

Abstract
Statins are 3-hydroxy-3-methylglutaryl-co-enzyme A reductase inhibitors of cholesterol biosynthesis, and have been reported to exert pleiotropic effects on cellular signalling and cellular functions involved in inflammation. Recent reports have demonstrated that previous statin therapy reduced the risk of pneumonia or increased survival in patients with community-acquired pneumonia. However, the precise mechanisms responsible for these effects are unclear. In the present study, we examined the effects of statins on cytokine production from lipopolysaccharide (LPS)-stimulated human bronchial epithelial cells (BEAS-2B). Interleukin (IL)-6 and IL-8 mRNA expression and protein secretion in LPS-stimulated cells were inhibited significantly by the lipophilic statin pitavastatin and the hydrophilic statin pravastatin. As these inhibitory effects of statin were negated by adding mevalonate, the anti-inflammatory effects of statins appear to be exerted via the mevalonic cascade. In addition, the activation levels of Ras homologue gene family A (RhoA) in BEAS-2B cells cultured with pitavastatin were significantly lower than those without the statin. These results suggest that statins have anti-inflammatory effects by reducing cytokine production through inhibition of the mevalonic cascade followed by RhoA activation in the lung.
Keywords: bronchial epithelial cell, inflammatory cytokine, mevaloate, RhoA, statin
Introduction
Statins are 3-hydroxy-3-methylglutaryl co-enzyme A (HMG-CoA) reductase inhibitors, which potently inhibit cholesterol biosynthesis and are used extensively in the treatment of patients with hypercholesterolaemia [1],[2]. Statins also have anti-inflammatory and immunomodulatory properties in patients with cardiovascular disease [3]. Recent reports have shown that these effects contribute to protection against cardiovascular events [4],[5], cerebrovascular disease [6] and kidney disease [7]. In addition, human observational studies demonstrated that statins prevent the mortality and deterioration associated with sepsis [8],[9] and that previous statin therapy reduces the risk of pneumonia [10],[11] or increases survival in patients with community-acquired pneumonia [12]–[16]. Although the mechanisms of beneficial effects of statins on patients with sepsis or pneumonia remain controversial, experimental studies have shown that statins decrease the release of proinflammatory cytokines from mononuclear cells or monocytes [17]–[19]. Clinical studies have also shown that statins decrease serum levels of proinflammatory cytokines in patients with hyperlipidaemia [20]–[22]. Recently, Wang et al. [23] reported statins to have an anti-inflammatory effect on primary human nasal epithelial cells exposed to ambient air particles. However, to our knowledge there have been no studies examining the effects of statins on the human respiratory tract in infectious states such as pneumonia. In this study, therefore, we evaluated the effects of both lipophilic and hydrophilic statins on cytokine production from human bronchial epithelial cells and their effects on intracellular signalling pathways.
Materials and methods
Reagents
Pitavastatin (lipophilic statin) and pravastatin (hydrophilic statin) were kindly provided by Kowa Co. Ltd (Nagoya, Japan) and Daiichi-sankyo Co. Ltd (Tokyo, Japan), respectively. Pitavastatin and pravastatin were dissolved in dimethylsulphoxide (DMSO; Wako Pure Chemical Industries Ltd, Osaka, Japan), and were maintained at concentrations of 100 nM, 10 nM and 1 nM and 10 µM, 1 µM and 100 nM, respectively. The final concentration of DMSO added to cells was < 0·2%. Mevalonate was purchased from Sigma-Aldrich (St Louis, MO, USA).
Cell culture
The bronchoepithelial cell line BEAS-2B (human bronchial epithelium, ECACC 95102433) was obtained from the European Collection of Cell Cultures (Porton Down, Salisbury, Wiltshire, UK). BEAS-2B cells (1 × 104 cells per well) were placed in 24-well plates (Nunc, Roskilde, Denmark) and were cultured in bronchial epithelial cell growth medium (BEGM) without hydrocortisone, with or without statins under a 5% CO2 humidified atmosphere at 37°C. The medium was replaced every 3 days. After the 6 days of culture, the cells were stimulated using lipopolysaccharide (LPS) from Pseudomonas aeruginosa (Sigma Chemical Co., St Louis, MO, USA) at a concentration of 100 ng/ml.
