

Background: An ER-associated chaperone protein, σ1 receptor (σ1R), regulates ER/mitochondrial Ca2+ mobilization through the IP3 receptor.
Results: We identify a novel short splicing variant of σ1R, termed σ1SR, and demonstrate its dominant negative function.
Conclusion: σ1SR interferes with σ1R function in mitochondrial Ca2+ mobilization and ATP production under ER stress conditions.
Significance: In contrast to σ1R function, σ1SR has detrimental effects on cell survival.
Keywords: Apoptosis; Calcium Imaging; Endoplasmic Reticulum Stress; Inositol 1,4,5-Trisphosphate; Mitochondria; Neuroblastoma; Neurodegeneration; σ Receptor
Abstract
The σ1 receptor (σ1R) regulates endoplasmic reticulum (ER)/mitochondrial interorganellar Ca2+ mobilization through the inositol 1,4,5-trisphosphate receptor (IP3R). Here, we observed that expression of a novel splice variant of σ1R, termed short form σ1R (σ1SR), has a detrimental effect on mitochondrial energy production and cell survival. σ1SR mRNA lacks 47 ribonucleotides encoding exon 2, resulting in a frameshift and formation of a truncated receptor. σ1SR localizes primarily in the ER at perinuclear regions and forms a complex with σ1R but not with IP3R in the mitochondrion-associated ER membrane. Overexpression of both σ1R and the truncated isoform promotes mitochondrial elongation with increased ER mitochondrial contact surface. σ1R overexpression increases the efficiency of mitochondrial Ca2+ uptake in response to IP3R-driven stimuli, whereas σ1SR overexpression reduces it. Most importantly, σ1R promotes ATP production via increased mitochondrial Ca2+ uptake, promoting cell survival in the presence of ER stress. By contrast, σ1SR suppresses ATP production following ER stress, enhancing cell death. Taken together, the newly identified σ1SR isoform interferes with σ1R function relevant to mitochondrial energy production under ER stress conditions, promoting cellular apoptosis.
Introduction
Endoplasmic reticulum (ER)2/mitochondrial Ca2+ transport contributes to many cellular processes, including ATP generation and cell survival (1, 2). ER inositol 1,4,5-trisphosphate receptors (IP3Rs) are localized in the mitochondrion-associated ER membrane (MAM) (3, 4), where the IP3R plays critical roles in mitochondrial Ca2+ transport. Ca2+ overload through IP3R promotes apoptosis under pathological conditions (5). By contrast, mitochondrial Ca2+ uptake through IP3R is crucial for basal mitochondrial ATP production required to maintain normal cellular biogenesis (6).
Mitochondrial Ca2+ uptake derived from IP3R-mediated Ca2+ release is facilitated by interaction between IP3R and voltage-dependent anion channels in the MAM (4, 7, 8), and several IP3R-binding molecular chaperones regulate mitochondrial Ca2+ influx from the ER (9). Among them, the σ1 receptor (σ1R), which was cloned by Hanner et al. (10), was recently identified as an ER-associated chaperone protein (11). σ1R can also translocate from the ER to the MAM or plasma membrane to modulate diverse cellular activities, including lipid metabolism (12) and N-methyl-d-aspartate receptor activity (13). Functional analyses in Chinese hamster ovary (CHO) cells reveal that the σ1R stabilizes the conformation of the MAM-associated IP3R type-3 in the ER, positively regulating Ca2+ influx into mitochondria. In addition, σ1R knockdown in CHO cells facilitates ER stress-induced cell death (11).
Ca2+ transport between the ER and mitochondria plays important roles in neurodegenerative diseases, such as Alzheimer, Parkinson, and Huntington disease (14). Indeed, σ1R ligands are considered therapeutic targets for several psychiatric and neurodegenerative diseases (15–17). However, how these ligands mediate neuroprotective effects remains unclear. More recently, σ1R mutations have been observed in patients with dementia attributable to frontotemporal lobar degeneration (18) or juvenile amyotrophic lateral sclerosis (19). Interestingly, the latter apparently promotes nuclear transport of mutant forms of σ1R.
