture of dense and fine fibres. (B) Four-month-old mstn−/− TA shows mostly fine fibres. (C) Nineteen-month-old mstn+/+ TA shows a dense honeycomb pattern of connective tissue. (D) Nineteen-month-old mstn−/− TA contains larger connective tissue compartments made of low-density fibres. (E) Four-month-old mstn+/+ BB shows a densely arranged regular network of collagen fibrils dense fibre. (F) Four-month-old mstn−/− BB shows less dense fibres that are often irregularly oriented. (G) Quantification of endomysial thickness of TA. (H) Quantification of endomysial thickness of BB. (I) Quantification of perimysial thickness of TA. (J) Quantification of perimysial thickness of BB. *Significant changes in thickness (P < 0.001).
We measured the thickness of the endomysium and perimysium of TA and BB muscles at 4 months old and 19 months old, and found that both were significantly thicker in the mstn+/+ compared with their mstn−/− counterparts at a comparable age (Fig. 1G–J; Table 1). The quantitative analysis also revealed an interesting temporal feature regarding connective tissue development. The thickness of the endomysium in TA increased significantly between 4 months and 19 months in mstn+/+ animals, but not in the mstn−/− (Fig. 1G). Likewise, the perimysium of the TA muscle increased significantly in the mstn+/+ and not in the mstn−/− over the same period (Fig. 1I). The endomysium from the mstn+/+ BB also increased in thickness (Fig. 1H).
Table 1.
Quantification of SEM images for endomysium and perimysium connective content of 4-month-old and 19-month-old mstn+/+ and mstn−/− mice.
| Endomysium thickness (μm) | Perimysium thickness (μm) | |||||||
|---|---|---|---|---|---|---|---|---|
| mstn+/+ | mstn−/− | mstn+/+ | mstn−/− | |||||
| 4 months | 19 months | 4 months | 19 months | 4 months | 19 months | 4 months | 19 months | |
| Tibialis anterior | 3.30 ± 0.1 | 4.49 ± 0.1 | 1.38 ± 0.03 | 1.66 ± 0.03 | 11.11 ± 0.4 | 15.11 ± 0.3 | 9.85 ± 0.4 | 8.99 ± 0.2 |
| Biceps brachii | 3.89 ± 0.1 | 4.48 ± 0.1 | 1.47 ± 0.03 | 1.42 ± 0.04 | 15.44 ± 0.4 | 16.57 ± 0.4 | 8.98 ± 0.2 | 8.76 ± 0.2 |
Immunohistological analysis of muscle connective tissue components of myostatin null mice
Our ultra-structural examination showed a reduction in the quantity of connective tissue in mstn−/− muscle. We next investigated the distribution of key components of the connective tissue in mstn−/− muscle. Examination of the distribution of laminin, a key component of the basement membrane, showed differences at the quantitative and qualitative levels between 4-month-old mstn−/− and age-matched mstn+/+ mice (Fig. 2A–D). Immunohistology revealed relatively uniform laminin expression around 4-month-old mstn+/+ BB muscle fibres (Fig. 2A). In contrast, laminin expression was at lower levels and varied in thickness, often showing discontinuity in BB fibres from 4-month-old mstn−/− (Fig. 2C). Examination of laminin expression in older animals revealed that intensity of immunofluoresence had increased in 19-month-old mstn+/+, but not in mstn−/− (Fig. 2B,D). Furthermore, we were able to detect extracellular spaces between laminin domains in mstn−/− that were not present in mstn+/+ muscle (Fig. 2D). We evaluated the distribution of the non-fibrillar type IV collagen, another major component of the basal lamina. Immunohistology showed that the expression level of collagen IV was high and uniform in 4-month-old mstn+/+ in contrast to the low expression profile in age-matched mstn−/− muscle (Fig. 2E,G). During ageing, an increase in collagen IV immune-fluorescence intensity was detected in the BB muscle from 19-month-old mstn+/+, but not in BB muscles from 19-month-old mstn-/- mice (Fig. 2F,H).
Fig. 2.