Real-time polymerase chain reaction (PCR)
After incubation with LPS for an additional 3 h, RNA was isolated from harvested BEAS-2B cells using TaqMan Gene Expression Cells-to-Ct™ Kit (Ambion Inc., RNA Company, Austin, TX, USA), according to the manufacturer's instructions. Cytokine mRNA levels [interleukin (IL)-6, IL-8 and granulocyte-macrophage colony-stimulating factor (GM-CSF)] were determined using the StepOnePlusTM Real-Time PCR System (Applied Biosystems Inc., Foster City, CA, USA). Results were normalized against the expression of an internal control [glyceraldehyde-3-phosphate dehydrogenase (GAPDH)], and relative gene expression levels were calculated using the ΔΔCt-method.
Enzyme-linked immunosorbent assay (ELISA)
After incubation with LPS for an additional 24 h, IL-6, IL-8 and GM-CSF levels in supernatants from BEAS-2B cells were measured by Multiscan JX (Thermo Fisher Scientific Inc., Waltham, MA, USA) using an ELISA kit (Quantikine; R&D Systems, Minneapolis, MN, USA).
Treatment with mevalonate
The effects of statins are related to their ability to competitively block HMG-CoA reductase and decrease the production of multiple intermediates in the cholesterol biosynthetic pathway [24]. Mevalonate is the initial product of HMG-CoA reductase, and the levels of mevalonate are reduced by statin therapy. To determine whether pitavastatin acts through an HMG-CoA reductase-dependent pathway (mevalonic cascade), BEAS-2B cells were cultured in BEGM with pitavastatin (10 nM) plus mevalonate (10 mM) under the same conditions as described above for 1 week before LPS stimulation. The culture duration was established in line with the criteria used in previous reports, with the cells being subjected to the same length of culture as that for both statins and mevalonate [25],[26].
Ras homologue gene family A (RhoA) activation assay
Inhibition of mevalonate synthesis by statins also prevents the synthesis of other important isoprenoid intermediates on the cholesterol biosynthetic pathway, such as farnesylpyrophosphate and geranylgeranylpyrophosphate. These intermediates serve as important lipid attachments for the post-translational modification of various cell-signalling proteins, such as Rho GTPases. To assess the signalling pathways of downstream of the mevalonic cascade, LPS-induced activation of RhoA was analysed. BEAS-2B cells (0·3 × 104 cells per well) were cultured with or without pitavastatin (100 nM). Medium was replaced every 2 days. After 5 days of culture, the cells were stimulated with LPS. After incubation with LPS for an additional 3 min, whole cell lysates were collected using the lysis buffer provided by ELISA-based RhoA activity assay (G-LISA; Cytoskeleton, Denver, CO, USA). Lysates were incubated in microwells to which the rhotekin binding domain peptide was bound, and active RhoA was measured using indirect immunodetection followed by a colorimetric reaction at 490 nm.
Statistical analysis
Comparisons of data were made utilizing the unpaired t-test. Two-tailed P-values of less than 0·05 were considered to be statistically significant.
Results
Inhibitory effects of pitavastatin
IL-6, IL-8 and GM-CSF mRNA expression in LPS-stimulated BEAS-2B cells was inhibited significantly by pitavastatin (1 nM, 10 nM and 100 nM) in a dose-dependent manner (Fig. 1a,c,e). IL-6 and IL-8 secretion by LPS-stimulated BEAS-2B cells was also reduced significantly by pitavastatin in a dose-dependent manner (Fig. 1b,d).
Fig. 1.