In this study, we observed a novel σ1R splicing variant in mouse brain, namely a short form (σ1SR) lacking 47 bp of exon 2. σ1SR protein is primarily expressed in the ER, where it interacts with σ1R. Interestingly, σ1SR overexpression decreased mitochondrial Ca2+ uptake in response to IP3R-mediated stimulation, indicating that it antagonizes σ1R activity. Moreover, σ1SR overexpression promoted autophagic apoptosis, consistent with IP3R destabilization and decreased ATP production attributable to reduced mitochondrial Ca2+ uptake. Our results show for the first time that σ1SR interferes with mitochondrial ATP biogenesis by inhibiting σ1R function following ER stress.
EXPERIMENTAL PROCEDURES
Sequencing and Construction of Expression Vectors Encoding σ1R Isoforms
Total RNAs were prepared from mouse brain hippocampus using TRIzol LS reagent (Invitrogen) according to the manufacturer's protocol. mRNA was reverse-transcribed into single-stranded cDNA using an oligo(dT) primer (Promega, Madison, WI) and Moloney murine leukemia virus-reverse transcriptase (Invitrogen). DNA sequences of σ1R isoforms amplified by PCR using specific 5′- and 3′-primers and primer sequences are shown in supplemental Table 1. All DNA sequences were determined at the Fasmac DNA Sequence Service (FASMAC Co., Ltd., Atsugi, Japan). To construct expression vectors, PCR amplification products were digested with XhoI and BamHI and ligated with purified XhoI- and BamHI-digested pmCherry-N1 or pEGFP-N1 vector (Clontech) in the sense orientation.
Cell Culture, Transfection, and Production of Stably Transfected Cell Lines
Neuro-2a cells from a mouse neuroblastoma C1300 tumor were obtained from the Human Science Research Resources Bank (IFO50081) (Osaka, Japan). Neuro-2a cells were grown in Dulbecco's minimal essential medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and penicillin/streptomycin (100 units/100 μg/ml) in a 5% CO2 incubator at 37 °C. Cells were transfected with expression vectors using Lipofectamine 2000 (Invitrogen), and experiments were performed 48 h later as described (20). σ1R small interfering (si)RNA (sense, 5′-ACACGTGGATGGTGGAGTA-3′, and antisense, 5′-TACTCCACCATCCACGTGT-3′) was purchased from Exigen (Tokyo, Japan). Transfections were performed with 100 nm σ1Rs siRNA according to the methods of Ref. 21. Stably transfected Neuro-2a cells were prepared as described (22). Briefly, cells were transfected with σ1R or σ1SR cDNA in pmCherry-N1 expression vectors as described above. Transfected cells were plated in medium containing 1000 μg G418/ml, and G418-resistant colonies were isolated.
