

Abstract
Interleukin (IL)-22 is a cytokine involved in inflammatory and wound healing processes that is secreted primarily by T helper type 17 (Th17) cells. IL-22 receptor (IL-22R) expression is limited to epithelial cells of the digestive organs, respiratory tract and skin. Most tumours originating in these sites over-express IL-22R. Interestingly, there is an increase in Th17 frequency within the peripheral blood and tumour microenvironment of advanced cancer patients. Subsequently, IL-17 has been shown to display both pro-tumour and anti-tumour functions. Because many tumours lack expression of the IL-17 receptor, the effects of IL-17 on tumour growth are generated by cells that surround the tumour cells. Like IL-17, high levels of IL-22 have been detected in tumour tissues and the peripheral blood of cancer patients; however, the direct effect of IL-22 on tumour cells has remained largely unknown. In this report, we show that IL-22 stimulated production of vascular endothelial growth factor (VEGF) and the anti-apoptotic factor Bcl-XL in IL-22R-positive HPAFII human pancreatic cancer cells. Additionally, IL-22 augmented HPAFII cell production of immunosuppressive cytokines. We show further that IL-22 activation of HPAFII cells diminished T cell production of interferon (IFN)-γ through the action of IL-10. Strikingly, we show for the first time that IL-22 can fully protect cancer cells from natural killer (NK) cell-mediated cytotoxicity by stimulating tumour production of IL-10 and transforming growth factor (TGF)-β1. Our data support the idea that IL-22 may act to promote the pathogenesis of cancers rather than function in anti-tumour immunity.
Keywords: IL-22, NK cells, pancreatic cancer, Th17, STAT-3
Introduction
Chronic inflammation is a general feature of actively growing tumours by serving as a mechanism for vascularization in order to supply the rapidly growing cells with nutrients and a conduit for metastasis. Additionally, the intratumoural vasculature provides populations of Th17 cells access to the tumour mass, where these helper T cells have been shown to accumulate during tumour progression [1]. Currently, IL-17 derived from these tumour-resident T cells has been shown to have both pro-tumour and anti-tumour effects [2]–[10]. That is, IL-17 can stimulate production of angiogenesis factors for survival and metastasis or recruit neutrophils secreting proinflammatory cytokines for tumour eradication. However, tumours do not generally express the IL-17R, thereby limiting the effects mediated to the responding population of cells resident within the tumour microenvironment [11],[12]. While the role of IL-17 in tumour growth and development has become clearer in recent years, studies of other relevant Th17-associated molecules are quite limited.
Another major product of Th17 cells is the cytokine IL-22. The receptor for IL-22 consists of a heterodimeric complex composed of the IL-22R alpha chain and IL-10 receptor 2 (IL-10R2) [13]. IL-22R is expressed in non-immune cells, such as the kidney, liver, skin, colon, lungs and pancreas [14]–[20]. Interestingly, the highest expression of IL-22R has been demonstrated in the colon, skin and pancreas as well as many tumour lines derived from these tissues [13]. Because these tumours preserve, and in many cases increase, expression of the IL-22R, it is thus likely that signalling induced by IL-22 ligation results in pro-tumour effects. Accordingly, the pro-tumour effects currently attributed to infiltrating Th17 cells may be due to the biological activities of secreted IL-22. To this end, we have employed an IL-22R-positive human pancreatic adenocarcinoma cell line HPAFII [17] and evaluated the outcome of IL-22 signalling upon tumour production of growth and survival factors as well as susceptibility to lymphocyte-mediated cytotoxicity.
Materials and methods
Cell lines and reagents
Human HPAFII pancreatic adenocarcinoma (CRl-1997) E6·1 CD4+ (Jurkat T) cells (TIB-152) and NK-92MI natural killer-like cells (CRL-2408) were obtained from the American Type Culture Collection (Manassas, VA, USA). The HPAFII cells were maintained in Eagle's minimum essential medium containing 10% fetal bovine serum and gentamicin. Jurkat T cells were maintained in RPMI-1640 medium containing 10% fetal bovine serum and gentamicin. NK-92MI cells were maintained in alpha minimum essential medium containing 12·5% fetal bovine serum, 12·5% horse serum, 0·1 mM 2-mercaptoethanol, 0·02 mM folic acid, 0·2 mM inositol and gentamicin. All cell lines were cultured and maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
IL-22 (Peprotech, Rocky Hill, NJ, USA) was reconstituted in the complete media used for HPAFII cells and added to cultures at a concentration of 200 ng/ml. The signal tranducer and activator of transcription (STAT) signalling inhibitor, Tyrphostin AG490 (Sigma-Aldrich, St Louis, MO, USA), was dissolved in dimethylsulphoxide (DMSO) and added to cultures at a concentration of 100 µM. Antibody against human IL-10 receptor (αIL-10R), clone 90220 (R&D Systems, Minneapolis, MN, USA) was used at a concentration of 2 µg/ml. Antibody against transforming growth factor (TGF)-β1 (αTGF-β), clone TB21 (Abcam, Cambridge, MA, USA), was used at a concentration of 2 µg/ml. For blockade experiments, mouse immunoglobulin (Ig)G1 (mIgG1), clone 11711 (R&D Systems) served as an isotype control and was used at a concentration of 2 µg/ml.
