

Abstract
Clinically available anti-tumour necrosis factor (TNF) biologics, which inhibit both soluble (sTNF) and transmembrane forms (tmTNF) of TNF, eliminating all TNF signalling, have successfully treated autoimmune diseases including uveitis. These have potentially serious side effects such as reactivation of latent Mycobacterium tuberculosis and, therefore, more specific inhibition of TNF signalling pathways may maintain clinical efficacy while reducing adverse effects. To determine the effects of specific pharmacological inhibition of sTNF on macrophage activation and migration, we used a mouse model of uveitis (experimental autoimmune uveoretinitis; EAU). We show that selective inhibition of sTNF is sufficient to suppress EAU by limiting inflammatory CD11b+ macrophages and CD4+ T cell migration into the eye. However, inhibition of both sTNF and tmTNF is required to inhibit interferon-γ-induced chemokine receptor 2, CD40, major histocompatibility complex class II and nitric oxide (NO) up-regulation, and signalling via tmTNF is sufficient to mediate tissue damage. In confirmation, intravitreal inhibition of sTNF alone did not suppress disease, and inflammatory cells that migrated into the eye were activated, generating NO, thus causing structural damage to the retina. In contrast, intravitreal inhibition of both sTNF and tmTNF suppressed macrophage activation and therefore disease. We conclude that sTNF is required for inflammatory cell infiltration into target tissue, but at the tissue site inhibition of both sTNF and tmTNF is required to inhibit macrophage activation and to protect from tissue damage.
Keywords: autoimmunity, eye (ocular) immunology disease, immune regulation, macrophages/monocytes, nitric oxide
Introduction
Tumour necrosis factor (TNF) is produced during autoimmune inflammatory diseases, where it contributes to leucocyte emigration [1] and tissue damage [2],[3]. A central role of TNF is facilitating monocyte trafficking to the target organ [4]. In a murine model of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE), TNF−/− mice have delayed onset of disease [5],[6]. TNF production by haematopoietic cells from the bone marrow is required for these inflammatory cells to infiltrate the target organ during EAE, as shown by studies of mice reconstituted with bone marrow from TNF−/− or wild-type (WT) donors [7]. The use of anti-TNF biologics in models of non-infectious posterior uveitis (experimental autoimmune uveoretinitis; EAU) has shown that eliminating TNF signalling delays cell trafficking into the retina [8]–[12]. As a model, EAU offers the ability to distinguish clinical disease from structural damage as a consequence of inflammatory cell infiltration and immunopathogenic mechanisms involved. In the context of EAU, our previous data demonstrated that signalling via TNFR1 controls macrophage activation as well as trafficking to the site of inflammation [4],[13],[14]. Using mixed bone marrow chimeras from both WT and TNFR1−/− donors, we showed that WT TNFR1+/+ monocytes, but not T cells, had a selective advantage and were recruited preferentially to the inflamed eye [4].
Other studies have used transmembrane (tm)TNF transgenic mice that cannot express soluble (s)TNF to show that these mice still develop EAE, although disease is delayed and cell movement within the central nervous system (CNS) is impaired. One study showed that tmTNF knock-in mice did not succumb to EAE [15], while another showed that, although tmTNF transgenic mice had comparable levels of EAE to WT, when the concentration of the Mycobacterium tuberculosis in the complete Freud's adjuvant (CFA) was decreased EAE in the tmTNF transgenics was delayed and suppressed [16]. This study shows the importance of inflammatory cell activation, presumably by sTNF, in the initiation of EAE. Such observations may be due to tmTNF mice inducing lower levels of chemokines [16]. These mice lacking sTNF expression have abnormal B cell follicle formation, and such differences could affect disease outcomes [16] and, therefore, inhibition of sTNF is required after the mice have developed normally. XPro1595 is a recently developed molecule that specifically targets and inhibits sTNF with no effect on tmTNF, and is an effective tool that has facilitated investigation into the function and role played by sTNF in disease. Previously it has been shown that specific blockade of sTNF with XPro1595 can suppress murine arthritis to the same extent as pan-blocking TNF agents such as etanercept, infliximab and adalimumab [17].
In this study, we show that sTNF is therefore necessary to mobilize cells for recruitment to the target organ during inflammation, but tmTNF signalling is required for local activation of these cells and subsequent tissue damage.
Methods and materials
Animals
B10.RIII mice were obtained from Harlan UK Limited (Oxford, UK), C57BL/6 Ly.5 (CD45.1) congenic mice were obtained from Charles River Laboratory (Kent, UK) and TNFRp55-deficient mice [TNF receptor 1 (TNFR1−/−)] of background strain C57BL/6 were obtained from The Jackson Laboratory. Breeding colonies were established within the Animal Services Unit (ASU) (Bristol, UK). Specific pathogen-free, isolator-reared female mice were maintained in accordance with Home Office Regulations for Animal Experimentation, UK, and conformed to the Association of Research in Vision and Ophthalmology statement of the use of animals in ophthalmic and vision research.
Reagents
Recombinant murine interferon (IFN)-γ (Peprotech, London, UK), lipopolysaccharide (LPS) (Sigma-Aldrich, Dorset, UK), human immunoglobulin (Ig)G (Genscript USA, Inc., Piscataway, NJ, USA), sTNFR-Ig [9] (a gift from Herman Waldmann, University of Oxford, UK) and XPro1595 [18] (a gift from Xencor, Monrovia, CA, USA). The fusion protein sTNFR-Ig is a human Ig Fc, and therefore the control for this was a human isotype IgG. XPro1595 is an engineered protein that can form heterotrimers with native sTNF to provide complexes that cannot bind to the TNF receptors. The control we used for this was inactivated XPro-1595 (I-XPro), where XPro1595 had been denatured (using trypsin and heat denaturing treatment, with repeated freeze/thaw cycles) but was still in the same vehicle (Fig. 2a).
Bone marrow-derived macrophages (BMDMϕ)
Bone marrow cells were obtained by flushing the femurs of female mice and the cells were cultured as described previously [19].
EAU induction and disease scoring
Female B10.RIII mice (6–9 weeks old) were immunized subcutaneously in one flank with 50 µg human retinol-binding protein 3 (hRBP-3)161–180 (SGIPYIISYLHPGNTILHVD) in 2% dimethylsulphoxide (DMSO) in emulsion with CFA (1 mg/ml, 1:1 v/v) supplemented with 1·5 mg/ml M. tuberculosis complete H37 Ra (Difco Laboratories, Detroit, MI, USA). Mice were also given an intraperitoneal injection of 1 µg Bordetella pertussis toxin (Tocris, Bristol, UK). Mice were treated with 10 mg/kg sTNFR-Ig, XPro1595, I-XPro or human IgG either intraperiteanally (i.p.) or 10 µg/eye intravitreally. Eyes were enucleated at various time-points and serial 12-µm sections cut and stained with rat anti-mouse monoclonal anti-CD45 antibody (Serotec, Oxford, UK) and counterstained with haematoxylin (ThermoShandon, Pittsburgh, PA, USA). Sections were scored for inflammatory infiltrate (presence of CD45+ cells) and structural disease (disruption of morphology), as described previously [13]. For immunofluorescence, nitrotyrosine was detected using an anti-nitrotyrosine antibody (Sigma-Aldrich, Dorset, UK), followed by a biotinylated anti-mouse antibody (Vector, Peterborough, UK) and streptavidin–rhodamine red-X (Jackson Immuno-Research, Newmarket, UK).