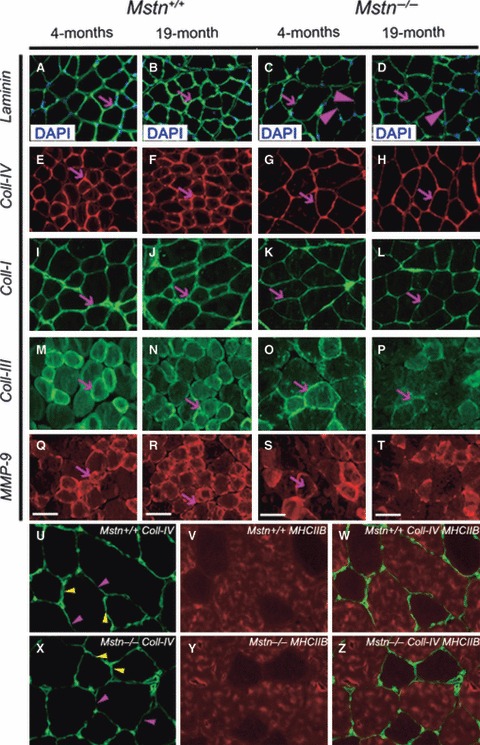
Myostatin deletion changes expression of extracellular matrix proteins in the mouse muscles. Representative images of mid-belly transverse section of BB muscle of 4-month-old and 19-month-old mstn+/+ and mstn−/− mice. Nuclei were visualised using DAPI. (C) Laminin distribution in the endomysium of 4-month-old mstn−/− was confined to a thin (arrow) and discontinuous layer (arrowhead) of basal lamina surrounding the muscle fibre, compared with (A) age-matched mstn+/+(arrow). (B) Nineteen-month-old mstn+/+ showed an increase in the thickness and density of laminin (arrow) compared with 4-month-old mstn+/+. (D) Nineteen-month-old mstn−/− demonstrated no difference in the thickness of the laminin domain compared with 4-month-old mstn−/−. Discontinuity was still present in aged mstn−/− tissue (arrowhead). (E, F) Collagen IV expression domain underwent an expansion in mstn+/+ muscle between 4 and 19 months (arrows). (G, H) In contrast, collagen thickness remained constant in muscle mstn−/−. (I, J) Collagen I expression domain also underwent an expansion in mstn+/+ muscle between 4 and 19 months (arrows). (K, L) In contrast, collagen thickness failed to increase in thickness with time in mstn−/− muscle (arrows). (M, N) Collagen III thickness remained relatively constant over time in mstn+/+ muscle (arrows). (O, P) In contrast, abundance of collagen III decreased over time in mstn−/− muscle (arrows). (Q, R) MMP-9 expression increased with time mstn+/+ in muscle (arrows). (S, T) MMP-9 abundance decreased in mstn−/− over time (arrows). Scale bar: 20 μm. (U–W) Collagen IV (green), MHCIIB (red) and dual stain for collagen IV (green) and MHCIIB (red) in mstn+/+ muscle, respectively. Note thinner layer of collagen between MHCIIB-expressing fibres (purple arrowheads) compared with thicker collagen layer adjacent to non-MHCIIB fibres (yellow arrowheads). (X–Z) Collagen IV (green), MHCIIB (red) and dual stain for collagen IV (green) and MHCIIB (red) in mstn−/− muscle, respectively. As in the case for mstn+/+, collagen between MHCIIB-expressing fibres (purple arrowheads) appeared thinner compared with thicker collagen layer adjacent to non-MHCIIB fibres (yellow arrowheads).
Next we examined the distribution of collagen type I and collagen type III; the most abundant fibrillar collagen types in the endomysium and perimysium of the skeletal muscle. We found that collagen I was highly expressed in the perimysium compared with endomysium of 4-month-old mstn+/+. The expression level of collagen I was lower in 4-month-old mstn−/− compared with age-matched mstn+/+ (Fig. 2I,K). Furthermore, the abundance of collagen I increased between 4 and 19 months in mstn+/+ but not in mstn−/− (Fig. 2J,L). Collagen III was primarily localised in the endomysium connective tissue of 4-month-old mstn+/+ BB (Fig. 2M,N). Furthermore, we observed that the concentration of collagen III was higher around small fibres compared with the large fibres in mstn+/+ BB (Fig. 2M,N). On the other hand, 4-month-old mstn−/− displayed reduced and discontinuous expression of collagen III in the endomysium (Fig. 2O,P). Moreover, the endomysium formed a thin film around the myofibre. Interestingly, in wild-type the number of fibres expressing collagen III and its thickness had increased in 19 months (Fig. 2N). In contrast, 19-month-old mstn−/− displayed a reduction in the expression of collagen III compared with 4-month-old mstn−/− animals (Fig. 2P).