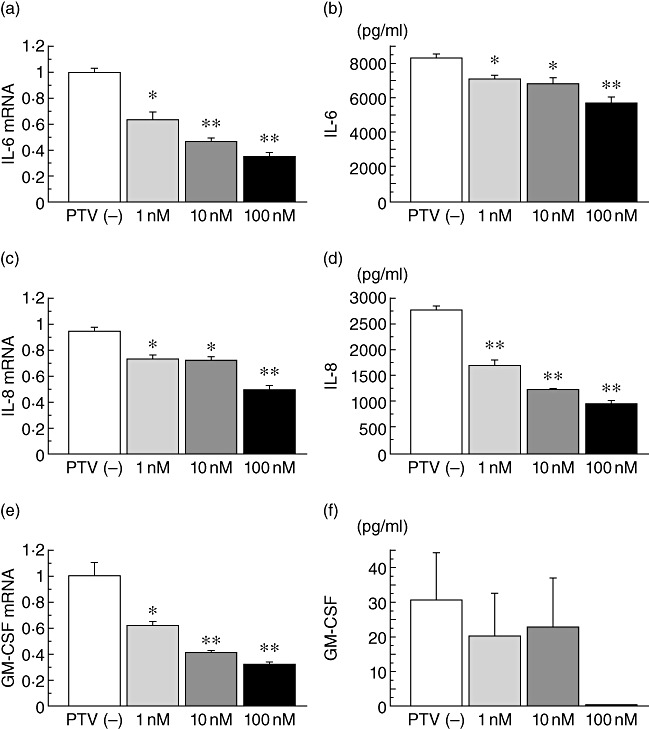
The inhibitory effects of pitavastatin (PTV) on inflammatory cytokine production. Interleukin (IL)-6, IL-8 and granulocyte–macrophage colony-stimulating factor (GM-CSF) mRNA expression in lipopolysaccharide (LPS)-stimulated BEAS-2B cells was inhibited significantly by PTV (1 nM, 10 nM, 100 nM) in a dose-dependent manner (a,c,e). IL-6 and IL-8 secretion, but not GM-CSF secretion, by LPS-stimulated BEAS-2B cells was also reduced significantly by PTV in a dose-dependent manner (b,d,f); *P < 0·05, **P < 0·001, compared with the cells cultured without PTV (white column).
Inhibitory effects of pravastatin
IL-6 and IL-8 mRNA expression in LPS-stimulated BEAS-2B cells was inhibited significantly at various concentrations of pravastatin (Fig. 2a,c). IL-6 and IL-8 secretion by LPS-stimulated BEAS-2B cells was also reduced significantly by pravastatin in a dose-dependent manner (Fig. 2b,d). GM-CSF secretion was reduced only by a high dose of pravastatin (Fig. 2f).
Fig. 2.
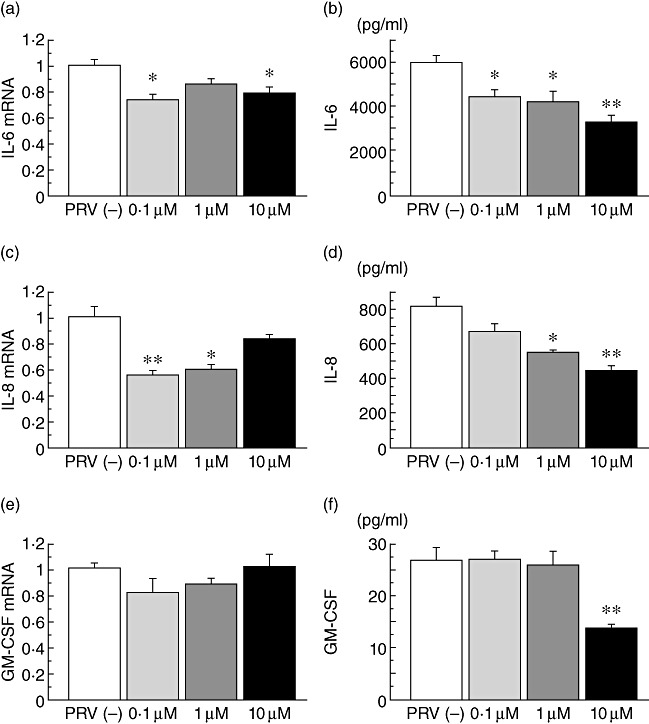
The effects of exogenous mevalonate on the cytokine production. Interleukin (IL)-6 and IL-8 mRNA expression, but not granulocyte–macrophage colony-stimulating factor (GM-CSF) mRNA expression, in lipopolysaccharide (LPS)-stimulated BEAS-2B cells was inhibited significantly at various concentrations of pravastatin (PRV) (a,c,e). IL-6 and IL-8 secretion by LPS-stimulated BEAS-2B cells was also reduced significantly by PRV in a dose-dependent manner (b,d). GM-CSF secretion was reduced only by 10 µM PRV (f); *P < 0·05, **P < 0·001, compared with the cells cultured without PRV (white column).