Topical endoscopic fundus imaging (TEFI)
The TEFI method used has been described previously [20]. Fundal images were blind-scored on inflammatory changes of the optic disc, retinal vessels, retinal lesions and structural damage. All scores were added together to make a final clinical disease score. This clinical grading system was adapted from Xu et al. [21] (Table 1).
Table 1.
Topical endoscopic fundus imaging (TEFI) scoring criteria.
| Score | Optic disc | Retinal vessels | Retinal tissue infiltrate | Structural damage |
|---|---|---|---|---|
| 1 | Minimal inflammation | 1–4 mild cuffings | 1–4 small lesions or 1 linear lesion | Retinal lesions or retinal atrophy involving 1/4 to 3/4 retina area |
| 2 | Mild inflammation | >4 mild cuffings or 1–3 moderate cuffings | 5–10 small lesions or 2–3 linear lesions | Pan retinal atrophy with multiple small lesions (scars) or <3 linear lesions (scars) |
| 3 | Moderate inflammation | >3 moderate cuffings | >10 small lesions or >3 linear lesions | Pan-retinal atrophy with >3 linear lesions or confluent lesions (scars) |
| 4 | Severe inflammation | >1 severe cuffing | Linear lesion confluent | Retinal detachment with folding |
| 5 | Not visible (white-out or extreme detachment) | Not visible (white-out or extreme detachment) | Not visible (white-out or extreme detachment) | Not visible |
Flow cytometry
The eyes were dissected and the retina extracted as described previously [22]. A spleen that had been taken at the same time was also prepared for flow cytometry to equate event counts with cell numbers. The retinal cells were stained with rat anti-mouse allophycocyanin (APC)-CD4 (clone RM4-5; BD Biosciences, Oxford, UK), rat anti-mouse phycoerythrin cyanin 7 (PE.Cy7)-CD45 (clone 30-F11; BD Biosciences, Oxford, UK) and rat anti-mouse APC.Cy7-CD11b (clone M1/70; BD Biosciences, Oxford, UK). 7-Aminoactinomycin D (7AAD) was used to eliminate dead cells. BMDMϕ were stimulated as in the figure legends, washed and stained with the live/dead near IR kit (Invitrogen, Paisley, UK) for 30 min. The cells were then washed and incubated with anti-CD45.1 (clone A20; BD Biosciences, Oxford, UK), anti-I-Ab (clone KH74; BD Biosciences, Oxford, UK), anti-CD40 (clone 3/23; BD Biosciences, Oxford, UK) and anti-chemokine receptor 2 (CCR2) (Abcam, Cambridge, UK) for 20 min on ice, followed by another wash. The cells were then incubated with anti-rabbit fluorescein isothiocyanate (FITC) (Invitrogen) and streptavidin–APC (BD Biosciences, Oxford, UK) for 30 min on ice prior to analysis. The BD FACS Canto II with two lasers and six colours and the FACS DiVa version 5.0.2 software (BD Biosciences, San Jose, CA, USA) were used for acquisition and FlowJo for analysis. BMDMϕ were stimulated, washed and stained with the live/dead near IR kit (Invitrogen) for 30 min.
Nitric oxide (NO) quantification
Twenty-four h after cell stimulation, each cell-free supernatant was incubated in Griess reagent (v/v) (Sigma-Aldrich, Dorset, UK) in 96-well flat-bottomed plates for 10 min at room temperature. To quantify NO production by splenic macrophages, spleen were mashed and put through a cell strainer to form a single-cell suspension. The red blood cells were lysed and the CD11b+-positive selection magnetic affinity cell sorting (MACS) kit (Miltenyi Biotec Ltd, Bisley, Surrey, UK) used to isolate macrophages, according to the kit's instructions.
Proliferation assays
Spleens were collected from immunized mice at various time-points. Cells were then stimulated with the immunizing peptide and incubated for 2 days at 37°C in 5% CO2 in a humidified atmosphere. The plates were pulsed with 18·5-kBq titrated thymidine (GE Healthcare, Bucks, UK) per well for the next 18 h of incubation. Thymidine uptake (counts per minute; cpm) was determined by liquid scintillation with a microbeta liquid scintillation counter (Wallac 1450; PerkinElmer Life Sciences, Cambridge, UK).
Enzyme-linked immunosorbent assays (ELISAs)
Interleukin (IL)-6 sandwich ELISA capture antibody (15 pg/ml limit, MP5-20F3; BD Biosciences, Oxford, UK) was applied in carbonate buffer, pH 9·6. The detection antibody (MP5-32C11; BD Biosciences, Oxford, UK) was added for 1 h at room temperature. The manufacturer's instructions were followed for the sandwich TNF ELISA kit (30 pg/ml limit; R&D Systems, Abingdon, UK).
Statistical analysis
The Mann–Whitney U-test (two-tailed) was used for statistical analysis and P < 0·05 was considered significant. All in vitro experiments were carried out at least three times. The in vivo experiments were carried out multiple times (>2) and the results combined. The mean ± standard error is shown graphically. Each eye was treated as an independent variable.
Results
XPro1595 inhibits sTNF but not IL-6 production from LPS-stimulated macrophages
Consistent with published data [13],[17], XPro1595 and sTNFR-Ig can inhibit production of sTNF by macrophages (Fig. 1a,b). However, in contrast to sTNFR-Ig (32·4% decrease, P = 0·0495), XPro1595 failed to suppress IL-6 production (Fig. 1c,d). The data demonstrate that both sTNFRIg and XPro1595 can suppress sTNF production in vitro while establishing that tmTNF alone is sufficient to induce IL-6 production by BMDMϕ.
Fig. 1.
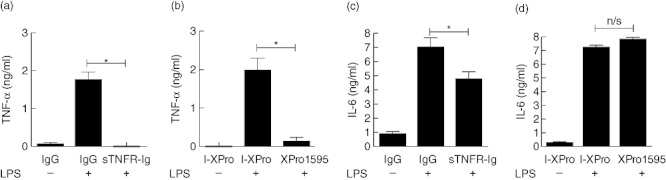
Both soluble tumour necrosis factor receptor-immunoglobulin (sTNFR-Ig) and XPro1595 can inhibit sTNF production but only sTNFR-Ig can suppress interleukin (IL)-6 secretion. Bone marrow-derived macrophages (BMDMϕ) from wild-type mice were incubated with 100 ng/ml lipopolysaccharide (LPS) for 24 h in the presence of 10 µg/ml human IgG or sTNFR-Ig (a,c) or 10 µg/ml I-XPro or XPro1595 (b,d). TNF (a,b) or IL-6 (c,d) production was quantified by enzyme-linked immunosorbent assay. Mean ± standard error shown, n = 3, *P < 0·05.
Systemic inhibition of sTNF alone can significantly suppress EAU
Previous work has shown that inhibition of TNF signalling using the sTNFR-Ig fusion protein (which inhibits pan-TNF) can suppress murine EAU [8]–[12]. Given the potency of XPro1595 (which inhibits sTNF only) in suppressing sTNF secretion from macrophages, it was important to assess the in vivo efficacy of XPro1595 treatment compared to the efficacy of sTNFR-Ig treatment. I-XPro-treated mice have comparable disease to phosphate-buffered saline (PBS)-treated mice (Fig. 3a). Progression of clinical disease [20],[21] (Fig. 3b) is slower in both the treated groups in comparison to the control group. While the control mice developed severe EAU, with synechia, vitritis, pan-vasculitis and retinal lesions (Fig. 2), both the treated groups only showed signs of mild EAU with an inflamed optic disc and mild vasculitis in some mice. Other treated mice showed no clinical signs of EAU, but a few developed severe EAU. By day 18 post-immunization (p.i.), the control I-XPro-treated mice had obvious signs of severe disease, therefore the mice were killed and eyes enucleated for flow cytometric (Fig. 3c,d) and histological (Fig. 3e) analysis. Treatment with sTNFR-Ig led to a 83·0% reduction in CD11b+ cells (P = 0·002) and a 94·5% reduction in CD4+ cells (P = 0·003) compared to the I-XPro control. In the XPro1595-treated group, there was a 77·9% reduction in CD11b+ cells (P = 0·040) and a 68·7% reduction in CD4+ cells (P = 0·040) compared to the I-XPro control. There was a significant increase in the CD11b+ : CD4+ cell populations (P = 0·03) in the sTNFR-Ig-treated mice versus the control I-XPro-treated group; there was no significant difference between the XPro1595 treatment group and the sTNFR-Ig or the XPro1595 and I-XPro control groups.