We examined the distribution of MMP-9, an enzyme that has been shown to either decrease extra-cellular matrix (ECM) content in muscle through direct proteolytic action on collagen or promote ECM development by activating TGF-β1. High levels of MMP-9 expression were detected in 4-month-old mstn+/+ compared with age-matched mstn−/− (Fig. 2Q–S). Interestingly, the small fibres showed stronger expression compared with larger fibres. Expression of MMP-9 increased with age in mstn+/+, but not in mstn−/− muscle (Fig. 2R,T).
We noted that the distribution of collagen I and IV was heterogeneous in skeletal muscle, and explored whether these findings were related to muscle fibre type. To that end we determined the distribution of collagen IV in relation to fast muscle fibre myosin heavy chain isoform IIB (MHCIIB). We found in both mstn+/+ and mstn−/− that the abundance of collagen IV appeared to be greater around non-MHCIIB fibres than between MHCIIB fibres (Fig. 2U–Z).
Quantification of connective tissue components of myostatin null skeletal muscle
In order to quantify the levels of collagens I, III, IV and MMP-9, we carried out Western blotting. Collagens I, III and IV were detected as discrete bands on Western blots with molecular weights of 160, 250 and 130 kDa, respectively. A major band at 120 kDa was detected using a MMP-9 antibody with one smaller and one larger species. Quantification of band intensity revealed a consistent pattern for all proteins. Western blots showed that all the proteins examined increased with age from 4 to 19 months in mstn+/+ (Fig. 3a, left). Furthermore, quantitative analysis showed that there was significantly more of each protein at 4 months in mstn+/+ compared with mstn−/− (Fig. 3b–e). In contrast to mstn+/+, the expression of collagen I, III and IV did not increase with age in mstn−/− mice (Fig. 3b–e). These results are consistent with our finding from the ultra-structural examination of muscle connective tissue and from the immunohistological examination that shows a reduction in ECM proteins in muscle from mstn−/− compared with age-matched mstn+/+ muscle. In addition, consistent with immunohistochemistry data, mstn−/− muscle failed to display an increase in ECM protein content over time as shown by wild-type muscle.
Fig. 3.
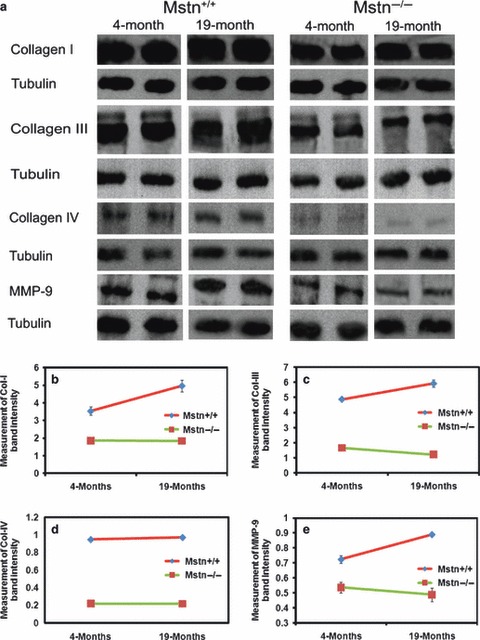
Quantification of muscle collagens and MMP-9 in myostatin null muscle. (a) Western blots of collagen I, III, IV and MMP-9 expression in BB muscle of 4-month-old and 19-month-old mstn+/+ and mstn−/− mice. Collagen I, III, IV and MMP-9 were recognised as bands of 160 kDa, 250 kDa, 130 kDa, 120 kDa, respectively. Tubulin was used as an internal loading control. (b, c) Quantification of collagen I and III Western blot bands from BB muscle of 4-month-old and 19-month-old mstn+/+ (red) and mstn−/− (green) mice. Two-way anova revealed significant reduction in the amount of the collagen I and III bands in 4-month-old mstn−/− compared with age-matched mstn+/+ (P = 0.001 and P < 0.001), respectively. A significant increase in protein levels in 19-month-old mstn+/+ compared with 4-month-old mstn+/+ was detected for collagen I and III (P < 0.05 and P = 0.08), respectively. Significant interaction (age vs. genotype) indicates the difference between both genotypes with ageing for collagen I and III (P < 0.05 and P = 0.001), respectively. (d) Quantification