Immunoprecipitation and Immunoblotting
Immunoprecipitation and immunoblotting analysis were performed as described (20). 12-Week-old male C57BL/6J mouse brains were immediately removed from euthanized mice and perfused in ice-cold buffer for 3 min (0.32 m sucrose, 20 mm Tris-HCl, pH 7.4), and olfactory bulb, cortex, hippocampus, striatum, and brainstem were dissected. These samples and Neuro-2a cells were homogenized in buffer containing 50 mm Tris-HCl, pH 7.4, 0.5% Triton X-100, 0.5 m NaCl, 4 mm EDTA, 4 mm EGTA, 1 mm Na3VO4, 50 mm NaF, 1 mm DTT, 2 μg/ml pepstatin A, and 1 μg/ml leupeptin, then treated with Laemmli sample solution, and boiled for 3 min. Antibodies included the following: rabbit polyclonal antibodies against the N-terminal cytosolic domain (52–69 amino acids) (1:1000) and C-terminal lumenal domain (143–165 amino acids) of σ1R (1:1000; a kind gift of Dr. Teruo Hayashi, NIDA, Baltimore); GFP (1:2000; Clontech); calcineurin (1:1000) (23); voltage-dependent anion channel (1:1000; Cell Signaling Technology, Beverly, MA); CREB-2 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA); LC-3 (PM036, 1:1000; MBL, Nagoya, Japan); rabbit monoclonal antibody against phospho-PERK (1:1000; Cell Signaling Technology); mouse monoclonal antibodies against C/EBP homologous protein (1:200; Santa Cruz Biotechnology); β-tubulin (1:1000; Sigma); and pan-IP3R (1:500; Millipore, Bedford, MA). For subcellular fractionation, cells were washed twice with ice-cold phosphate-buffered saline (PBS), separated into cytosolic, ER, and mitochondrial membranes and nuclear fractions using the subcellular protein fractionation kit (Thermo Fisher Scientific Inc., Waltham, MA) following the manufacturer's protocol, and analyzed by Western blot analysis.
Immunohistochemistry
Fluorescence immunohistochemical studies were performed as described (24). Cells were fixed in 4% paraformaldehyde in phosphate buffer for 30 min at room temperature and washed in PBS. Cells were blocked with 3% bovine serum albumin in PBS for 1 h. First antibodies included mouse monoclonal antibodies against GM130 (1:1000; BD Biosciences) and rabbit polyclonal antibody against LC-3 (PM036, 1:1000; MBL). After thorough washes in PBS, sections were incubated with secondary antibodies in blocking solution at 20 °C for 24 h. Antibodies included Alexa 594-labeled anti-mouse IgG and Alexa 448-labeled anti-rabbit IgG (1:500; Invitrogen) in blocking solution at 20 °C for 3 h. After several washes in PBS, sections were mounted on slides with Vectashield (Vector Laboratories Inc., Burlingame, CA). For nuclear staining, sections were incubated with DAPI (Vector Laboratories, Burlingame, CA). Immunofluorescent images were analyzed using a confocal laser scanning microscope (LSM700; Zeiss, Thornwood, NY). To detect ER and mitochondrial distribution, Neuro-2a cells were transfected with erRFP or mitochondrial GFP (mtGFP) (Bacman 2.0, Invitrogen) and analyzed using confocal microscopy of fixed cells.
Morphometric and Contact Analysis
Morphometric and contact analysis was performed as described previously (25) with modifications. For morphometric analysis of mitochondria, the major axis length of each identified object was calculated. Cells were scored as showing elongated mitochondria when >50% of the objects in the image displayed a major axis longer than 3 μm. For morphometric analysis of ER, major axis length and elongation index of each identified object were calculated. Cells were scored as showing reticular ER when the major axis was longer than 5 μm, and the elongation index exceeded 4 in more than 50% of the identified objects. For mitochondrial network and ER co-localization, one focal plane was analyzed. Images were deconvoluted, and background was subtracted using ImageJ software. Co-localization of organelles was quantified using the co-localization coefficient of Manders et al. (26).
Quantification of Neurite Sprouting
Neurite sprouting by Neuro-2a cells was assessed by staining with βIII-tubulin (1:1000, Promega, Madison, WI) and use of the VECTASTAIN ABC kit (Vector Laboratories, Burlingame, CA), according to the manufacturer's protocol. Images were acquired using the 40× objective of a microscope (BX51WI, Olympus, Tokyo, Japan) equipped with a digital camera (Micropublisher 5.0, QIMAGING, Burnaby, British Columbia, Canada). Sprouting was quantified as the percentage of cells with neurites longer than twice the body diameter. Neurite length was determined using ImageJ software. Six fields (100 cells per field) in each condition were chosen randomly and photographed.