Fig. 2.
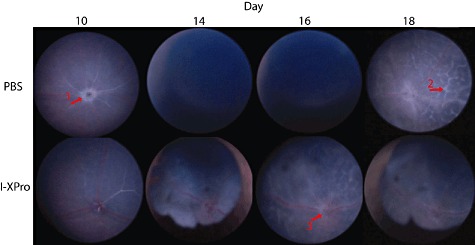
Topical endoscopic fundal imaging (TEFI). Experimental autoimmune uveoretinitis (EAU) was induced in B10.RIII mice, and on day 10 post-immunization (p.i.) the mice were treated intraperitoneally (i.p.) with 10 mg/kg [in 100 µl phosphate-buffered saline (PBS)] I-XPro or 100 µ1 PBS. Clinical progression of disease was assessed by TEFI from days 8 to 18; n = 4 mice per group and representative images are shown. In the PBS day 10 image, a raised and swollen optic disc is clearly visible (arrow 1). In the PBS-treated group on days 14 and 16, severe vitritis is causing a vitreous haze, precluding fundal imaging. On day 18 in the PBS-treated group, lesions (arrow 2) that correspond to histological retinal folds are present. In the I-XPro-treated mice on days 14 and 18, development of synechia was noted. On day 16 in the I-XPro-treated group, the optic disc is inflamed (raised and blurred margins; arrow 3) alongside widely distributed perivascular inflammation (vasculitis) and exudative retinal detachment as well as development of inflammatory retinal infiltrates.
Fig. 3.
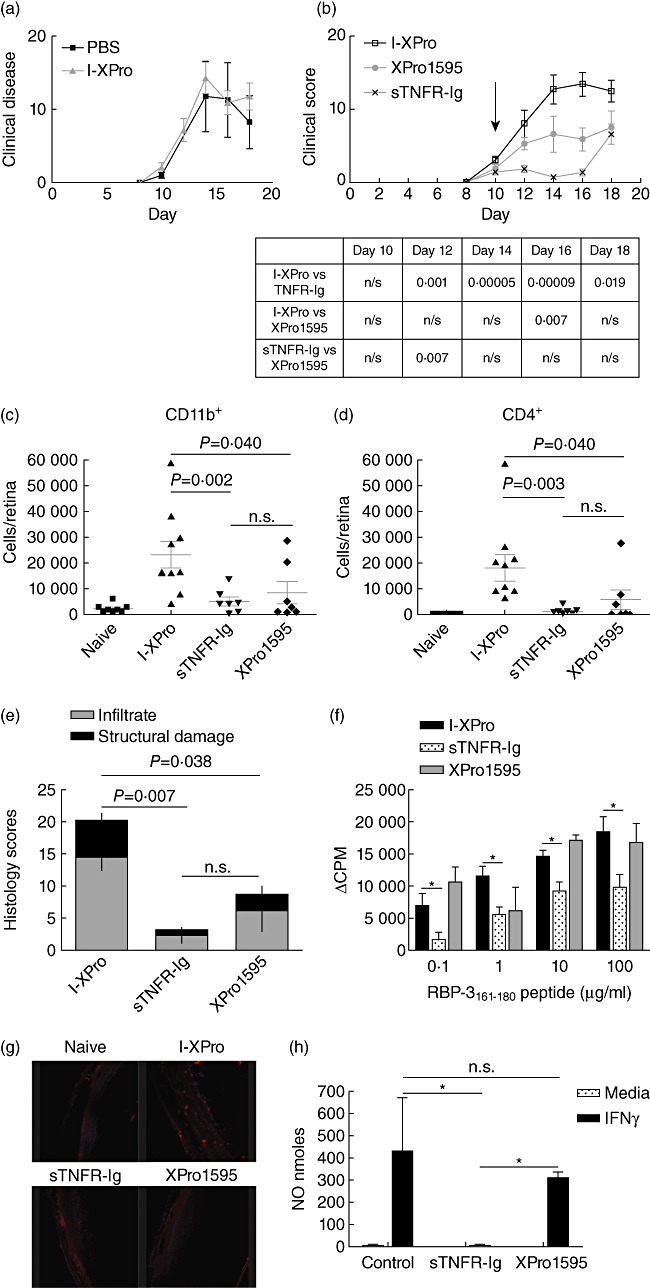
Inhibition of soluble tumour necrosis factor (sTNF) can suppress experimental autoimmune uveoretinitis (EAU). EAU was induced in B10.RIII mice, and on day 10 post-immunization (p.i.) the mice were treated intraperitoneally (i.p.) with 10 mg/kg [in 100 µl phosphate-buffered saline (PBS)] I-XPro or 100 µ1 PBS. Clinical progression of disease with assessed by topical endoscopic fundus imaging (TEFI) from days 8 to 18; n = 4 mice per group (a). EAU was induced in B10.RIII mice, and on day 10 p.i. the mice were treated i.p. with 10 mg/kg of either I-XPro, sTNF receptor-immunoglobulin (sTNFR-Ig) or XPro1595 and the clinical progression of disease with assessed by TEFI from days 8 to 18. Treatment day is indicated by the arrow (b). The P-values for each day are tabulated below the graph. On day 18, the mice were killed and eyes taken for flow cytometric analysis of CD11b+ (c) and CD4+ (d) cells and histological analysis (e); mean ± standard error shown, n = 7–9 mice per group. On day 18 post-immunization, splenocytes were also used for a proliferation assay using 0·1–100 µg retinol-binding protein 3 (RBP-3)161–181 peptide (f); mean ± standard error shown. Eye sections from each mouse were also processed to detect nitrotryosine (red) and nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI) (blue), ×20 magnification (g). Naive mice were injected (i.p.) with 200 µg/mouse of I-XPro, sTNFR-Ig or XPro1595 and after 3 days the spleens removed and macrophages isolated using magnetic affinity cell sorting (MACS). The macrophages were stimulated in vitro with 100 U/ml interferon (IFN)-γ for 72 h prior to nitric oxide (NO) quantification; n = 3 (h).
Histological analysis showed that on day 18 p.i., the scores decreased by 85·1% (P = 0·007) in the sTNFR-Ig group and 41·1% (P = 0·038) in the XPro1595-treated group compared to the I-XPro control group. These data demonstrate that selective inhibition of sTNF alone is sufficient to suppress infiltration to the retina of both CD11b+ macrophages and CD4+ T cells subsets during EAU, with reduced retinal damage.
On day 18 p.i., splenocytes were obtained to assess potential differences in antigen-specific (RBP-3161–180) T cell proliferation (Fig. 3f). Treatment with sTNFR-Ig suppressed T cell proliferation significantly, while XPro1595 left T cell proliferation unimpaired.