of the band of collagen IV showed a significant reduction in the amount of collagen IV (P < 0.001) in 4-month-old mstn−/− compared with age-matched mstn+/+. Two-way anova demonstrated a significant increase in collagen IV band of 19-month-old mstn+/+ compared with age-matched mstn+/+ (P < 0.05). No significant difference was observed in 19-month-old mstn−/− compared with 4-month-old mstn−/−. (e) Quantification of Western blot bands showed a significant reduction in the amount of MMP-9 (P < 0.001) in 4-month-old mstn−/− (green) compared with age-matched mstn+/+ (red). A significant increase in the MMP-9 levels was found in 19-month-old mstn+/+ compared with 4-month-old mstn+/+ (P < 0.001). A significant interaction (genotype vs. age) was found between both genotypes with ageing (P < 0.001). Band intensities were plotted relative to tubulin. All data presented as mean ± SEM.
Discussion
Skeletal muscles can be viewed as a composite structure composed of muscle fibres, nerves, blood vessels and, importantly for this study, connective tissue. It is necessary that these differing components develop in a coordinated manner to generate an optimally functional organ. The mechanism underpinning interdependency between components of skeletal muscle has fascinated biologists for hundreds of years, and considerable advances have been made primarily in explaining the coordinated development of muscle fibre and nerves.
The relationship between muscle fibre development and its connective tissue is not well understood. Myostatin is a potent inhibitor of muscle development. Loss of function mutations have been described in numerous vertebrate species including humans that result in enhanced skeletal muscle development (Mcpherron et al. 1997) (Mcpherron & Lee, 1997; Schuelke et al. 2004; Shelton & Engvall, 2007). We have recently shown that skeletal muscle hypertrophy in the mstn−/− mouse is accompanied by a proportionate increase in motor neurons and proprioceptors that ensure the development of a normal functional organ (Elashry et al. 2011). In this study we investigated the development of connective tissue in the absence of myostatin in skeletal muscles. We show that the thickness of the perimysium and endomysium is reduced in mstn−/− muscle compared with mstn+/+. This is congruent with our findings that show decreased amounts of key components of the perimysium and endomysium including collagen I, II and IV in mstn−/−. Similar results have been found in cattle harbouring a mutation in the myostatin gene (Bailey et al. 1982).
Interestingly, we also showed that the age-related increase in MMP-9 expression in mstn+/+ does not occur in mstn−/− muscle. MMPs have been traditionally implicated in degradation of the components of the ECM (Vu & Werb, 2000). However, additional functions for these enzymes have been sought, as MMPs are often active in conditions that display increased rather than decreased levels of ECM deposition (Choi & Dalakas, 2000). Indeed, recent work has shown that MMP-9 activity can promote ECM development through its ability to process and activate TGF-β1, a predominant mediator of fibrosis (Li et al. 2009). Here, we propose that in wild-type mice, MMP-9 acts to promote collagen synthesis in skeletal muscle during ageing, whereas its activity decreases with time in mstn−/− muscle, which may explain the lack of connective tissue.