Reverse effects of exogenous mevalonate
All the inhibitory effects of pitavastatin on IL-6, IL-8 and GM-CSF cytokine mRNA expression and/or production, as shown in Fig. 1, were negated by treatment with mevalonate (Fig. 3), indicating that the inhibitory effects of statins act via the mevalonic cascade. The levels of LPS-induced GM-CSF protein secretion were very low (0–30 pg/ml in Fig. 1), and there were no significant inhibitory effects of pitavastatin (even at the high concentration of 100 nM pitavastatin). Therefore, the results for the GM-CSF protein were omitted from the analysis.
Fig. 3.
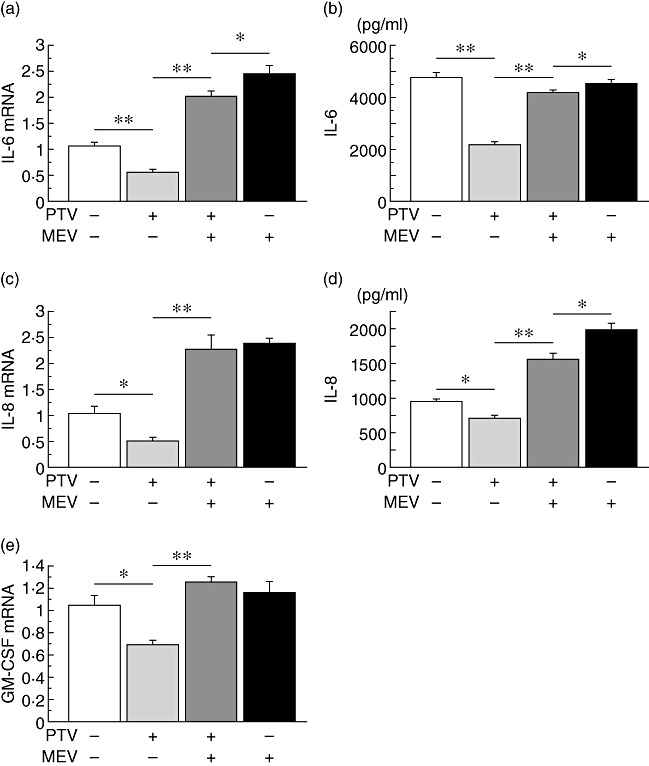
Reverse effects of exogenous mevalonate (MEV) to the pitavastatin (PTV) inhibition. The inhibitory effects of PTV (10 nM) on lipopolysaccharide (LPS)-induced interleukin (IL)-6, IL-8 and granulocyte–macrophage colony-stimulating factor (GM-CSF) cytokine mRNA expression and production, except GM-CSF production, were negated by treatment with 10 mM of MEV (a–e); *P < 0·05, **P < 0·001, compared between the groups.
Reduction of RhoA activation by pitavastatin
The LPS-induced activation levels of RhoA in BEAS-2B cells cultured with pitavastatin were significantly lower than those without the statin (Fig. 4).
Fig. 4.