Fluorometric [Ca2+]c and [Ca2+]mt Measurements
Neuro-2a cells were grown on 0.01% poly-l-lysine (Sigma)-coated glass-bottom dishes. For [Ca2+]c measurement, cells were rinsed in Krebs buffer (135 mm NaCl, 6 mm KCl, 1.2 mm MgCl2, 12 mm glucose, 1.5 mm CaCl2, 12 mm HEPES, pH 7.3), loaded with 2.5 μm Fura-2/AM (Sigma) for 15 min at 37 °C in darkness, and washed with Krebs buffer for 15 min. Measurements were performed in Ca2+-free Krebs buffer including 2 mm EGTA. Ratio measurements were performed every 3 s by excitation at 340 and 380 nm and recording of the emission at 530 nm. Ratio values were derived by averaging fluorescence intensity from the entire cytosolic area. To measure [Ca2+]mt, cells were transfected with the ratiometric pericam (a kind gift of Dr. Atsushi Miyawaki, RIKEN Brain Science Institute, Wako-City, Japan) targeted to the mitochondrial matrix. Two days later, cells were exposed to externally applied 10 μm ATP in Ca2+-free Krebs buffer and imaged. Cells were permeabilized by exposure to intracellular like medium (ICM) (125 mm KCl, 19 mm NaCl, 10 mm HEPES-KOH, pH 7.3, 1 mm EGTA) and appropriate concentrations of CaCl2 (330 μm CaCl2 for 50 nm free Ca2+) (27) containing 250 μg/ml (w/v) saponin (MP Biomedicals, Ohio) for 3 min. Permeabilized cells were washed with ICM and then exposed to ICM containing 1 μm inositol 1,4,5-trisphosphate (IP3) (Biomol, Plymouth Meeting, PA). Dual excitation imaging with ratiometric pericam-mt required two filters (excitation 482/35, dichroic mirror 506, emission 536/40 and excitation 414/46, dichroic mirror 510, emission 527/20). [Ca2+]c and [Ca2+]mt, which were monitored during the assay on an inverted microscope (Leica DM IRB, Japan), were equipped with CCD cameras (ORCA-ER; Hamamatsu, Japan). Captured images were analyzed using the Metafluor imaging system (Molecular Devices, Sunnyvale, CA).
Drug Treatments
ER stress was induced by treatment of cells with 2 μg/ml tunicamycin (Sigma) for 4 or 24 h. For cell viability experiments, tunicamycin treatment continued for 48 h. Mitochondrial Ca2+ uptake was inhibited by 10 μm Ru360 (Calbiochem).
ATP Measurement
Cells were plated in 6-cm plates, and ATP content was determined using luciferin and a luciferase assay kit (Toyo B-net, Tokyo, Japan) following the manufacturer's protocol.
TUNEL Staining
DNA fragmentation and apoptotic bodies were detected by the TUNEL method using an in situ apoptosis detection kit (Takara Bio Inc., Shiga, Japan), as described (28). One hundred cells from 13 randomly selected fields were counted in each experiment.
Quantification of mRNA by Real Time PCR
Real time PCR analysis was performed as described (24) in 48-well plates (Mini Opticon Real Time PCR System, Bio-Rad) using iQ SYBR Green Supermix 2× (Bio-Rad). Primer sequences are shown in supplemental Table 1. Relative quantities of target mRNAs were determined by the comparative threshold cycle (ΔCT) method and normalized to GAPDH quantity. Product purity and specificity were confirmed by omitting the template and performing a standard melting curve analysis.
Statistical Evaluation
All values were expressed as means ± S.E. Comparison between two experimental groups was made using the unpaired Student's t test. Statistical significance for differences among groups was tested by one-way analysis of variance, followed by multiple comparisons between control and other groups using Dunnett's multiple comparison test. p < 0.05 was considered significant.