A number of elegant studies have dissected the molecular mechanisms by which myostatin controls connective tissue development, which have highlighted the tissue-specific deployment of signalling pathways. Smad- and Akt-mediated signalling inhibits myogenesis, whereas the same pathways promote proliferation of myofibroblasts and development of connective tissue (Langley et al. 2002; Zhang et al. 2006; Li et al. 2008). Therefore, it seems paradoxical in the context of developing skeletal muscle as an organ that a molecule that increases connective tissue should at the same time decrease the amount of muscle. This apparent inconsistency can be resolved by viewing the myostatin phenotype from a qualitative perspective rather than a quantitative point of view. Key to our explanation is the fact that the loss of myostatin results in the development of fast contracting muscle. The concentration of collagen and other components of the ECM are lower in fast muscle compared with slow muscle (Kovanen et al. 1980). This correlation extends to the level of single myofibres, where it has been shown that the concentration of collagen associated with fast fibres is half that of slow fibres (Kovanen et al. 1984a). The high levels of connective tissue associated with slow contracting muscle enables them to store more energy in the collagenous compartment compared with muscles with low connective tissue content. Fast contracting muscle with its decreased capacity to store energy in connective tissues uses contraction more directly to move skeletal elements (Kovanen et al. 1984b). We suggest that in the absence of myostatin, the type of connective tissue (thin) that develops is ideally suited to the contractile profile of the muscle (fast). However, myostatin-regulated signalling does not control the initial step in the formation of connective tissue. This is also the case for skeletal muscle, as Matsakas et al. have shown primary myogenesis proceeds normally in the absence of myostatin, which is subsequently modified (Matsakas et al. 2010). Therefore, myostatin signalling ensures that the correct type of connective tissue develops for specific contractile muscle types.
The interplay between the development of connective tissue and myogenic compartment has been of considerable interest to embryologists for decades, and it is generally believed that muscle patterning (size and form) is controlled by the surrounding connective tissue (Kardon et al. 2003; Valasek et al. 2011). Our data imply that the initial formation of connective tissue and muscle occurs independently of myostatin. However, myostatin then may dictate the expansion of these two tissue types through paracrine- and autocrine-positive feedback mechanisms that have been demonstrated in fibroblasts and myogenic cells (Zhu et al. 2007). In such a scenario, high levels of myostatin originating from connective tissues (through its central role in patterning) would initiate high levels of myostatin in muscle cells. The outcome here would be robust myofibroblast proliferation and attenuated muscle development. Furthermore, high levels of myostatin signalling in muscle cells ensure the development of slow phenotype through the induction of MEF2C (Hennebry et al. 2009).
Concluding remarks
The results from this study showed that myostatin signalling is required for post-natal age-related increase in endomysium and perimysium connective tissue. A number of studies have shown that myostatin promotes the proliferation of not only muscle fibroblasts (Zhu et al. 2007) but also other populations of fibroblasts, including those from tendons (Mendias et al. 2008; Fulzele et al. 2010). The fact that myostatin promotes myofibroblast development could be exploited in a clinical setting to control fibrosis, which is often associated with conditions that involve persistent muscle damage, such as Duchenne muscular dystrophy (DMD; Mcloon, 2008; Zhou & Lu, 2010). Fibrosis is postulated to inhibit not only muscle regeneration but also muscle function by decreasing its strength and elasticity as well as impeding the diffusion of nutrients (Kaariainen et al. 2000). Inhibition of myostatin signalling using epigenetic approaches has already been shown to increase muscle growth in adult animals and to increase muscle mass in mouse models of DMD (Bogdanovich et al. 2002; Matsakas et al. 2009). Therefore, inhibition of myostatin signalling not only stimulates the formation of muscle, but inhibits fibrosis (Bogdanovich et al. 2002; Wagner et al. 2002). In addition, because myostatin is produced by fibroblasts resident in the connective tissue compartment of most organs (Li et al. 2008), we suggest that myostatin antagonists could be useful in controlling fibrosis in conditions such as pulmonary fibrosis (Laurent & Tetley, 1984).
Acknowledgments
We are indebted to the Government of Egypt for financial support permitting this study. We would also like to thank Sajjida Jaffer and three anonymous reviewers for valuable comments during the preparation of the manuscript, and Professor Ashok Kumar (Louisville, USA) for technical information regarding Western blotting and the collagen antibodies.