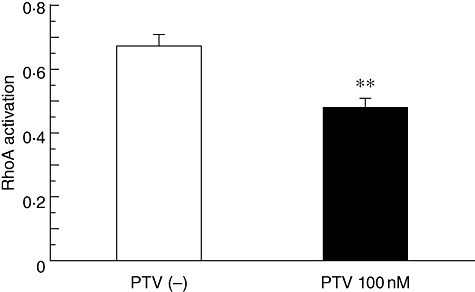
The reduction of Ras homologue gene family A (RhoA) activation by pitavastatin. The lipopolysaccaride (LPS)-induced activation levels of RhoA in BEAS-2B cells cultured with pitavastatin (PTV) (100 nM) were significantly lower than those without the statin; **P < 0·001, compared with the cells cultured without PTV (white column).
Discussion
The present study demonstrated that both lipophilic and hydrophilic statins inhibited the inflammatory response of BEAS-2B cells by suppressing the production of cytokines, independent of the cholesterol synthesis pathway. It is certain that the anti-inflammatory effects of statins are exerted via the mevalonic cascade, as the inhibition of cytokine production was blocked by addition of mevalonate. Previous studies have shown that statins alter the expression of various cytokines in monocytes, macrophages or vascular endothelial cells [17],[19],[27],[28]. Wang et al. [23] have recently reported the expression of CCL5, CCL11 and IL13RA in the nasal epithelial cells, which were obtained from patients with chronic rhinosinusitis, exposed to ambient air pollution particulates, was inhibited by statins. However, this is the first study to show that statins also alter the expression of cytokines in bronchial epithelial cells, although the hydrophilic statin required a higher dose to exert similar effects as the lipophilic statin. In the clinical setting, the concentrations (Cmax) of pivastatin in the blood are 16·8–26·1 ng/ml (2-mg single administration) and are 16·5 ± 6·9 ng/ml (10-mg single administration) for pravastatin. Therefore, the concentrations of pravastatin used in our experiments were higher than those achieved in the clinical setting. Although the actual concentration of these agents in the human airway is unclear in the clinical setting, this is one of the limitations of our in-vitro experiment.
In this study, to test the effects of statins on the human respiratory tract in infectious states such as pneumonia, we examined the expression and production of IL-6, IL-8, and GM-CSF from BEAS-2B cells stimulated with LPS. IL-6 is a pleiotropic cytokine, and also a critical inflammatory mediator in inflammatory lung diseases, including bacterial pneumonia. IL-8 is known to be a potent activator of neutrophils. In addition, GM-CSF is known to prime leucocytes for inflammatory stimuli in vitro, and a previous study [29] suggested that GM-CSF represents an endogenous enhancer of LPS-induced organ injury by potentiating the release of proinflammatory cytokines. The IL-8 and GM-CSF mRNA levels appeared to be increased by pravastatin in a dose-dependent manner in this study (Fig. 2), although they were not found to be statistically significant increases compared with the cells cultured without pravastatin. This phenomenon was not seen in the experiment using pitavastatin (Fig. 1). Lipophilic statins (e.g. pitavastatin) have higher cell permeabilities and better stabilities in terms of their pharmacological efficacy, in comparison to hydrophilic statins (e.g. pravastatin) [30]. Although the clear mechanism is unknown, especially in the circumstances of the airway, there is a possibility that the lower cell membrane permeability and instability of pravastatin might cause the discrepancy between mRNA and protein inhibition concerning IL-8 and GM-CSF.
Several clinical studies have shown that previous use of statins is able to reduce the risk of pneumonia or increase survival rate in patients, particularly those with severe community-acquired pneumonia [10]–[16]. Although the precise mechanisms are unknown, some biological mechanisms of statins that modify the humoral immune response and inhibit endothelial cell dysfunction may contribute to achieving beneficial outcomes [16]. A previous cohort study showed that C-reactive protein levels at admission tend to be lower in statin users than in non-users, and this may be due to the anti-inflammatory effects of statins [16]. In fact, our study demonstrated that statins inhibited the production of IL-6, which plays a role in the production of C-reactive protein. C-reactive protein is involved in organ dysfunction and death in critically ill patients [24], thus the inhibition of IL-6 production may be a mechanism to reduce the risk of pneumonia.