RESULTS
Identification of a Novel σ1R Splicing Variant
σ1R, a nonopioid receptor (29, 30), was previously cloned from guinea pigs (10), humans (31), and mice (32). All forms showed greater than 80% amino acid homology but no structural homology with any other receptor family. We screened a mouse hippocampal cDNA library by PCR using primer sets derived from the coding region of mouse Sigmar1 (Fig. 1A, p1 and p2) and identified the Sigmar1-coding sequence fragment (Fig. 1B, lane 1) and a novel smaller fragment (Fig. 1B, lane 2). When we amplified what would be an ∼200-bp fragment spanning σ1R exon 2 to exon 3 (Fig. 1A, p3 and p4) from the Sigmar1-coding sequence fragment, we detected the expected 200-bp fragment (Fig. 1B, lane 3) and also an ∼150-bp fragment (Fig. 1B, lane 4). Sequencing analysis revealed that the short fragment lacks 47 bp from exon 2 of the full-length gene. We subsequently identified a novel 231-bp splicing variant of σ1R and called it σ1SR (Fig. 1B, lane 5). A human sequence corresponding to mouse σ1SR had been deposited in GenBankTM under accession number BC007839.2. (Fig. 1C, left), but protein expression and function had not been evaluated. Our analysis indicated that the novel splice conformed to the G(T/A)G rule (Fig 1D) and that splicing of the 47-bp fragment caused a frameshift starting at amino acid 103 and translation termination due to a newly generated stop codon. The predicted primary structure of σ1SR contains 106 rather than the 223 amino acids seen in σ1R. The first 102 amino acids of both proteins are identical, but the C-terminal four residues of σ1SR differ from the longer form, because they are derived from translation of the frame-shifted σ1R exon 3 (Fig. 1, C and D).
FIGURE 1.

σ1R alternative splicing. A, position of PCR primers relative to receptor sequences (transmembrane domain). B, PCR of an adult mouse brain hippocampus cDNA library was performed using the indicated oligonucleotide primers. C, sequence of coding region of human SIGMAR1 (left) and mouse Sigmar1 (right) cDNA. The deduced amino acid sequence is presented using the one-letter code. Differences between homologues are shown in bold. Two putative transmembrane domains (TM1 and TM2) are shown in gray underlines, and the 47-bp of sequences deleted from full-length σ1R are shown in black underlines. Their deletion results in a frameshift, giving rise to four novel amino acids in σ1SR (boxed). D, proposed splicing mechanism of σ1Rs. Exons are outlined, and coding sequence is denoted by capital letters. Intronic sequence is denoted by lowercase letters.
Tissue Distribution and Localization of Both σ1R Isoforms
To investigate tissue distribution and relative expression levels of σ1R and σ1SR, we performed immunoblot analysis in adult mouse brain using antibodies against the σ1R N-terminal cytosolic domain (amino acids 52–69) or the C-terminal lumenal domain (amino acids 143–165). To confirm endogenous expression of σ1R and σ1SR, we observed immunoreactive bands in cell extracts from neuroblastoma Neuro-2a cells transfected with both cDNAs (Fig. 2A, lanes 3–8). Using an N-terminal cytosolic domain-specific antibody, we observed marked variation in the ratio of endogenous σ1SR (seen as a 12-kDa band) to σ1R (seen as a 26-kDa band) protein in the mouse brain regions tested (Fig. 2A, upper panel, lanes 9–13). An unidentified protein of the 19-kDa protein was observed in the hippocampus and brainstem lysates. As expected, a C-terminal lumenal domain antibody detected an immunoreactive σ1R band but not σ1SR (Fig. 2A, lower panel, lanes 9–13). σ1SR protein levels were high in cortex, hippocampus, and striatum (σ1SR/σ1R ratio of 0.4) but barely detectable in olfactory bulb and brainstem (Fig. 2B). To further investigate differences in subcellular localization, we fractionated lysates of Neuro-2a cells transfected with 3′ eGFP-tagged σ1R (σ1R-eGFP) or σ1SR (σ1SR-eGFP) cDNA into cytosolic, ER, and mitochondrial