The presence of nitrotyrosine in the histological sections was also investigated (Fig. 3g). No nitrotyrosine is present in naive eyes. Nitrotyrosine is present in eyes from mice treated with I-XPro, in contrast to eyes from mice treated with sTNFR-Ig or XPro1595, suggesting that NO is not present in the eyes of treated mice. Using macrophages isolated from spleens of naive mice that were treated with I-XPro, sTNFR-Ig and XPro1595, we showed that IFN-γ-induced NO is suppressed significantly when the cells have been treated previously with sTNFR-Ig but not when they have been treated with XPro1595 (Fig. 3h).
These results demonstrate that systemic inhibition of sTNF alone, with a single dose of either sTNFR-Ig or XPro1595, can reduce clinical disease, structural tissue damage and CD4+ and CD11b+ cell infiltration. Systemic treatment reduces the number of infiltrating cells in the eye, therefore nitrotyrosine production is absent or very low. However, splenic macrophages in the XPro1595-treated group can still produce NO, unlike splenic macrophages from the sTNFR-Ig-treated group.
XPro1595 can suppress established EAU
Clearly, inhibition of sTNF alone is sufficient to prevent the progression of EAU when treatment was initiated before clinical signs developed, with no significant differences between sTNFR-Ig [8]–[12] and XPro1595 [17]; therefore, it was necessary to determine whether similar efficacy could be achieved in established disease when only inhibiting sTNF. To this end, on day 12 p.i., only animals that had developed initial signs of clinical disease by TEFI were selected and randomized into different groups. Groups of mice were then treated continuously with either XPro1595 or I-XPro and monitored clinically (Fig. 4a). XPro1595 suppressed clinical disease significantly throughout the course of the experiment. Treatment retarded EAU, with delayed development of vasculitis and lesions. On day 23 p.i., the mice were killed and eyes enucleated for flow cytometric (Fig. 4b,c) and histological (Fig. 4d) analysis. Inhibition of sTNF after clinical disease had been established still led to 58·5% fewer CD11b+ macrophages (P = 0·023) and 77·3% fewer CD4+ T cells (P = 0·0064) in the retinas of XPro1595-treated mice.
Fig. 4.
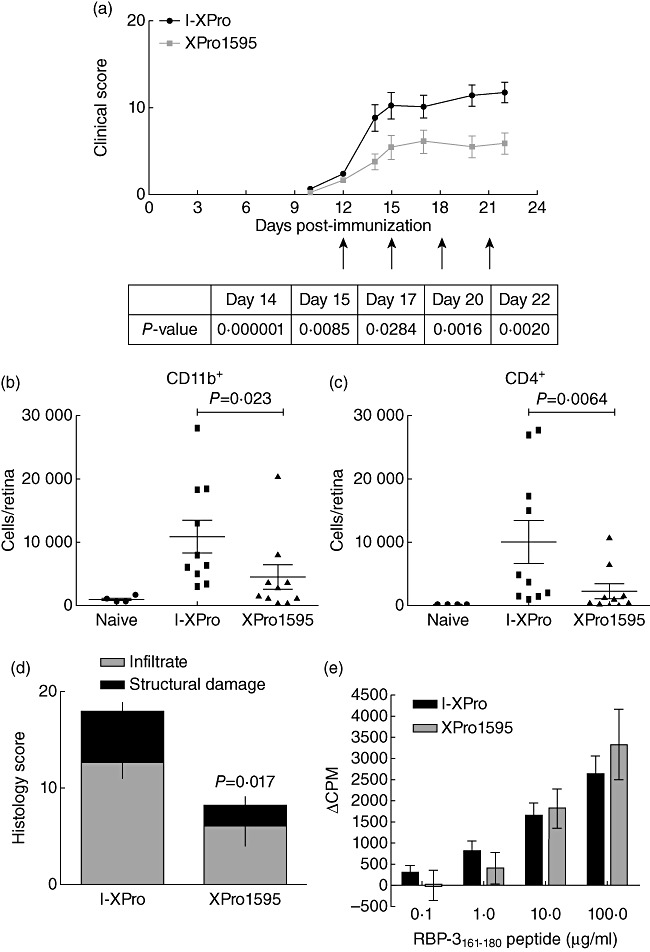
Inhibition of soluble tumour necrosis factor (sTNF) can also suppress established experimental autoimmune uveoretinitis (EAU). EAU was induced in B10.RIII mice and on day 12 post-immunization (p.i.); only mice that showed signs of clinical disease by topical endoscopic fundus imaging (TEFI) were used in the experiment. These mice were randomized prior to intraperitoneal (i.p.) treatment with 10 mg/kg of either I-XPro or XPro1595 every 3 days from day 12 (indicated by arrows). Clinical progression of disease with assessed by TEFI from days 8 to 22 (a). The P-value for each day is stated in the table under the graph. On day 23, the mice were killed and eyes taken for flow cytometric analysis of CD11b+ (b) and CD4+ (c) cells and histological analysis (d); mean ± standard error shown, n = 10 mice per group. On day 18 p.i., splenocytes were also used in a proliferation assay stimulated with a range of 0·1–100 µg retinol-binding protein 3 (RBP-3)161–181 peptide (e); mean ± standard error shown.
Histological scores showed a 54·7% (P = 0·017) decrease in disease in the XPro1595-treated group. Proliferation assays were performed using spleens taken on day 23 p.i., and confirmed the previous observation that inhibition of sTNF does not alter T cell proliferation (Fig. 3e).
The results thus far show that systemic inhibition of sTNF in the periphery can suppress established EAU with a concomitant reduction in extent and number of infiltrating macrophages and T cells into the retina.
Inhibition of both sTNF and tmTNF is required to inhibit macrophage activation
TNF plays a central role in macrophage activation, disease expression and ultimately tissue damage in EAU [4],[9],[13],[14],[23]–[25]. Inhibiting TNF activity via sTNFR-Ig reduces EAU severity, suppresses NO expression in infiltrating myeloid cells [25] and reduces the activation status of both T cell and myeloid cells isolated from the retina [26]. Here we show that, in agreement with published data [13], and as seen in the rat [24], sTNFR-Ig can inhibit IFN-γ-induced NO production by WT BMDMϕ effectively (Fig. 5a), whereas XPro1595 does not suppress IFN-γ-induced NO by WT BMDMϕ (Fig. 5b). WT BMDMϕ still produced NO in the presence of XPro1595, while BMDMϕ from TNFR1−/− mice did not (Fig. 5c). These results show that tmTNF signalling via TNFR1 is sufficient to induce NO from IFN-γ stimulated macrophages. LPS-induced NO production is not TNF-dependent and was not suppressed by either sTNFR-Ig (Fig. 5d) or XPro1595 (Fig. 5e).
Fig. 5.