Detection of phosphorylated STAT-3
All procedures were carried out according to the manufacturer's instructions (R&D Systems). Briefly, HPAFII cells (3–5×106) were cultured at the given times with either IL-22, AG490 or IL-22 + AG490. Following incubation, cells were isolated by centrifugation, washed in cold phosphate-buffered saline (PBS) and resuspended at 10 × 106 cells/ml in lysis buffer (50 mM Tris (ph7·4), 150 mM NaCl, 1% NP-40 alternative, 0·5% sodium deoxycholate, 0·1% sodium dodecyl sulphate and 100 µl protease inhibitor cocktail). After incubation on ice for 5 min, supernatant was collected and total protein concentration determined by spectrophotometry. Subsequently, phosphorylated STAT-3 was quantitated by enzyme-linked immunosorbent assay (ELISA) according to R&D Systems' standard protocol from 10–30 µg of total cell protein in 100 µl of reagent diluent [5% bovine serum albumin (BSA), 0·5% Tween20 in PBS]. The capture antibody was mouse anti-human STAT-3 (Y705) (R&D Systems) antibody and detection antibody was biotinylated rabbit anti-human phospho-STAT-3 (R&D Systems) antibody. Graded amounts of recombinant human phospho-STAT-3 (R&D Systems) were included for construction of standard curves. The phosphorylated STAT-3 concentration in cell lysates was extrapolated from the linear portion of the standard curve. All ELISA procedures were read on a spectrophotometer (Beckman Coulter Inc., Indianapolis, IN, USA) and analysed using Multimode Detection Software version 2·0.0·12.
Detection of Bcl-XL protein
All procedures were carried out according to the manufacturer's instructions (R&D Systems). Briefly, HPAFII cells (3–5×106) were cultured for 20 h, as for measurement of phosphorylated STAT-3. Subsequently, the cells were isolated, washed and resuspended at 10×106 cells/ml in lysis buffer [1 mM ethylenediamine teraacetic acid (EDTA), 0·5% Triton X-100, 10 µg/ml leupeptin, 10 µg/ml pepstatin, 3 µg/ml aprotinin, 100 µM phenylmethylsulphonylfluoride]. After incubation on ice for 15 min, supernatant was collected and total protein concentration determined. Thereafter, Bcl-XL was quantitated by ELISA according to R&D Systems' standard protocol from 10–30 µg of total cell protein in 100 µl of reagent diluent (1 mM EDTA, 0·5% Triton X-100 in PBS). The capture antibody was rat anti-mouse/human Bcl-XL (R&D Systems) antibody and detection antibody was biotinylated rat anti-mouse/human Bcl-XL (R&D Systems) antibody. Graded quantities of recombinant human Bcl-XL (R&D Systems) were included for standard curve generation and Bcl-XL concentrations were extrapolated from the linear portion of the standard curve.
Stimulation of HPAFII and Jurkat T cell cytokine production
HPAFII cells (2 × 105 cells/well) were stimulated with 200 ng/ml of IL-22 for 24 h. Jurkat T cells (1 × 105 cells/well) were stimulated with 100 ng/ml of phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich) for 24 h in the presence of 100 µl of supernatant derived from IL-22-stimulated HPAFII cells. Subsequently, cytokine production by HPAFII cells and Jurkat T cells was assessed by ELISA from 100 µl of the culture supernatant.
Detection of cytokines in cell cultures
Detection of IL-10 (R&D Systems), VEGF (R&D Systems) and TGF-β1 (R&D Systems) was achieved by ELISA according to R&D Systems' reagents and standard protocols. IFN-γ (BD Pharmingen, San Diego, CA, USA) was detected using BD Pharmingen's reagents and recommended ELISA procedure. The capture antibodies were mouse anti-human IL-10 (841825), mouse anti-human VEGF (841495), mouse anti-human TGF-β1 (840116) and mouse anti-human IFN-γ clone NIB42. Detection biotinylated antibodies were as follows: goat anti-human IL-10 (840196), goat anti-human VEGF (840163), chicken anti-human TGF-β1 (840117) and mouse anti-human IFN-γ clone 4S.B3. Detection of TGF-β1 required activation of latent TGF-β1 by acidification (1 N HCl was added to cell culture supernatant, incubated for 10 min then the sample was neutralized by adding 1·2 N NaOH/0·5 M HEPES). Recombinant IL-10 (841532), VEGF (840164), TGF-β1 (840118) and IFN-γ (554616) were included in the experiments in order to generate standard curves.