membrane (including MAM) and nuclear fractions. High levels of σ1R-eGFP were found in ER and mitochondrial membrane fractions, and relatively low levels were seen in other fractions, whereas σ1SR-eGFP was detected in membrane and nuclear fractions (Fig. 2C). To determine subcellular localization of these proteins, we undertook confocal microscopy of fluorescence-tagged σ1R and σ1SR in Neuro-2a cells. σ1R-eGFP and σ1SR-eGFP were primarily observed in perinuclear regions, although low levels of fluorescence were seen in the nuclei of σ1SR-eGFP-expressing cells (Fig. 2D). A punctate perinuclear staining pattern of erRFP, which targets and serves as a marker of endoplasmic reticulum, largely co-localized with σ1R-eGFP (Fig. 2E) and σ1SR-eGFP (Fig. 2F). Interestingly, the Golgi marker GM130 showed considerable co-localization with σ1SR-eGFP (Fig. 2H) but not with σ1R-eGFP (Fig. 2G). Both mCherry-tagged σ1R isoforms were detected in some overlays with the mitochondrial marker mtGFP, suggesting that σ1SR protein also localizes on MAMs, as reported for σ1R (Fig. 2, I and J) (11). These results suggest that the subcellular distribution of both σ1R isoforms is similar, although σ1SR is also seen in the Golgi apparatus and nucleus.
FIGURE 2.

Tissue distribution and localization of σ1Rs. A, representative immunoblots probed with antibodies against the σ1R N-terminal cytosolic domain (upper panel) or the C-terminal lumenal domain (lower panel). As control bands, extracts from Neuro-2a cells transfected with σ1R and σ1SR constructs (lanes 1–8) are shown. B, quantitative densitometry shows the ratio of σ1SR to σ1R protein expression among different brain regions. OB, olfactory bulb; CX, cortex; HP, hippocampus; ST, striatum; BS, brainstem. n = 4 in each group. C, cytosolic (Cyto), ER, mitochondrial membrane (Mem), and nuclear (Nuc) fractions from eGFP tagged-σ1R isoform-transfected Neuro-2a cells were blotted with antibodies against GFP, calcineurin (CaN, cytosolic marker), voltage-dependent anion channel (ER and mitochondrial membrane marker), and CREB-2 (nuclear marker). D–I, σ1R localization in Neuro-2a cells. Confocal images show co-localization of fluorescence (eGFP or mCherry)-tagged-σ1R isoforms and markers of ER (erRFP), Golgi apparatus (GM130), and mitochondria (mtGFP). D, σ1R-eGFP (left) and σ1SR-eGFP (right) are mainly expressed in perinuclear regions. A small number of GFP aggregates are detected in nuclei of σ1SR-eGFP-expressing cells (shown by arrow). Right panels are high magnification images. E and F, immunoreactivity of both σ1R-eGFP isoforms (green) and erRFP (red) almost completely merge. G and H, GM130 (green) immunoreactivity co-localizes with σ1SR-mCherry (H) (red) but not with σ1R-mCherry (G) (red). I and J, both mCherry-tagged σ1R isoforms (red) were detected in a few overlays with the mitochondrial marker mtGFP (green). Lower panels in I and J are high magnification images. Scale bars, 10 μm.
Overexpression of Either σ1R Isoform Promotes Mitochondrial Elongation and Increases ER/Mitochondrial Contact Space
Based on their subcellular location, we analyzed the role of both σ1R isoforms in ER and mitochondrial morphology and in ER/mitochondria interaction. σ1R or σ1SR overexpression did not alter the reticular ER structure of erRFP-infected Neuro-2a cells (Fig. 3B; 89.1 ± 2.8% in control, 88.7 ± 2.9% in σ1R-, and 83.6 ± 3.6% in σ1SR-transfected cells). However, expression of either isoform alone promoted mitochondrial elongation, as shown by three-dimensional reconstruction of mitochondrion-targeted green fluorescent protein (Fig. 3C; 41.6 ± 7.5% in control, 77.4 ± 7.0% in σ1R-, and 67.3 ± 8.9% in σ1SR-transfected cells). In addition, confocal semiquantitative analysis of ER/mitochondria juxtaposition showed that tethering of ER and mitochondria increased either isoform expressed in Neuro-2a cells (Fig. 3D; 2.6 ± 0.3% in controls, 4.6 ± 0.9% in σ1R-, and 3.8 ± 0.7% in σ1SR-transfected cells).