When bacteria invade the alveolar space, pneumonia is accompanied by inflammation induced by various inflammatory cytokines, which are produced by bronchial epithelial cells or alveolar macrophages in order to attract or activate inflammatory cells such as neutrophils or lymphocytes. This response is useful for killing bacteria, while causing tissue injury via the excess accumulation of neutrophils or lymphocytes from blood vessels to the site of infection. In particular, severe pneumonia tends to be concurrent with acute lung injury, acute respiratory distress syndrome and bacteraemia or sepsis, which are related to neutrophil inflammation. In-vivo studies have shown that statins inhibit lung inflammation by suppressing myeloperoxidase activity and reducing the accumulation of neutrophils in a murine inflammatory model of acute lung injury [25],[31]. Furthermore, animal and human observational studies suggest that statins may prevent the morbidity and mortality associated with the sepsis [24]. As in our study, statins inhibit the secretion of IL-8, a neutrophil chemoattractant from bronchoepithelial cells, and this effect might lead to the suppression of severe inflammation by neutrophils, followed by protecting severe pneumonia with bacteraemia or sepsis.
In innate immunity, Toll-like receptors (TLRs) play a crucial role by recognizing and reacting to microbial pathogens. LPS is the major component of the outer membrane of Gram-negative bacteria and is recognized by TLR-4. TLR-4 activation by LPS is followed by signal pathways dependent upon/independent of myeloid differentiation factor, which elicits the activation of nuclear factor (NF)-kappa B or activating protein 1 (AP-1) in inflammatory pathways [32]. AP-1 activation in TLR signalling is mediated mainly by mitogen-activated protein kinases (MAPKs), such as c-Jun N-terminal kinase, p38 and extracellular signal-regulated kinase [32]. Conversely, Rho-dependent signalling pathways are also considered to be part of TLR-4 signalling pathways, and may regulate MAPKs [33]. Moreover, the Rho signal transduction pathway is downstream of the mevalonic pathway [26]. As the inhibition of HMG-CoA reductase leads to a decrease in farnesylated and geranylgeranylated small G-proteins such as Rho, its inhibition subsequently blocks Rho signalling [26]. In this study, the level of RhoA activation in the LPS-stimulated BEAS-2B cells was decreased by adding statin, therefore the anti-inflammatory effects of statins may be related in part to the Rho-dependent signalling pathway. In addition, mevalonate itself appeared to increase the cytokine expression and production by BEAS-2B cells (Fig. 3). Assuming that the LPS-induced cytokine production from the cells is partly involved in the mevalonic cascade, there is a possibility that mevalonate directly accelerated this pathway and stimulated the production of farnesylated and geranylgeranylated pyrophosphates such as Rho, and subsequently increased the cytokine production from the cells.
In conclusion, this study indicates the anti-inflammatory effects of statins by reducing cytokine production through inhibition of the mevalonic cascade in human bronchial epithelial cells. These effects possibly contribute, at least in part, to the decrease in mortality of patients with severe pneumonia, and suggest that statins could be another strategy in the treatment of critically ill patients with severe inflammation.
Disclosure
The authors have no disclosures in relation to the article.