References
- Bailey AJ, Enser MB, Dransfield E. In: Muscle hypertrophy of genetic origin and its use to improve beef production. King JWB, Ménissier F, et al., editors. The Hague: Martinus Nijhoff Publishers; 1982. pp. 179–202. [Google Scholar]
- Bogdanovich S, Krag TO, Barton ER, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- Choi YC, Dalakas MC. Expression of matrix metalloproteinases in the muscle of patients with inflammatory myopathies. Neurology. 2000;54:65–71. doi: 10.1212/wnl.54.1.65. [DOI] [PubMed] [Google Scholar]
- Elashry MI, Otto A, Matsakas A, et al. Axon and muscle spindle hyperplasia in the myostatin null mouse. J Anat. 2011;218:173–184. doi: 10.1111/j.1469-7580.2010.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulzele S, Arounleut P, Cain M, et al. Role of myostatin (GDF-8) signaling in the human anterior cruciate ligament. J Orthop Res. 2010;28:1113–1118. doi: 10.1002/jor.21112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass DJ. Molecular mechanisms modulating muscle mass. Trends Mol Med. 2003;9:344–350. doi: 10.1016/s1471-4914(03)00138-2. [DOI] [PubMed] [Google Scholar]
- Hennebry A, Berry C, Siriett V, et al. Myostatin regulates fiber-type composition of skeletal muscle by regulating MEF2 and MyoD gene expression. Am J Physiol Cell Physiol. 2009;296:C525–C534. doi: 10.1152/ajpcell.00259.2007. [DOI] [PubMed] [Google Scholar]
- Kaariainen M, Jarvinen T, Jarvinen M, et al. Relation between myofibers and connective tissue during muscle injury repair. Scand J Med Sci Sports. 2000;10:332–337. doi: 10.1034/j.1600-0838.2000.010006332.x. [DOI] [PubMed] [Google Scholar]
- Kardon G, Harfe BD, Tabin CJ. A Tcf4-positive mesodermal population provides a prepattern for vertebrate limb muscle patterning. Dev Cell. 2003;5:937–944. doi: 10.1016/s1534-5807(03)00360-5. [DOI] [PubMed] [Google Scholar]
- Kovanen V, Suominen H, Heikkinen E. Connective tissue of “fast” and “slow” skeletal muscle in rats – effects of endurance training. Acta Physiol Scand. 1980;108:173–180. doi: 10.1111/j.1748-1716.1980.tb06515.x. [DOI] [PubMed] [Google Scholar]
- Kovanen V, Suominen H, Heikkinen E. Collagen of slow twitch and fast twitch muscle fibres in different types of rat skeletal muscle. Eur J Appl Physiol Occup Physiol. 1984a;52:235–242. doi: 10.1007/BF00433399. [DOI] [PubMed] [Google Scholar]
- Kovanen V, Suominen H, Heikkinen E. Mechanical properties of fast and slow skeletal muscle with special reference to collagen and endurance training. J Biomech. 1984b;17:725–735. doi: 10.1016/0021-9290(84)90103-9. [DOI] [PubMed] [Google Scholar]
- Langley B, Thomas M, Bishop A, et al. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002;277:49 831–49 840. doi: 10.1074/jbc.M204291200. [DOI] [PubMed] [Google Scholar]
- Laurent GJ, Tetley TD. Pulmonary fibrosis and emphysema: connective tissue disorders of the lung. Eur J Clin Invest. 1984;14:411–413. doi: 10.1111/j.1365-2362.1984.tb01204.x. [DOI] [PubMed] [Google Scholar]
- Li ZB, Kollias HD, Wagner KR. Myostatin directly regulates skeletal muscle fibrosis. J Biol Chem. 2008;283:19 371–19 378. doi: 10.1074/jbc.M802585200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Mittal A, Makonchuk DY, et al. Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum Mol Genet. 2009;18:2584–2598. doi: 10.1093/hmg/ddp191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsakas A, Patel K. Intracellular signalling pathways regulating the adaptation of skeletal muscle to exercise and nutritional changes. Histol Histopathol. 2009a;24:209–222. doi: 10.14670/HH-24.209. [DOI] [PubMed] [Google Scholar]
- Matsakas A, Patel K. Skeletal muscle fibre plasticity in response to selected environmental and physiological stimuli. Histol Histopathol. 2009b;24:611–629. doi: 10.14670/HH-24.611. [DOI] [PubMed] [Google Scholar]