FIGURE 3.

Both σ1R isoforms regulate mitochondrial morphology and juxtaposition of the ER to mitochondria. A, representative images of three-dimensional reconstructions of ER (erRFP; left), mitochondria (mtGFP; middle), and merged images showing ER and mitochondria co-localization (yellow; right). B, morphometric analysis of ER shape in cells transfected with erRFP (n = 3, 18 cells per experiment). C, quantitative analysis of mitochondrial shape in cells transfected with mtGFP (n = 4, 12 cells per experiment). D, quantitative analysis of co-localization of ER and mitochondria (as a percentage of total mitochondrial volume) in cells co-transfected with erRFP and mtGFP (n = 4, 20 cells per experiment). Each bar represents the mean ± S.E. *, p < 0.05; **, p < 0.01 versus control cells. Scale bar, 10 μm.
σ1SR Forms a Complex with σ1R but Not with IP3Rs
Because both σ1R isoforms are similarly distributed, we asked whether either directly interacted with IP3Rs. To do so, we performed immunoprecipitation of extracts from σ1SR-mCherry-expressing cells co-expressing σ1R-eGFP using an eGFP antibody followed by immunoblotting with an anti-σ1R N-terminal cytosolic domain-specific antibody. We observed an immunoreactive σ1SR-mCherry band (41 kDa) (Fig. 4A, upper panel, lane 9) not seen in immunoprecipitates from extracts from eGFP-, σ1R-eGFP-, or σ1SR-mCherry-overexpressing cells (Fig. 4A, upper panel, lanes 6–8). Immunoreactive bands with molecular masses corresponding to two fluorescence-tagged σ1R isoforms of 55 and 41 kDa were detected in each cell extract (Fig. 4A, lanes 2–4). To confirm the specificity of fluorescence-tagged σ1SR bands, we used an anti-σ1R C-terminal lumenal domain-specific antibody. σ1SR-eGFP (37 kDa) and σ1SR-mCherry (41 kDa) bands were not detected (Fig. 4A, lower panel).
FIGURE 4.

σ1SR forms a complex with and co-localizes with σ1R. A, co-immunoprecipitation of fluorescence-tagged σ1SR and σ1R in Neuro-2a cells. Extracts were immunoprecipitated (IP) with anti-eGFP antibody, and immunoprecipitates were immunoblotted (WB) with anti-σ1R N-terminal cytosolic domain (upper panel) or with the C-terminal lumenal domain (lower panel) antibody. Cell extracts (Input) from Neuro-2a cells transfected with eGFP (lane 1), σ1R-eGFP (lane 2), σ1R-mCherry (lane 3), σ1SR-eGFP (lane 4), or σ1SR-mCherry (lane 5) constructs are shown as positive controls for σ1R isoform immunoreactive bands. B and C, co-immunoprecipitation of σ1Rs and IP3Rs in Neuro-2a cells. Extracts were immunoprecipitated with anti-IP3Rs (B) or anti-σ1R N-terminal cytosolic domain (C) antibody, and immunoprecipitates were immunoblotted (WB) with anti-IP3Rs (B) or anti-σ1R N-terminal (C) antibody. As a positive control for σ1Rs and IP3Rs immunoreactive bands, cell extracts (Input) are shown from intact (lane 1) or σ1R- (lane 2) or σ1SR (lane 3)-transfected cells. D, confocal images showing co-localization of σ1R-eGFP (green) and σ1SR-mCherry (red) in Neuro-2a cells. Scale bar, 10 μm.