Assessment of NK-92MI-mediated cytotoxicity
NK-92MI cells were co-cultured with HPAFII target cells (5 × 104 cells/well) that may or may not have been incubated with IL-22 at a 5 : 1 (effector : target) ratio for 4 h at 37°C and 5% CO2. Additionally, αIL-10R or αTGF-β was included in some wells. Cytotoxicity was determined by colorimetric assay through measurement of lactate dehydrogenase (LDH) release from the cytosol using an LDH release cytotoxicity detection kit (Roche Diagnostics, Indianapolis, IN, USA). The LDH release assay was performed according to the manufacturer's instructions. Assay media contained only 1% serum to reduce background levels of LDH. Optimal cell concentration for the LDH release was performed prior to experimentation with NK-92MI cells in order to derive the number of HPAFII target cells used in the assay. The following controls were assessed: background control (LDH activity in media alone); low control (spontaneous LDH release by target cells); high control (maximal LDH release by target cells induced by 1% Triton X-100); and effector control (spontaneous LDH release by effector cells). Background control absorbances were subtracted from all absorbance measures and resultant values substituted into the calculation for cell-mediated cytotoxicity. Percentage cytotoxicity was calculated from the mean absorbance values as follows: [(effector-target cell mix – effector control) – low control]/(high control – low control) × 100 (%).
Assessment of LDH release by HPAFII cells in low serum
HPAFII cells (2·5 × 106 cells/Petri dish) were incubated with or without IL-22 in media containing 0·1% serum (low serum media) for 24 h. Cells were harvested, washed with PBS and incubated 4 h in low serum media and LDH activity determined by ELISA. Percentages of LDH release were determined by comparing LDH activity absorbance values for untreated and IL-22 stimulated HPAFII cells to the following control: HPAFII cells incubated in media containing 0·1% serum and 1% Triton X-100 for 4 h. The additional 4 h incubation in low serum media permitted measurement of the control absorbances within the limits of the spectrophotometer. Background absorbances (media only) were subtracted from each absorbance measure and the subsequent corrected values were used in the calculation of LDH release. The HPAFII LDH release was represented as a percentage of the maximal release of LDH quantified by the control for which the absorbance was set at 100%.
Results
IL-22 stimulation of HPAFII cells triggers STAT-3 phosphorylation
IL-22 binding to IL-22R/IL-10R2 results in tyrosine phosphorylation of Janus kinase (Jak) and subsequent activation of STAT molecules [21],[22]. In humans, IL-22 binding to IL-22R activates STAT-3, and to a much lesser extent STAT-1 [22]. Phosphorylation of STAT-3 molecules results in dimerization and translocation to the nucleus where the dimer serves as a transcription factor for genes involved in cell survival and proliferation [23]. Interestingly, STAT-3 has been shown to support tumour angiogenesis, survival and immune evasion [24]–[26]. Given that IL-22 can activate STAT-3 in normal tissue cells, we sought to determine whether IL-22 could also promote phosphorylation of STAT-3 in the IL-22R-positive human pancreatic cancer line HPAFII. As shown in Fig. 1, when HPAFII cells were incubated with IL-22 a rapid and significant increase in phosphorylated STAT-3 was detected within 15 min. Phosphorylated STAT-3 protein concentration remained 10-fold higher compared to the control at 30 min post-exposure to IL-22. Of note, IL-22R-negative Jurkat T cells [17] served as a negative control for all experiments that assessed the direct effects of IL-22R signalling in HPAFII cells. Subsequently, no significant difference was measured between Jurkat T cells incubated with or without IL-22 (data not shown). Thus, IL-22 promoted rapid activation of STAT-3 in HPAFII cells.
Fig. 1.
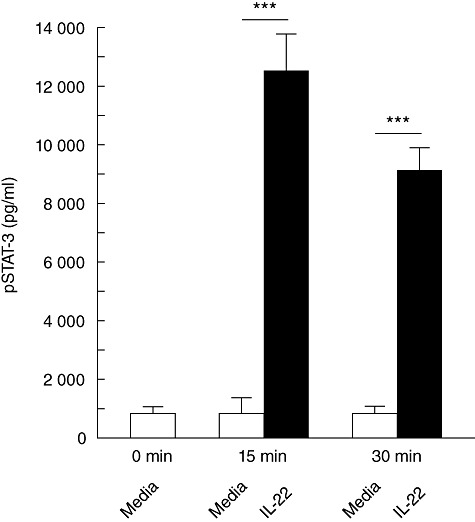
IL-22 activation of STAT-3 in HPAFII cells. HPAFII cells were stimulated with (black bars) or without (white bars) IL-22 up to 30 min. Cell lysates were prepared at 0-, 15- and 30-min time-points and cytoplasmic phosphorylated STAT-3 (pSTAT-3) levels were assessed by ELISA. Columns represent mean ± standard deviation. Comparable results were obtained from three independent experiments. ***P < 0·0001.