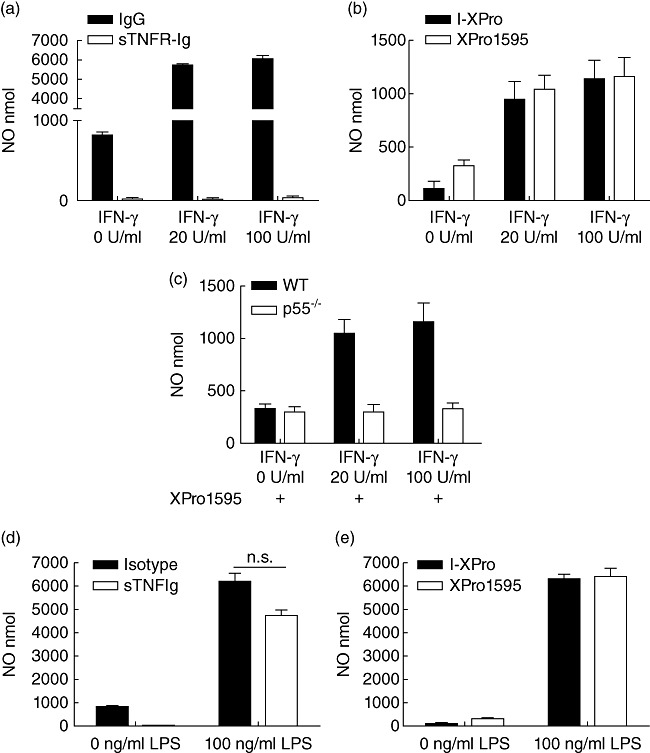
Transmembrane tumour necrosis factor (TNF) signalling via TNF receptor 1 (TNFR1) in interferon (IFN)-γ-induced macrophages is sufficient to induce nitric oxide. BMDMϕ from wild-type mice were incubated with 0 U/ml (media alone), 20 U/ml or 100 U/ml IFN-γ for 24 h in the presence of either 10 µg/ml human IgG or sTNFR-Ig (a) or 10 µg/ml I-XPro or XPro1595 (b). BMDMϕ from wild-type and TNFR1−/− were also incubated with 0–100 U/ml IFN-γ in the presence of XPro1595 (all samples, c). Nitric oxide in the supernatant was quantified. Bone marrow-derived macrophages (BMDMϕ) from wild-type mice were also incubated with media alone or 100 ng/ml lipopolysaccharide (LPS) for 24 h in the presence of 10 µg/ml immunoglobulin (Ig)G or soluble TNFR-Ig (sTNFR-Ig) (d) or 10 µg/ml I-XPro or XPro1595 (e). Mean ± standard error shown; n = 3–4.
IFN-γ-induced expression of both CD40 and major histocompatibility complex (MHC) class II (I-Ab) has been shown previously to be TNF-dependent [13]. CCR2 has also been implicated as a pivotal receptor required by inflammatory macrophages to infiltrate inflamed organs [27]–[29]. Therefore, the effects of sTNFR-Ig and XPro1595 on CD40, MHC class II and CCR2 cell surface expression were investigated. IFN-γ (Fig. 6a) and LPS (Fig. 6b) were used to stimulate WT and TNFR1−/− BMDMϕ. Stimulation with both LPS and IFN-γ induced CD40, MHC class II and CCR2 expression in WT macrophages but macrophages from TNFR1−/− mice expressed lower levels of CD40 after addition of IFN-γ compared to WT, in concordance with published data [13]. CCR2 expression was also reduced in macrophages from TNFR1−/− mice after stimulation with both IFN-γ and LPS. Addition of sTNFR-Ig reduced expression of IFN-γ-induced MHC class II, CD40 and CCR2 as well as LPS-induced MHC class II and CCR2, but not LPS-induced CD40. XPro1595, however, did not reduce the LPS- or IFN-γ-induced expression of MHC class II, CD40 or CCR2.
Fig. 6.
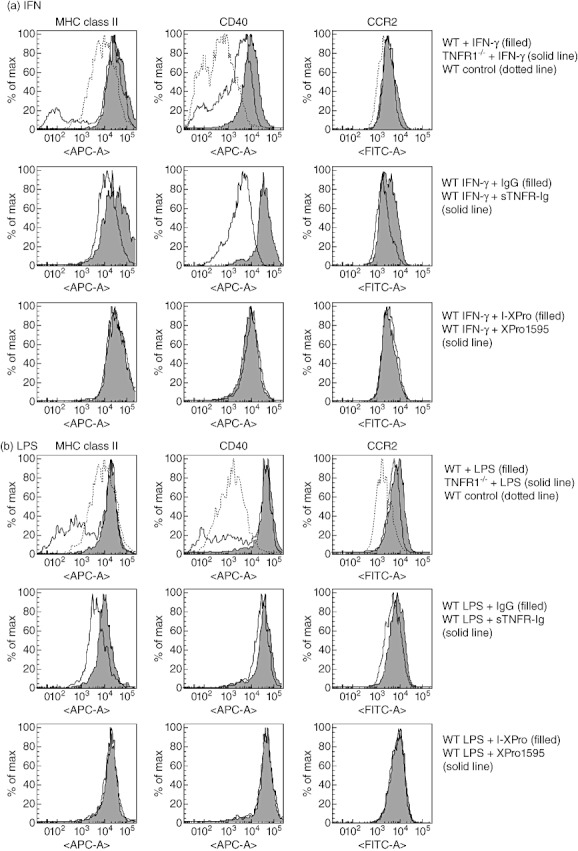
Inhibition of both transmembrane tumour necrosis factor (tmTNF) and soluble TNF (sTNF) is required to suppress interferon (IFN)-γ-induced CD40, major histocompatibility complex (MHC) class II and chemokine receptor 2 (CCR2) expression by macrophages. Bone marrow-derived macrophages (BMDMϕ) from wild-type or TNF receptor 1 (TNFR1−/−) mice were incubated with 100 U/ml IFN-γ for 24 h prior to analysis of cell surface expression of MHC class II, CD40 and CCR2 by fluorescence activated cell sorter (FACS). In some samples, IFN-γ and 10 µg/ml immunoglobulin (Ig)G, sTNFR-Ig or IFN-γ and 10 µg/ml I-XPro or XPro1595 were added to wild-type cells (a). BMDMϕ from wild-type or TNFR1−/− mice were also incubated with 100 ng/ml lipopolysaccharide (LPS) for 24 h prior to analysis of cell surface expression of MHC class II, CD40 and CCR2 by FACS. In some samples, LPS and 10 µg/ml IgG, sTNFR-Ig or LPS and 10 µg/ml I-XPro or XPro1595 were added to wild-type cells (b). Representative results from three independent experiments are shown.
Taking the data together, the results support the conclusion that inhibition of pan-TNF is necessary to inhibit NO production.
Local inhibition of sTNF cannot suppress EAU
Despite being dispensable for macrophage activation, selective systemic inhibition of sTNF alone suppressed EAU severity and reduced the extent of inflammatory cell infiltrate to the retina. We therefore investigated the effect of selective inhibition of sTNF within the target organ. The rationale for this approach was that treatment would not directly affect systemic cell emigration to the eye, but may still suppress activation of retinal infiltrating cells.
Treatments were administered by intravitreal injection on day 9 p.i., with XPro1595 or sTNFR-Ig in one eye and I-XPro or human IgG controls in the contralateral eye. In comparison to PBS, intravitreal injections of IgG can increase clinical disease significantly by 1·8-fold (P = 0·044), but I-XPro has no effect (data not shown). Clinical disease after treatment with sTNFR-Ig was suppressed significantly (Fig. 7a), but XPro1595 had no significant effect (Fig. 7b). Histological assessment on day 15 p.i. demonstrated that administration of sTNFR-Ig suppressed the total disease severity with 66·8% less structural damage (P = 0·036) (Fig. 7c). In contrast, in mice that received XPro1595, disease severity was not reduced, and was comparable to control I-XPro-treated eyes (Fig. 7d). Further analysis of the infiltrate using immunohistochemical stains demonstrated infiltrate that was present regardless of treatment, and consisted of both CD4+ T cells (Fig. 7e) and F4/80+ macrophages (Fig. 7f) within the vitreous and retinal layers.
Fig. 7.