References
- 1.Borghi C, Dormi A, Veronesi M, Immordino V, Ambrosioni E. Use of lipid-lowering drugs and blood pressure control in patients with arterial hypertension. J Clin Hypertens. 2002;4:277–85. doi: 10.1111/j.1524-6175.2002.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaw A. A new reality: achieving cholesterol-lowering goals in clinical practice. Atheroscler Suppl. 2002;2:5–8. doi: 10.1016/s1567-5688(01)00018-6. discussion 8–11. [DOI] [PubMed] [Google Scholar]
- 3.Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov. 2005;4:977–87. doi: 10.1038/nrd1901. [DOI] [PubMed] [Google Scholar]
- 4.Yamada T, Node K, Mine T, et al. Long-term effect of atorvastatin on neurohumoral activation and cardiac function in patients with chronic heart failure: a prospective randomized controlled study. Am Heart J. 2007;153:1055. doi: 10.1016/j.ahj.2007.03.027. e1051–8. [DOI] [PubMed] [Google Scholar]
- 5.Alber HF, Frick M, Sussenbacher A, et al. Effect of atorvastatin on peripheral endothelial function and systemic inflammatory markers in patients with stable coronary artery disease. Wien Med Wochenschr. 2007;157:73–8. doi: 10.1007/s10354-007-0377-y. [DOI] [PubMed] [Google Scholar]
- 6.Nassief A, Marsh JD. Statin therapy for stroke prevention. Stroke. 2008;39:1042–8. doi: 10.1161/STROKEAHA.107.501361. [DOI] [PubMed] [Google Scholar]
- 7.Campese VM, Park J. HMG-CoA reductase inhibitors and the kidney. Kidney Int. 2007;71:1215–22. doi: 10.1038/sj.ki.5002174. [DOI] [PubMed] [Google Scholar]
- 8.Almog Y, Shefer A, Novack V, et al. Prior statin therapy is associated with a decreased rate of severe sepsis. Circulation. 2004;110:880–5. doi: 10.1161/01.CIR.0000138932.17956.F1. [DOI] [PubMed] [Google Scholar]
- 9.Dobesh PP, Klepser DG, McGuire TR, Morgan CW, Olsen KM. Reduction in mortality associated with statin therapy in patients with severe sepsis. Pharmacotherapy. 2009;29:621–30. doi: 10.1592/phco.29.6.621. [DOI] [PubMed] [Google Scholar]
- 10.van de Garde EM, Hak E, Souverein PC, Hoes AW, van den Bosch JM, Leufkens HG. Statin treatment and reduced risk of pneumonia in patients with diabetes. Thorax. 2006;61:957–61. doi: 10.1136/thx.2006.062885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schlienger RG, Fedson DS, Jick SS, Jick H, Meier CR. Statins and the risk of pneumonia: a population-based, nested case–control study. Pharmacotherapy. 2007;27:325–32. doi: 10.1592/phco.27.3.325. [DOI] [PubMed] [Google Scholar]
- 12.Mortensen EM, Restrepo MI, Anzueto A, Pugh J. The effect of prior statin use on 30-day mortality for patients hospitalized with community-acquired pneumonia. Respir Res. 2005;6:82. doi: 10.1186/1465-9921-6-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falagas ME, Makris GC, Matthaiou DK, Rafailidis PI. Statins for infection and sepsis: a systematic review of the clinical evidence. J Antimicrob Chemother. 2008;61:774–85. doi: 10.1093/jac/dkn019. [DOI] [PubMed] [Google Scholar]
- 14.Mortensen EM, Pugh MJ, Copeland LA, et al. Impact of statins and angiotensin-converting enzyme inhibitors on mortality of subjects hospitalised with pneumonia. Eur Respir J. 2008;31:611–7. doi: 10.1183/09031936.00162006. [DOI] [PubMed] [Google Scholar]
- 15.Chalmers JD, Singanayagam A, Murray MP, Hill AT. Prior statin use is associated with improved outcomes in community-acquired pneumonia. Am J Med. 2008;121:1002–7. doi: 10.1016/j.amjmed.2008.06.030. e1001. [DOI] [PubMed] [Google Scholar]
- 16.Thomsen RW, Riis A, Kornum JB, Christensen S, Johnsen SP, Sorensen HT. Preadmission use of statins and outcomes after hospitalization with pneumonia: population-based cohort study of 29 900 patients. Arch Intern Med. 2008;168:2081–7. doi: 10.1001/archinte.168.19.2081. [DOI] [PubMed] [Google Scholar]
- 17.Li JJ, Chen XJ. Simvastatin inhibits interleukin-6 release in human monocytes stimulated by C-reactive protein and lipopolysaccharide. Coron Artery Dis. 2003;14:329–34. doi: 10.1097/01.mca.0000078062.22445.60. [DOI] [PubMed] [Google Scholar]