IL-22-mediated production of angiogenic and anti-apoptotic factors in HPAFII cells
STAT-3 activation in tumours has been associated with increased angiogenesis [24],[27],[28], thus we sought to determine whether IL-22-mediated STAT-3 activation would increase VEGF expression in HPAFII cells. In Fig. 2a, IL-22 stimulation of HPAFII cells resulted in a nearly fourfold increase in the production of VEGF compared to control (190 versus 49 pg/ml). IL-22-mediated production of VEGF was dependent upon STAT signalling, as the addition of AG490, a Jak-STAT signalling inhibitor [29], diminished VEGF expression (Fig. 2a). Activation of STAT-3 has also been associated with the regulation of genes that prevent apoptosis such as Bcl-XL[24],[28]. Accordingly, HPAFII cells were analysed for expression of Bcl-XL following stimulation with IL-22. As shown in Fig. 2b, IL-22 induced a significant increase in Bcl-XL compared to control (11 490 versus 8650 ng/ml). This increase in Bcl-XL expression correlated with reduced HPAFII cell apoptosis. Consequently, a significant release of the cytoplasmic enzyme LDH from HPAFII cells, incubated in the absence of IL-22, was measured when the cells were cultured in low serum media (Fig. 2c). However, HPAFII cells cultured in low serum in the presence of IL-22 displayed a nearly fourfold reduction in LDH activity (Fig. 2c). Interestingly, control levels of Bcl-XL are elevated in HPAFII cells in the absence of IL-22 (Fig. 2b), most probably a consequence of genetic modification such as constitutive STAT activation, as is common in cancer cells. Blockade of Jak-STAT signalling by AG490 in IL-22-stimulated HPAFII cells reduced Bcl-XL protein concentration to constitutive levels (Fig. 2b). Neither VEGF nor Bcl-XL production was significantly different from the control (media) when HPAFII cells (or Jurkat T cells) were incubated with AG490 alone (data not shown). Thus, IL-22-mediated STAT-3 activation resulted in significantly increased VEGF and Bcl-XL protein expression in HPAFII cells. Furthermore, IL-22 enhancement of Bcl-XL expression correlated with decreased cell death from nutrient deprivation.
Fig. 2.
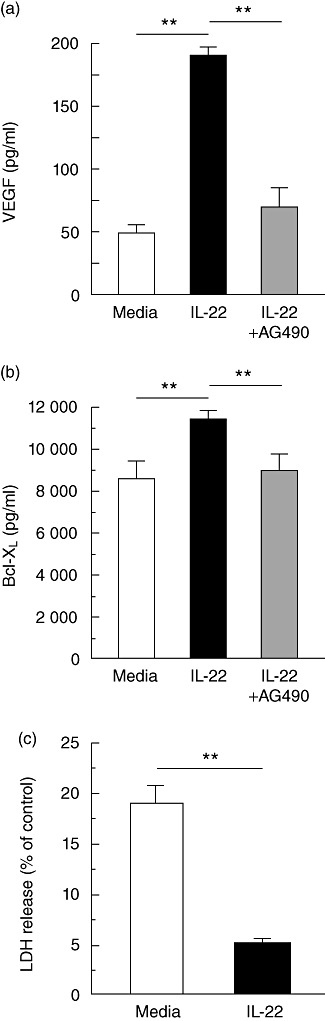
Increased HPAFII cell expression of VEGF and Bcl-XL upon IL-22 stimulation. (a) HPAFII cells were incubated with IL-22 (black bar) or IL-22 and AG490 (grey bar) for 24 h and VEGF secretion determined by ELISA. Wells containing only cells and media were included as a control (white bar). (b) HPAFII cells were incubated as in (a) (20 h) and assessed for cytoplasmic expression of Bcl-XL protein by ELISA from cell lysates. (c) HPAFII cells were incubated in the presence (black bar) or absence (white bar) of IL-22 in low serum media for 24 h. Subsequently, LDH activity was assessed by ELISA following an additional 4 h incubation in low serum media. HPAFII cells cultured for 4 h in low serum media and 1% Triton X-100 served as the control (maximum LDH release). Columns represent mean ± standard deviation. Comparable results for VEGF, Bcl-XL and LDH release were obtained from three independent experiments for each. **P < 0·001.
Suppression of T cell IFN-γ production and NK cell cytotoxicity by immunosuppressive cytokines derived from IL-22-stimulated HPAFII cells
Production of immunosuppressive cytokines such as IL-10 and TGF-β1 is but one mechanism employed by the tumour to frustrate an effective immune response [30]. Because activation of STAT-3 can increase production of both IL-10 and TGF-β1 [30]–[32], we assessed IL-22-stimulated HPAFII cells for cytokine production. As shown in Fig. 3, both IL-10 and TGF-β1 production increased at least twofold relative to the controls and inclusion of AG490. Therefore, IL-22 stimulation of HPAFII cells resulted in increased production of the immunosuppressive cytokines, IL-10 and TGF-β1.
Fig. 3.
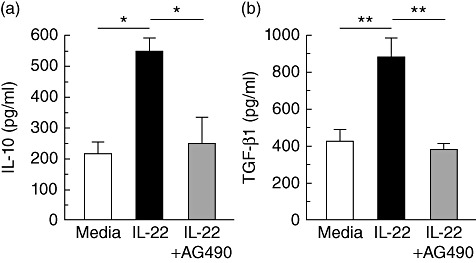
IL-22 augmentation of HPAFII IL-10 and TGF-β1 secretion. HPAFII cell production of IL-10 (a) and TGF-β1 (b) was assessed by ELISA following a 24-h incubation with IL-22 (black bars) or IL-22 and AG490 (grey bars). Wells containing cells and media only were included as a control (white bar). Columns represent mean ± standard deviation. Comparable results for IL-10 and TGF-β1 were obtained from three independent experiments for each. *P < 0·01; **P < 0·005.