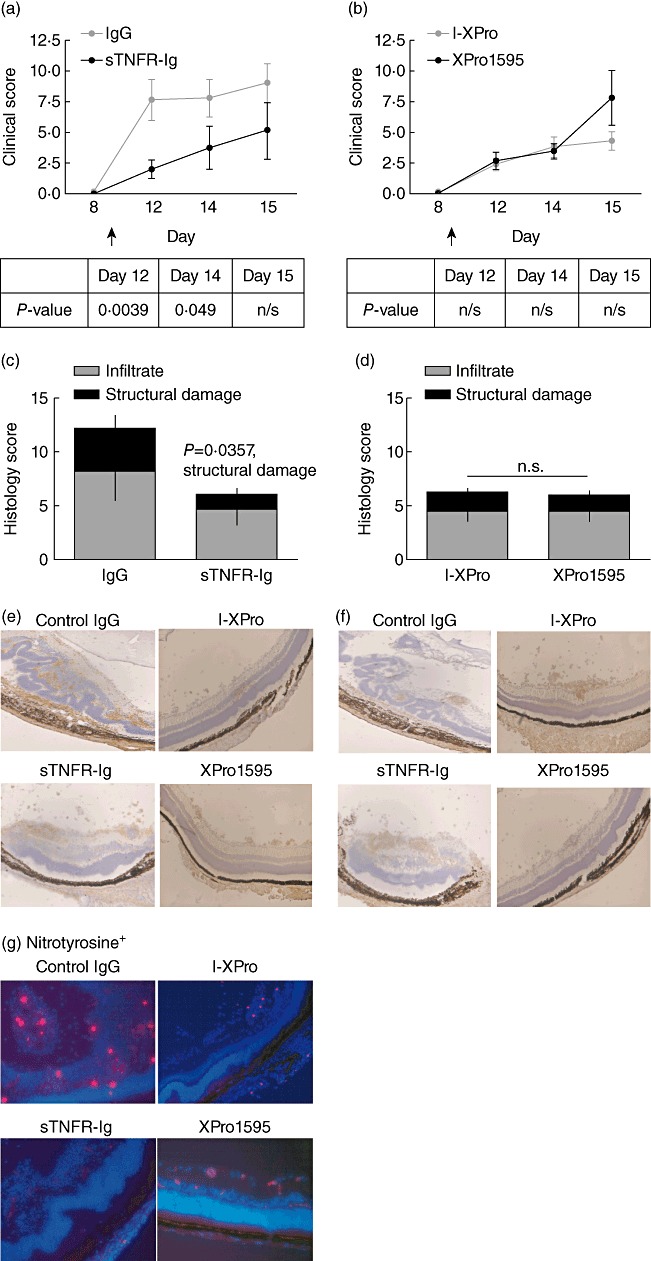
Local inhibition of soluble tumour necrosis factor (sTNF) and transmembrane (tm)TNF is required to suppress experimental autoimmune uveoretinitis (EAU). EAU was induced in B10.RIII mice, and on day 9 post-immunization (p.i.) mice were treated intravitreally with 10 µg/eye of human immunoglobulin (Ig)G or sTNFR-Ig (same mouse, contralateral eyes) or 10 µg/ml of I-XPro or XPro1595. Clinical progression of disease was assessed by topical endoscopic fundus imaging (TEFI) from days 8 to 15 p.i. (a,b). The P-value for each day is stated in the table beneath the graphs. On day 15, the mice were killed and eyes taken for histological analysis (c,d); mean ± standard error shown, n = 9–11 mice per group. The Mann–Whitney U-test was used for statistical analysis. Histological sections were used to assess F4/80+ (a mouse macrophage marker) (e) and CD4+ (f) cell infiltrate. Histological sections were also used to assess nitrotyrosine expression (red) in conjunction with 4′,6-diamidino-2-phenylindole (DAPI) (blue); ×20 magnification (g).
We then sought to determine whether disease suppression observed in the sTNFR-Ig animals following local administration was therefore as a direct result of reduced macrophage activation. A surrogate of myeloid-derived NO production via staining for the presence of nitrotyrosine (Fig. 7g) was investigated. Nitrotyrosine was clearly evident (red) within areas of retinal damage and disruption (Fig. 7g) in eyes from mice treated with XPro1595, I-XPro and IgG. In contrast, sections from eyes that received sTNFR-Ig were completely absent of nitrotyrosine.
These results show that pan-TNF inhibition within the eye decreases clinical disease significantly, with less structural damage but no effect on cell infiltration. Although there are macrophages and CD4+ T cells present in all the eyes, when pan-TNF is inhibited there is no evidence of classical macrophage activation, as corroborated by the absence of nitrotyrosine expression. Taken together, these results show that inhibition of both tmTNF and sTNF in the target organ is required to suppress inflammatory damage caused by activated macrophages.
Discussion
In this study, XPro1595 (an inhibitor of sTNF) and sTNFR-Ig (an inhibitor of sTNF and tmTNF) were used to investigate the contributions of sTNF versus tmTNF during autoimmune inflammation. The EAU model provides an excellent platform to dissect the immunopathogenic mechanisms controlling disease, as the target organ can be isolated easily to investigate the contribution of TNF action on myeloid cell recruitment and activation. Three different methods were used to assess EAU: TEFI to quantify clinical disease, flow cytometry to quantify and qualify cell infiltrate and histology to quantify morphological changes and infiltrate. The results of sTNF and pan-TNF blockade were assessed using all these methods. Blocking sTNF alone to suppress uveitis is potentially advantageous because the tmTNF response to infections such as tuberculosis (TB), which is a major concern in anti-TNF therapies, is maintained.
We have established here that for the full manifestation of disease, the release and action of sTNF is required for inflammatory cells to infiltrate the target organ during inflammation. The use of anti-TNF biologics in models of non-infectious posterior uveitis (EAU) has shown that eliminating TNF signalling delays cell trafficking into the retina [8]–[12]. We have shown previously that signalling via TNFR1 controls macrophage activation as well as trafficking to the site of inflammation [4],[13],[14]. TNFR1−/− mice have reduced disease index and incidence of disease [4],[13] and have fewer CD11b+ and CD4+ cells infiltrating the retina during EAU. However, structural damage is reduced because these cells cannot become fully activated, for example generating NO.
Together with these published studies, the current results show that sTNF signalling is required for macrophage trafficking into the eye and hence disease development. Other data in the EAU model have shown that inhibition of pan-TNF delays, but does not inhibit, infiltration of inflammatory cells into the eye [9]. In the eye, these cells are unable to cause damage due to the inability to become active. Thus, sTNF signalling via TNFR1 mediates cell trafficking into the eye. Once in the eye, both sTNF and tmTNF signalling via TNFR1 can lead to macrophage activation and subsequent tissue damage. Clinically, therefore, systemic treatment with both XPro1595 and sTNFR-Ig could suppress disease, but intravitreal delivery of sTNFR-Ig will be required to suppress tissue damage within the eye.
CCR2 has been implicated as a pivotal receptor for inflammatory cell migration to the target organs from bone marrow [27]–[29]. However, infliximab (an anti-TNF biologics) treatment of patients with rheumatoid arthritis decreases CCR2 expression by CD4+ cells [30], but clinical inhibition of CCR2 failed to suppress rheumatoid arthritis [31],[32]. Previous work in EAU has shown that macrophage migration into the eye is CCR2-independent [33]. Also, corneal inflammation (cauterization-induced) has been shown to be CCR2-independent [34]. We show here that LPS-induced CCR2 up-regulation is partially dependent on signalling via TNFR1. IFN-γ-induced CCR2 up-regulation, however, is fully dependent on TNF signalling, involving both receptors of TNF. Inhibition of sTNF did not suppress IFN-γ-induced CCR2 up-regulation, even though systemic inhibition of sTNF using XPro1595 suppressed CD11b+ and CD4+ cell infiltration into the eye. One theory from the data is that although CCR2 is not essential for myeloid migration to the eye during EAU, changes in expression represent altered macrophage activation and this is mediated in part by TNF.