- 18.Bessler H, Salman H, Bergman M, Straussberg R, Djaldetti M. In vitro effect of statins on cytokine production and mitogen response of human peripheral blood mononuclear cells. Clin Immunol. 2005;117:73–7. doi: 10.1016/j.clim.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 19.Okopien B, Krysiak R, Kowalski J, et al. Monocyte release of tumor necrosis factor-alpha and interleukin-1beta in primary type IIa and IIb dyslipidemic patients treated with statins or fibrates. J Cardiovasc Pharmacol. 2005;46:377–86. doi: 10.1097/01.fjc.0000175455.46245.c8. [DOI] [PubMed] [Google Scholar]
- 20.Rosenson RS, Tangney CC, Casey LC. Inhibition of proinflammatory cytokine production by pravastatin. Lancet. 1999;353:983–4. doi: 10.1016/S0140-6736(98)05917-0. [DOI] [PubMed] [Google Scholar]
- 21.Ascer E, Bertolami MC, Venturinelli ML, et al. Atorvastatin reduces proinflammatory markers in hypercholesterolemic patients. Atherosclerosis. 2004;177:161–6. doi: 10.1016/j.atherosclerosis.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 22.Tousoulis D, Antoniades C, Vassiliadou C, et al. Effects of combined administration of low dose atorvastatin and vitamin E on inflammatory markers and endothelial function in patients with heart failure. Eur J Heart Fail. 2005;7:1126–32. doi: 10.1016/j.ejheart.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 23.Wang W, Le W, Ahuja R, Cho DY, Hwang PH, Upadhyay D. Inhibition of inflammatory mediators: role of statins in airway inflammation. Otolaryngol Head Neck Surg. 2011;144:982–7. doi: 10.1177/0194599811400367. [DOI] [PubMed] [Google Scholar]
- 24.Terblanche M, Almog Y, Rosenson RS, Smith TS, Hackam DG. Statins and sepsis: multiple modifications at multiple levels. Lancet Infect Dis. 2007;7:358–68. doi: 10.1016/S1473-3099(07)70111-1. [DOI] [PubMed] [Google Scholar]
- 25.Kiener PA, Davis PM, Murray JL, Youssef S, Rankin BM, Kowala M. Stimulation of inflammatory responses in vitro and in vivo by lipophilic HMG-CoA reductase inhibitors. Int Immunopharmacol. 2001;1:105–18. doi: 10.1016/s0162-3109(00)00272-1. [DOI] [PubMed] [Google Scholar]
- 26.Ramasubbu K, Estep J, White DL, Deswal A, Mann DL. Experimental and clinical basis for the use of statins in patients with ischemic and nonischemic cardiomyopathy. J Am Coll Cardiol. 2008;51:415–26. doi: 10.1016/j.jacc.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 27.Shovman O, Levy Y, Gilburd B, Shoenfeld Y. Antiinflammatory and immunomodulatory properties of statins. Immunol Res. 2002;25:271–85. doi: 10.1385/IR:25:3:271. [DOI] [PubMed] [Google Scholar]
- 28.Greenwood J, Mason JC. Statins and the vascular endothelial inflammatory response. Trends Immunol. 2007;28:88–98. doi: 10.1016/j.it.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tiegs G, Barsig J, Matiba B, Uhlig S, Wendel A. Potentiation by granulocyte macrophage colony-stimulating factor of lipopolysaccharide toxicity in mice. J Clin Invest. 1994;93:2616–22. doi: 10.1172/JCI117274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodwell VW, Nordstrom JL, Mitschelen JJ. Regulation of HMG-CoA reductase. Adv Lipid Res. 1976;14:1–74. doi: 10.1016/b978-0-12-024914-5.50008-5. [DOI] [PubMed] [Google Scholar]
- 31.Jacobson JR, Barnard JW, Grigoryev DN, Ma SF, Tuder RM, Garcia JG. Simvastatin attenuates vascular leak and inflammation in murine inflammatory lung injury. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1026–32. doi: 10.1152/ajplung.00354.2004. [DOI] [PubMed] [Google Scholar]
- 32.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–25. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 33.Vojtek AB, Cooper JA. Rho family members: activators of MAP kinase cascades. Cell. 1995;82:527–9. doi: 10.1016/0092-8674(95)90023-3. [DOI] [PubMed] [Google Scholar]