As IL-22 can provoke HPAFII cell production of IL-10 and TGF-β1, we wanted to assess whether stimulated HPAFII cells could suppress immune responses in vitro. Hence, HPAFII cells were stimulated with IL-22 overnight and cell supernatant collected. The HPAFII supernatant was then added to a culture of Jurkat T cells concurrent with stimulation by PMA and assessed for IFN-γ secretion. In Fig. 4a, when unstimulated HPAFII cell supernatant (HPAF sup) was added, Jurkat T cells produced nearly twofold more IFN-γ compared to Jurkat T cells incubated with supernatant derived from IL-22-stimulated HPAFII cells (HPAF+IL-22 sup). We used supernatant from the cultured HPAFII cells to rule out any potential cell–cell inhibitory effects contributed by the tumour cells [30]. To confirm that the reduction of Jurkat T cell IFN-γ was due to the action of IL-10, we employed antibody to block the IL-10 receptor and re-examined IFN-γ production. Indeed, the addition of anti-IL-10 receptor (αIL-10R) antibody during activation of Jurkat T cells, co-cultured in the presence of IL-22-stimulated HPAFII supernatant, restored production of IFN-γ. Inclusion of the isotype control, mouse IgG1 (mIgG1), rather than αIL-10R, failed to re-establish IFN-γ production (Fig. 4a). Thus, IL-22-mediated HPAFII cell production of IL-10 operated to suppress IFN-γ production by activated Jurkat T cells.
Fig. 4.
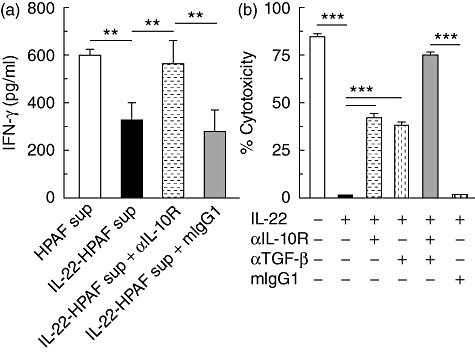
IL-22-mediated HPAFII suppression of Jurkat T cell IFN-γ production and NK-92MI cytotoxicity. (a) HPAFII cells were stimulated with IL-22 and supernatant collected after 24 h. Jurkat T cells were incubated with both PMA and supernatant derived from IL-22 activated HPAFII cells (black bar) for 24 h and IFN-γ secretion assessed by ELISA. Jurkat T cell IFN-γ production upon blockade of IL-10 signalling was determined by supplementation of cultures with αIL-10R (dashed horizontal bar). Jurkat T cells incubated with PMA and either supernatant from unstimulated HPAFII cells (white bar) or supernatant from IL-22-activated HPAFII cells and mouse immunoglobulin (Ig)G1 (grey bar) were included as controls. (b) NK-92MI cells were cultured for 4 h with IL-22-stimulated HPAFII target cells (black bar) at an effector : target ratio of 5 : 1 and cytotoxicity determined by LDH activity released by lysed cells. NK-92MI per cent cytotoxicity, upon cytokine blockade, was assessed by addition of αIL-10R (dashed horizontal bar), α-TGF-β (dashed vertical bar) or TGF-β (grey bar) antibodies. NK-92MI cells cultured with IL-22-stimulated HPAFII cells and mIgG1 (vertical hatched bar) or HPAFII cells preincubated in media alone (white bar) were included as controls. Columns represent the mean ± standard deviation. Comparable results for all conditions were obtained from three independent experiments for each. **P < 0·001; ***P < 0·0001.
Next, we investigated whether IL-22 would support HPAFII cell resistance to NK cell cytotoxicity, to our knowledge a hypothesis not tested previously. To this end, HPAFII cells stimulated with IL-22 served as target cells in a cytotoxicity assay using the NK cell line NK-92MI. The NK-92MI line, an IL-2-independent NK cell line highly cytotoxic to a wide range of malignant cells, however, has yet to be tested against HPAFII cells. As shown in Fig. 4b, HPAFII cells are highly susceptible to NK-92MI cytolysis (media), as 85% of the tumour cells are lysed during the 4-h assay. Strikingly, prior stimulation of HPAFII cells with IL-22 completely protected HPAFII cells from NK-92MI-mediated cytotoxicity (Fig. 4b, black bar). From this result, we postulated that resistance to cytotoxicity may be mediated by HPAFII secretion of IL-10 and/or TGF-β1. Therefore, αIL-10R and/or anti-TGF-β1 (αTGF-β) antibodies were included in the assays. As shown in Fig. 4b, when added separately, either antibody alone only partially restored NK-92MI cytotoxicity. However, addition of both antibodies enhanced tumour susceptibility by nearly 40% compared to either antibody alone. Therefore, IL-22 conferred to HPAFII cells absolute resistance to NK-92MI cell cytotoxicity through the actions of tumour-derived IL-10 and TGF-β1.