Current therapies targeting TNF are effective in patients with autoimmune and autoinflammatory disorders such as uveitis and rheumatoid arthritis [9],[24],[35]–[37], but it is recognized that long-term treatment is associated with reduced immunosurveillance leading to an enhanced risk of infections and potentially neoplasia [38],[39]. It has been shown that by sparing tmTNF signalling mice are able to recover from sepsis even though sTNF is blocked [40], because these mice can produce local NO which is sufficient to fight infection. More recent work has also shown that, in contrast to treatment with etanercept, which blocks all TNF, mice treated with XPro1595 did not succumb to infection but were able to recover from both Listeria monocytogenes and M. tuberculosis[17],[41].
Our data and these other reports in the literature therefore indicate that more selective targeting of sTNF using XPro1595 can be effective in treating uveitis, while sparing immunosurveillance, particularly in countries with a high incidence of latent TB. Inhibition of sTNF will reduce inflammatory cell migration into the eye but allow macrophages to maintain their ability to counter infection systemically, as they can still be classically activated. These results show that neutralizing the activity of both sTNF and tmTNF via local delivery to the site of inflammation, the intravitreal route (with anti-TNF biologics), inhibits macrophage activation and therefore suppresses structural and functional tissue damage. Moreover, in situations where the target organ can be accessed easily (such as by intravitreal injection), a combination of local therapy inhibiting pan-TNF actions via sTNFR-Ig or current anti-TNF biologics, combined with blockade of sTNF with XPro1595 systemically, might generate increased efficacy clinically alongside a more favourable adverse effect profile than offered by current systemic anti-TNF therapies. These results put forward a strong case for clinical trials of anti-TNF therapy as intravitreal injections to treat uveitis in combination or independently to clinical trials to establish the efficacy and safety of small biological inhibitors and potentially circumvent the side effects of pan-TNF blockade.
Acknowledgments
This work was supported by grants from the National Eye Research Centre (NERC), UK. The flow cytometric analysis was carried out in the Flow Cytometry Facility, Cellular and Molecular Medicine, Bristol University. We thank Herman Waldmann (University of Oxford, UK) for the sTNFR-Ig fusion protein.
Disclosure
D. E. Symkowski is an employee of Xencor Inc., and provided XPro-1595 to T. K Khera and L. B. Nicholson free of charge.
References
- 1.Sedgwick JD, Riminton DS, Cyster JG, Körner H. Tumor necrosis factor: a master-regulator of leukocyte movement. Immunol Today. 2000;21:110–3. doi: 10.1016/s0167-5699(99)01573-x. [DOI] [PubMed] [Google Scholar]
- 2.Clark IA. How TNF was recognized as a key mechanism of disease. Cytokine Growth Factor Rev. 2007;18:335–43. doi: 10.1016/j.cytogfr.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Tincani A, Andreoli L, Bazzani C, Bosiso D, Sozzani S. Inflammatory molecules: a target for treatment of systemic autoimmune diseases. Autoimmun Rev. 2007;7:1–7. doi: 10.1016/j.autrev.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Raveney BJE, Copland DA, Dick AD, Nicholson LB. TNFR1-dependent regulation of myeloid cell function in experimental autoimmune uveoretinitis. J Immunol. 2009;183:2321–9. doi: 10.4049/jimmunol.0901340. [DOI] [PubMed] [Google Scholar]
- 5.Korner H, Riminton DS, Strickland DH, Lemckert FA, Pollard JD, Sedgwick JD. Critical points of tumor necrosis factor action in central nervous system autoimmune inflammation defined by gene targeting. J Exp Med. 1997;186:1585–90. doi: 10.1084/jem.186.9.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riminton SD, Korner H, Strickland DH, Lemckert FA, Pollard JD, Sedgwick JD. Challenging cytokine redundancy: inflammatory cell movement and clinical course of experimental autoimmune encephalomyelitis are normal in lymphotoxin-deficient, but not tumor necrosis factor-alpha deficient, mice. J Exp Med. 1998;187:1517–28. doi: 10.1084/jem.187.9.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy CA, Hoek RM, Wiekowski MT, Lira SA, Sedgwick JD. Interactions between hemopoietically derived TNF and central nervous system-resident glial chemokines underlie initiation of autoimmune inflammation in the brain. J Immunol. 2002;169:7054–62. doi: 10.4049/jimmunol.169.12.7054. [DOI] [PubMed] [Google Scholar]
- 8.Dick AD, Duncan L, Hale G, Waldmann H, Isaacs J. Neutralizing TNF-alpha activity modulates T-cell phenotype and function in experimental autoimmune uveoretinitis. J Autoimmun. 1998;11:255–64. doi: 10.1006/jaut.1998.0197. [DOI] [PubMed] [Google Scholar]
- 9.Dick AD, McMenamin PG, Körner H, et al. Inhibition of tumor necrosis factor activity minimizes target organ damage in experimental autoimmune uveoretinitis despite quantitatively normal activated T cell traffic to the retina. Eur J Immunol. 1996;26:1018–25. doi: 10.1002/eji.1830260510. [DOI] [PubMed] [Google Scholar]
- 10.Hankey DJR, Lightman SL, Baker D. Interphotoreceptor retinoid binding protein peptide-induced uveitis in B10.RIII mice: characterization of disease parameters and immunomodulation. Exp Eye Res. 2001;72:341–50. doi: 10.1006/exer.2000.0957. [DOI] [PubMed] [Google Scholar]
- 11.Kowalczuk L, Touchard E, Camelo S, et al. Local ocular immunomodulation resulting from electrotransfer of plasmid encoding soluble TNF receptors in the ciliary muscle. Invest Ophthalmol Vis Sci. 2009;50:1761–8. doi: 10.1167/iovs.08-3027. [DOI] [PubMed] [Google Scholar]
- 12.Sartani G, Silver PB, Rizzo LV, et al. Anti-tumor necrosis factor alpha therapy suppresses the induction of experimental autoimmune uveoretinitis in mice by inhibiting antigen priming. Invest Ophthalmol Vis Sci. 1996;37:2211–8. [PubMed] [Google Scholar]
- 13.Calder CJ, Nicholson LB, Dick AD. A selective role for the TNF p55 receptor in autocrine signaling following IFN-gamma stimulation in experimental autoimmune uveoretinitis. J Immunol. 2005;175:6286–93. doi: 10.4049/jimmunol.175.10.6286. [DOI] [PubMed] [Google Scholar]
- 14.Dick AD, Forrester JV, Liversidge J, Cope AP. The role of tumour necrosis factor (TNF-alpha) in experimental autoimmune uveoretinitis (EAU) Prog Retin Eye Res. 2004;23:617–37. doi: 10.1016/j.preteyeres.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 15.Alexopoulou L, Kranidioti K, Xanthoulea S, et al. Transmembrane TNF protects mutant mice against intracellular bacterial infections, chronic inflammation and autoimmunity. Eur J Immunol. 2006;36:2768–80. doi: 10.1002/eji.200635921. [DOI] [PubMed] [Google Scholar]