- Matsakas A, Foster K, Otto A, et al. Molecular, cellular and physiological investigation of myostatin propeptide-mediated muscle growth in adult mice. Neuromuscul Disord. 2009;19:489–499. doi: 10.1016/j.nmd.2009.06.367. [DOI] [PubMed] [Google Scholar]
- Matsakas A, Otto A, Elashry MI, et al. Altered primary and secondary myogenesis in the myostatin-null mouse. Rejuvenation Res. 2010;13:717–727. doi: 10.1089/rej.2010.1065. [DOI] [PubMed] [Google Scholar]
- Mccroskery S, Thomas M, Platt L, et al. Improved muscle healing through enhanced regeneration and reduced fibrosis in myostatin-null mice. J Cell Sci. 2005;118:3531–3541. doi: 10.1242/jcs.02482. [DOI] [PubMed] [Google Scholar]
- Mcloon LK. Focusing on fibrosis: halofuginone-induced functional improvement in the mdx mouse model of Duchenne muscular dystrophy. Am J Physiol Heart Circ Physiol. 2008;294:H1505–H1507. doi: 10.1152/ajpheart.00176.2008. [DOI] [PubMed] [Google Scholar]
- Mcpherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA. 1997;94:12 457–12 461. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcpherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- Mendias CL, Marcin JE, Calerdon DR, et al. Contractile properties of EDL and soleus muscles of myostatin-deficient mice. J Appl Physiol. 2006;101:898–905. doi: 10.1152/japplphysiol.00126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendias CL, Bakhurin KI, Faulkner JA. Tendons of myostatin-deficient mice are small, brittle, and hypocellular. Proc Natl Acad Sci USA. 2008;105:388–393. doi: 10.1073/pnas.0707069105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nindl BC, Pierce JR. Insulin-like growth factor I as a biomarker of health, fitness, and training status. Med Sci Sports Exerc. 2010;42:39–49. doi: 10.1249/MSS.0b013e3181b07c4d. [DOI] [PubMed] [Google Scholar]
- Schuelke M, Wagner KR, Stolz LE, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- Shelton GD, Engvall E. Gross muscle hypertrophy in whippet dogs is caused by a mutation in the myostatin gene. Neuromuscul Disord. 2007;17:721–722. doi: 10.1016/j.nmd.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Srikanthan P, Karlamangla AS. Relative muscle mass is inversely associated with insulin resistance and prediabetes. Findings from the Third National Health and Nutrition Examination Survey. J Clin Endocrinol Metab. 2011;96:2898–2903. doi: 10.1210/jc.2011-0435. [DOI] [PubMed] [Google Scholar]
- Valasek P, Theis S, Delaurier A, et al. Cellular and molecular investigations into the development of the pectoral girdle. Dev Biol. 2011;357:108–116. doi: 10.1016/j.ydbio.2011.06.031. [DOI] [PubMed] [Google Scholar]
- Vu TH, Werb Z. Matrix metalloproteinases: effectors of development and normal physiology. Genes Dev. 2000;14:2123–2133. doi: 10.1101/gad.815400. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Mcpherron AC, Winik N, et al. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002;52:832–836. doi: 10.1002/ana.10385. [DOI] [PubMed] [Google Scholar]
- Zhang D, Liu M, Ding F, et al. Expression of myostatin RNA transcript and protein in gastrocnemius muscle of rats after sciatic nerve resection. J Muscle Res Cell Motil. 2006;27:37–44. doi: 10.1007/s10974-005-9050-5. [DOI] [PubMed] [Google Scholar]
- Zhou L, Lu H. Targeting fibrosis in Duchenne muscular dystrophy. J Neuropathol Exp Neurol. 2010;69:771–776. doi: 10.1097/NEN.0b013e3181e9a34b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Li Y, Shen W, et al. Relationships between transforming growth factor-beta1, myostatin, and decorin: implications for skeletal muscle fibrosis. J Biol Chem. 2007;282:25 852–25 863. doi: 10.1074/jbc.M704146200. [DOI] [PubMed] [Google Scholar]