Discussion
Few studies to date have addressed the role of IL-22 in cancer. In this report, we endeavoured to assess the outcome of IL-22 signalling in an IL-22R-positive pancreatic cancer cell line HPAFII. IL-22 can promote phosphorylation of STAT-3 and activation of this STAT has been shown to occur in many human tumours, leading to production of proteins involved in cell proliferation and survival as well as cancer vascularization and immune evasion [24]–[26]. Indeed, HPAFII cells stimulated with IL-22 secreted VEGF, IL-10 and TGF-β1 and increased cytoplasmic expression of Bcl-XL. IL-22-mediated increase in Bcl-XL expression correlated with enhanced HPAFII cell survival during culture in low serum. We attempted to link HPAFII production of IL-10 and TGF-β1 with a capacity to suppress immune responses in vitro. Neutralization of IL-10 signalling in activated Jurkat T cells restored IFN-γ production when co-incubated with supernatant from IL-22-stimulated HPAFII cells. We also showed that HPAFII-derived IL-10 and TGF-β1 directly inhibited the cytolytic activity of a potent NK cell line NK-92MI. To our knowledge, this is the first in-vitro demonstration of a role for IL-22 in tumour cell resistance to lymphocyte-mediated cytotoxicity. Thus, IL-22 stimulation of HPAFII cells leads to production of angiogenic and survival factors as well as immunosuppressive cytokines, which offers insight into the functioning of IL-22 in tumour growth and immune evasion in vivo.
Recently, IL-22 was shown to support the growth of mantle cell lymphoma (MCL) [33], anaplastic large cell lymphoma (ALCL) [34] and colon carcinoma cell lines [17] through STAT-3 activation. Moreover, increased serum and tumour tissue levels of IL-22 were detected in both non-small cell lung carcinoma (NSCLC) [35] and multiple myeloma patients [36]. Interestingly, IL-22 protected NSCLC cell lines from starvation- and chemotherapeutic-induced apoptosis through STAT-3 up-regulation of the anti-apoptotic factors, Bcl-2 and Bcl-XL[35]. This is in agreement with our findings involving IL-22 augmentation of HPAFII expression of Bcl-XL and correlation with protection from cell death induced during nutrient deprivation. Thus, IL-22 may facilitate growth in a wide range of IL-22R-positive tumours by increasing anti-apoptotic factor expression.
Activated STAT-3 has also been shown to stimulate tumour angiogenesis through the up-regulation of VEGF [24],[27],[28],[37]. Mice transplanted with the human hepatocellular carcinoma cell line MHCC-97H displayed enhanced growth and metastasis of the tumour upon co-transplantation with IL-22-producing tumour infiltrating lymphocytes [38]. IL-22 production augmented STAT-3 activation which increased VEGF expression, thereby promoting the MHCC-97H growth and metastasis in vivo. This is in agreement with our data using the HPAFII pancreatic cancer line, as increased VEGF secretion was demonstrable upon IL-22 stimulation. Hence, IL-22 may support the growth of tumours through VEGF-mediated angiogenesis.
Using an in-vitro model, we have suggested that IL-22 may play a role in tumour immune evasion by indirectly suppressing both T cell IFN-γ production and NK cytotoxicity. Previously, Nagalakshmi et al. showed that IL-22 could stimulate IL-10 production by Colo205 colon epithelial cells [17]. If HPAFII cells could also produce IL-10 in response to IL-22, this would provide a mechanism whereby tumour cells might suppress inflammatory immune responses. In this study we show that IL-22 was able to stimulate HPAFII cells to secrete IL-10 which, in turn, diminished Jurkat T cell IFN-γ production, as confirmed by IL-10R blockade. In support of this mechanism, Halak et al. has shown that tumour-induced IL-10 could inhibit anti-tumour Th1 response development, as well as approaches to generate an IFN-γ-producing T cell population using immunogenic non-tumour antigen at the tumour site [39]. In terms of lymphocyte-mediated killing, IL-22 imparted to HPAFII cells complete resistance to NK-92MI cell cytotoxicity through the action of both IL-10 and TGF-β1. While TGF-β1 has been shown previously to impact NK cell activity negatively both in vitro and in vivo[40],[41], this is the first demonstration that a Th17-derived cytokine, IL-22, can promote the production of TGF-β1 by a human tumour line and such TGF-β1 can operate to impair NK cell function in vitro. Therefore, IL-22 signalling may serve as a mechanism for IL-22R-positive tumours to escape anti-tumour immunity mediated by Th1 and NK cells; however, subsequent in-vivo studies are needed to confirm this proposal.
Interestingly, administration of IL-22 small interfering RNA to Balb/c nude mice, which had been challenged previously with IL-22-expressing NSCLC cells, was shown to decrease tumour volume and weight over time [35]. Our work provides a possible explanation for this result. As nude mice have increased numbers of functional NK cells, we suggest that the observed anti-tumour response was due to alleviation of IL-22 pro-tumour effects, thereby permitting effective NK cell-mediated killing of the xenograft tumours.