- 16.Ruuls SR, Hoek RM, Ngo VN, et al. Membrane-bound TNF supports secondary lymphoid organ structure but is subservient to secreted TNF in driving autoimmune inflammation. Immunity. 2001;15:533–43. doi: 10.1016/s1074-7613(01)00215-1. [DOI] [PubMed] [Google Scholar]
- 17.Zalevsky J, Secher T, Ezhevsky SA, et al. Dominant-negative inhibitors of soluble TNF attenuate experimental arthritis without suppressing innate immunity to infection. J Immunol. 2007;179:1872–83. doi: 10.4049/jimmunol.179.3.1872. [DOI] [PubMed] [Google Scholar]
- 18.Steed PM, Tansey MG, Zalevsky J, et al. Inactivation of TNF signaling by rationally designed dominant-negative TNF variants. Science. 2003;301:1895–8. doi: 10.1126/science.1081297. [DOI] [PubMed] [Google Scholar]
- 19.Munder M, Eichmann K, Modolell M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol. 1998;160:5347–54. [PubMed] [Google Scholar]
- 20.Copland DA, Wertheim MS, Armitage WJ, Nicholson LB, Raveney BJE, Dick AD. The clinical time–course of experimental autoimmune uveoretinitis using topical endoscopic fundal imaging with histologic and cellular infiltrate correlation. Invest Ophthalmol Vis Sci. 2008;49:5458–65. doi: 10.1167/iovs.08-2348. [DOI] [PubMed] [Google Scholar]
- 21.Xu H, Koch P, Chen M, Lau A, Reid DM, Forrester JV. A clinical grading system for retinal inflammation in the chronic model of experimental autoimmune uveoretinitis using digital fundus images. Exp Eye Res. 2008;87:319–26. doi: 10.1016/j.exer.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 22.Raveney BJE, Richards CM, Aknin M-L, et al. The B subunit of Escherichia coli heat-labile enterotoxin inhibits Th1 but not Th17 cell responses in established autoimmune uveoretinitis. Invest Ophthalmol Vis Sci. 2008;49:4008–17. doi: 10.1167/iovs.08-1848. [DOI] [PubMed] [Google Scholar]
- 23.Forrester JV, Huitinga I, Lumsden L, Dijkstra CD. Marrow-derived activated macrophages are required during the effector phase of experimental autoimmune uveoretinitis in rats. Lisse, Pays-Bas: Swets & Zeitlinger; 1998. [DOI] [PubMed] [Google Scholar]
- 24.Robertson M, Liversidge J, Forrester JV, Dick AD. Neutralizing tumor necrosis factor-alpha activity suppresses activation of infiltrating macrophages in experimental autoimmune uveoretinitis. Invest Ophthalmol Vis Sci. 2003;44:3034–41. doi: 10.1167/iovs.02-1156. [DOI] [PubMed] [Google Scholar]
- 25.Robertson MJ, Erwig LP, Liversidge J, Forrester JV, Rees AJ, Dick AD. Retinal microenvironment controls resident and infiltrating macrophage function during uveoretinitis. Invest Ophthalmol Vis Sci. 2002;43:2250–7. [PubMed] [Google Scholar]
- 26.Kerr EC, Raveney BJE, Copland DA, Dick AD, Nicholson LB. Analysis of retinal cellular infiltrate in experimental autoimmune uveoretinitis reveals multiple regulatory cell populations. J Autoimmun. 2008;31:354–61. doi: 10.1016/j.jaut.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Maus UA, Waelsch K, Kuziel WA, et al. Monocytes are potent facilitators of alveolar neutrophil emigration during lung inflammation: role of the CCL2-CCR2 axis. J Immunol. 2003;170:3273–8. doi: 10.4049/jimmunol.170.6.3273. [DOI] [PubMed] [Google Scholar]
- 28.Platt AM, Bain CC, Bordon Y, Sester DP, Mowat AM. An independent subset of TLR expressing CCR2-dependent macrophages promotes colonic inflammation. J Immunol. 2010;184:6843–54. doi: 10.4049/jimmunol.0903987. [DOI] [PubMed] [Google Scholar]
- 29.Si Y, Tsou C-L, Croft K, Charo IF. CCR2 mediates hematopoietic stem and progenitor cell trafficking to sites of inflammation in mice. J Clin Invest. 2010;120:1192–203. doi: 10.1172/JCI40310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xia L, Lu J, Xiao W. Blockage of TNF-alpha by infliximab reduces CCL2 and CCR2 levels in patients with rheumatoid arthritis. J Invest Med. 2011;59:961–3. doi: 10.2310/JIM.0b013e31821c0242. [DOI] [PubMed] [Google Scholar]
- 31.Lebre MC, Vergunst CE, Choi IYK, et al. Why CCR2 and CCR5 blockade failed and why CCR1 blockade might still be effective in the treatment of rheumatoid arthritis. PLoS ONE. 2011;6:e21772. doi: 10.1371/journal.pone.0021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vergunst CE, Gerlag DM, Lopatinskaya L, et al. Modulation of CCR2 in rheumatoid arthritis: a double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum. 2008;58:1931–9. doi: 10.1002/art.23591. [DOI] [PubMed] [Google Scholar]
- 33.Dagkalis A, Wallace C, Xu H, et al. Development of experimental autoimmune uveitis: efficient recruitment of monocytes is independent of CCR2. Invest Ophthalmol Vis Sci. 2009;50:4288–94. doi: 10.1167/iovs.09-3434. [DOI] [PubMed] [Google Scholar]
- 34.Oshima T, Sonoda KH, Tsutsumi-Miyahara C, et al. Analysis of corneal inflammation induced by cauterisation in CCR2 and MCP-1 knockout mice. Br J Ophthalmol. 2006;90:218–22. doi: 10.1136/bjo.2005.077875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shakoor N, Michalska M, Harris CA, Block JA. Drug-induced systemic lupus erythematosus associated with etanercept therapy. Lancet. 2002;359:579–80. doi: 10.1016/S0140-6736(02)07714-0. [DOI] [PubMed] [Google Scholar]
- 36.Criscione LG, St. Clair EW. Tumor necrosis factor-alpha antagonists for the treatment of rheumatic diseases. Curr Opin Rheumatol. 2002;14:204–11. doi: 10.1097/00002281-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 37.Greiner K, Murphy CC, Willermain F, et al. Anti-TNFalpha therapy modulates the phenotype of peripheral blood CD4+ T cells in patients with posterior segment intraocular inflammation. Invest Ophthalmol Vis Sci. 2004;45:170–6. doi: 10.1167/iovs.03-0659. [DOI] [PubMed] [Google Scholar]
- 38.Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor-alpha neutralizing agent. N Engl J Med. 2001;345:1098–104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]
- 39.Kempen JH, Daniel E, Dunn JP, et al. Overall and cancer related mortality among patients with ocular inflammation treated with immunosuppressive drugs: retrospective cohort study. BMJ. 2009;339:b2480. doi: 10.1136/bmj.b2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olleros ML, Guler R, Vesin D, et al. Contribution of transmembrane tumor necrosis factor to host defense against Mycobacterium bovis bacillus Calmette–Guérin and Mycobacterium tuberculosis infections. Am J Pathol. 2005;166:1109–20. doi: 10.1016/S0002-9440(10)62331-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olleros ML, Vesin D, Lambou AF, et al. Dominant-negative tumor necrosis factor protects from Mycobacterium bovis bacillus Calmette–Guérin (BCG) and endotoxin-induced liver injury without compromising host immunity to BCG and Mycobacterium tuberculosis. J Infect Dis. 2009;199:1053–63. doi: 10.1086/597204. [DOI] [PubMed] [Google Scholar]