In summary, we have shown that IL-22 can promote pro-tumour effects in a human pancreatic cancer cell line, which sheds light upon the possible role of IL-22 in tumour growth and development in vivo. Given that IL-22R is restricted to tissues of epithelial origin, blockade of endogenous IL-22 might be a useful approach in the treatment of varying types of IL-22R-positive cancers.
Acknowledgments
We would like to thank Laurel Ip for her technical assistance with the cell lines. In addition, we also thank Siri Chirumamilla for critical reading of the manuscript. This work was supported by funds provided through the Department of Basic Sciences, Georgia Campus – Philadelphia College of Osteopathic Medicine.
Disclosure
The authors have no financial conflicts of interest.
References
- 1.Su X, Ye J, Hsueh EC, Zhang Y, Hoft DF, Peng G. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J Immunol. 2010;184:1630–41. doi: 10.4049/jimmunol.0902813. [DOI] [PubMed] [Google Scholar]
- 2.Ankathatti MM, Deng Y, Mulligan SH, et al. Th17 and Th17-stimulated CD8(+) T cells play a distinct role in Th17-induced preventive and therapeutic antitumor immunity. Cancer Immunol Immunother. 2011;60:1473–84. doi: 10.1007/s00262-011-1054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gnerlich JL, Mitchem JB, Weir JS, et al. Induction of Th17 cells in the tumor microenvironment improves survival in a murine model of pancreatic cancer. J Immunol. 2010;185:4063–71. doi: 10.4049/jimmunol.0902609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin-Orozco N, Muranski P, Chung Y, et al. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31:787–98. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muranski P, Boni A, Antony PA, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–73. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L, Yi T, Kortylewski M, et al. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206:1457–64. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, Duan Y, Cheng X, et al. IL-17 is associated with poor prognosis and promotes angiogenesis via stimulating VEGF production of cancer cells in colorectal carcinoma. Biochem Biophys Res Commun. 2011;407:348–54. doi: 10.1016/j.bbrc.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 8.Iida T, Iwahashi M, Katsuda M, et al. Tumor-infiltrating CD4+ Th17 cells produce IL-17 in tumor microenvironment and promote tumor progression in human gastric cancer. Oncol Rep. 2011;25:1271–7. doi: 10.3892/or.2011.1201. [DOI] [PubMed] [Google Scholar]
- 9.Zhou P, Sha H, Zhu J. The role of T-helper 17 (Th17) cells in patients with medulloblastoma. J Int Med Res. 2010;38:611–19. doi: 10.1177/147323001003800223. [DOI] [PubMed] [Google Scholar]
- 10.Zhang JP, Yan J, Xu J, et al. Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol. 2009;50:980–9. doi: 10.1016/j.jhep.2008.12.033. [DOI] [PubMed] [Google Scholar]
- 11.Tartour E, Fossiez I, Joyeux A, et al. Interleukin 17, a T-cell-derived cytokine, promotes tumorigenicity of human cervical tumors in nude mice. Cancer Res. 1999;59:3698–704. [PubMed] [Google Scholar]
- 12.Numasaki M, Watanabe M, Suzuki T, et al. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J Immunol. 2005;175:6177–89. doi: 10.4049/jimmunol.175.9.6177. [DOI] [PubMed] [Google Scholar]
- 13.Xie MH, Aggarwal S, Ho WH, et al. Interleukin (IL)-22, a novel human cytokine that signals through the interferon receptor-related proteins CRF2-4 and IL-22R. J Biol Chem. 2000;275:31335–9. doi: 10.1074/jbc.M005304200. [DOI] [PubMed] [Google Scholar]
- 14.Kotenko SV, Izotova LS, Mirochnitchenko OV, et al. Identification of the functional interleukin-22 (IL-22) receptor complex: the IL-10R2 chain (IL-10Rbeta) is a common chain of both the IL-10 and IL-22 (IL-10-related T cell-derived inducible factor, IL-TIF) receptor complexes. J Biol Chem. 2001;276:2725–32. doi: 10.1074/jbc.M007837200. [DOI] [PubMed] [Google Scholar]
- 15.Radaeva S, Sun R, Pan HN, et al. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332–42. doi: 10.1002/hep.20184. [DOI] [PubMed] [Google Scholar]
- 16.Sonnenberg GF, Fouser LA, Artis D. Functional biology of the IL-22-IL-22R pathway in regulating immunity and inflammation at barrier surfaces. Adv Immunol. 2010;107:1–29. doi: 10.1016/B978-0-12-381300-8.00001-0. [DOI] [PubMed] [Google Scholar]
- 17.Nagalakshmi ML, Rascle A, Zurawski S, et al. Interleukin-22 activates STAT3 and induces IL-10 by colon epithelial cells. Int Immunopharmacol. 2004;4:679–91. doi: 10.1016/j.intimp.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Hoegl S, Bachmann M, Scheiermann P, et al. Protective properties of inhaled IL-22 in a model of ventilator-induced lung injury. Am J Respir Cell Mol Biol. 2011;44:369–76. doi: 10.1165/rcmb.2009-0440OC. [DOI] [PubMed] [Google Scholar]
- 19.Shioya M, Andoh A, Kakinoki S, et al. Interleukin 22 receptor 1 expression in pancreas islets. Pancreas. 2008;36:197–9. doi: 10.1097/MPA.0b013e3181594258. [DOI] [PubMed] [Google Scholar]
- 20.Aggarwal S, Xie MH, Maruoka M, et al. Acinar cells of the pancreas are a target of interleukin-22. J Interferon Cytokine Res. 2001;21:1047–53. doi: 10.1089/107999001317205178. [DOI] [PubMed] [Google Scholar]
- 21.Dumoutier L, Louahed J, Renauld JC. Cloning and characterization of IL-10-related T cell-derived inducible factor (IL-TIF), a novel cytokine structurally related to IL-10 and inducible by IL-9. J Immunol. 2000;164:1814–19. doi: 10.4049/jimmunol.164.4.1814. [DOI] [PubMed] [Google Scholar]
- 22.Dumoutier L, Van Roost E, Colau D, Renauld JC. Human interleukin-10 T cell-derived inducible factor: molecular cloning and functional characterization as an hepatocyte-stimulating factor. Proc Natl Acad Sci USA. 2000;97:10144–9. doi: 10.1073/pnas.170291697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levy DE, Darnell JE. STATs: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 24.Aggarwall BB, Sethi G, Ahn KS, et al. Targeting signal-transducer-and-activator-of-transcription-3 for prevention and therapy of cancer: modern target but ancient solution. Ann NY Acad Sci. 2006;1091:151–69. doi: 10.1196/annals.1378.063. [DOI] [PubMed] [Google Scholar]
- 25.Wang T, Niu G, Kortylewski M, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 26.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT-3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 27.Rivat C, Rodrigues S, Bruyneel E, et al. Implication of STAT3 signaling in human colonic cancer cells during intestinal trefoil factor 3 (TFF3) – and vascular endothelial growth factor-mediated cellular invasion and tumor growth. Cancer Res. 2005;65:195–202. [PubMed] [Google Scholar]
- 28.Aggarwal BB, Kunnumakkara AB, Harikumar KB, et al. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann NY Acad Sci. 2009;1171:59–76. doi: 10.1111/j.1749-6632.2009.04911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eriksen KW, Kaltoft K, Mikkelsen G, et al. Constitutive STAT3-activation in Sezary syndrome: tyrphostin AG490 inhibits STAT3-activation, interleukin-2 receptor expression and growth of leukemia Sezary cells. Leukemia. 2001;15:787–93. doi: 10.1038/sj.leu.2402093. [DOI] [PubMed] [Google Scholar]
- 30.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogata H, Chinen T, Yoshida T, et al. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-β1 production. Oncogene. 2006;25:2520–30. doi: 10.1038/sj.onc.1209281. [DOI] [PubMed] [Google Scholar]
- 32.Kinjyo I, Inoue H, Hamano S, et al. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-beta 1. J Exp Med. 2006;203:1021–31. doi: 10.1084/jem.20052333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gelebart P, Zak Z, Dien-Bard J, et al. Interleukin 22 signaling promotes cell growth in mantle cell lymphoma. Transl Oncol. 2011;4:9–19. doi: 10.1593/tlo.10172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bard JD, Gelebart P, Anand M, et al. Aberrant expression of IL-22 receptor 1 and autocrine IL-22 stimulation contribute to tumorigenicity in ALK+ anaplastic large cell lymphoma. Leukemia. 2008;22:1595–603. doi: 10.1038/leu.2008.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang W, Chen Y, Wei H, et al. Antiapoptotic activity of autocrine interleukin-22 and therapeutic effects of interleukin-22-small interfering RNA on human lung cancer xenografts. Clin Cancer Res. 2008;14:6432–9. doi: 10.1158/1078-0432.CCR-07-4401. [DOI] [PubMed] [Google Scholar]
- 36.Prabhala RH, Pelluru D, Fulciniti M, et al. Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood. 2010;115:5385–92. doi: 10.1182/blood-2009-10-246660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niu G, Wright KL, Huang M, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–8. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 38.Jiang R, Tan Z, Deng L, et al. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. 2011;54:900–9. doi: 10.1002/hep.24486. [DOI] [PubMed] [Google Scholar]
- 39.Halak BK, Maguire HC, Jr, Lattime EC. Tumor-induced interleukin-10 inhibits type 1 immune responses directed at a tumor antigen as well as non-tumor antigen present at the tumor site. Cancer Res. 1999;59:911–17. [PubMed] [Google Scholar]
- 40.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10:554–67. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JC, Lee KM, Ahn YO, et al. A possible mechanism of impaired NK cytotoxicity in cancer patients: down-regulation of DAP10 by TGF-beta1. Tumori. 2011;97:350–7. doi: 10.1177/030089161109700316. [DOI] [PubMed] [Google Scholar]