Because σ1R forms a complex with IP3R type-3 in CHO cells (11), we examined potential binding of σ1R and σ1SR with IP3R in Neuro-2a cells. After immunoprecipitation with an IP3R antibody, we observed only a 26-kDa σ1R immunoreactive band in σ1R- or σ1SR-overexpressing cells (Fig. 4B, lanes 4–6). Next, after σ1R immunoprecipitation with an anti-σ1R N-terminal specific antibody, we undertook immunoblotting using an anti-IP3Rs antibody. As expected, increased levels of an IP3R immunoreactive band were seen in σ1R-overexpressing (Fig. 4C, lane 5) but not σ1SR-overexpressing (Fig. 4C, lanes 4 and 6) cells relative to controls. No significant differences in IP3R protein expression were observed between control cells and cells expressing σ1R isoforms (Fig. 4C, lanes 1–3). Confocal microscopy of Neuro-2a cells indicated that σ1SR-mCherry coincided with σ1R-GFP immunofluorescence in the ER (Fig. 4D).
σ1SR Expression Reduces Mitochondrial Ca2+ Uptake in Response to IP3R-driven Stimuli
IP3Rs are important for ER-mitochondrial Ca2+ transport, which is regulated by σ1R in the MAM of CHO cells (11). Because σ1SR forms a complex with σ1R, we assessed a potential role for σ1SR in mitochondrial Ca2+ uptake. To assay mitochondrial Ca2+ uptake in Neuro-2a cell lines stably expressing σ1R-mCherry (σ1R-mCh cells) or σ1SR-mCherry (σ1SR-mCh cells), we conducted Ca2+ imaging using ratiometric pericam-mt Ca2+ probes, which localize to mitochondria (Fig. 5A) (33). We confirmed that σ1R- and σ1SR-mCherry mRNA levels in each line were equivalent, and we also found that levels showed an approximate 20-fold increase over parental Neuro-2a cells (supplemental Fig. 1A).
FIGURE 5.

σ1SR expression reduces mitochondrial Ca2+ uptake in response to IP3R-driven stimuli. A, pseudo-colored images of Neuro-2a cells expressing the mitochondrial Ca2+ indicator, ratiometric pericam-mt, at 30 s (left) and 60 s (right), corresponding to a control cell in B. Regions of interest for [Ca2+]mt measurement is indicated by the circles in A. B, C, and E, relative fluorescence intensity changes following treatment with 10 μm ATP in Neuro-2a cells (B and E) or 1 μm IP3 in permeabilized Neuro-2a cells (C) after transfection of a mitochondrial pericam probe. D and F, relative Fura-2/AM fluorescence intensity changes following 10 μm ATP stimulation. Top line indicates incubation period. The ratio of the ratiometric pericam-mt and Fura-2 fluorescence peak value of the release phase relative to the preceding base line is shown on the right. Each bar represents the mean ± S.E. *, p < 0.05; **, p < 0.01 versus control cells. #, p < 0.05; ##, p < 0.01 versus σ1SR-mCh cells. n = 12 cells per experiment, performed in triplicate from four preparations. Scale bar, 10 μm.
We then asked whether mitochondrial Ca2+ elevation elicited by ATP, acting on receptors coupled with Gq protein to stimulate IP3 production. For this experiment, Ca2+-free Krebs buffer containing 2 mm EGTA was used to assess intracellular Ca2+ mobilization and mitochondrial Ca2+ elevation through the ER IP3R. As shown in Fig. 5B, ATP stimulation caused a rapid rise in [Ca2+]mt, followed by a gradual decline with a sustained plateau phase in control cells (black line). In σ1R-mCh cells (Fig. 5B, red line), the [Ca2+]mt increase was markedly enhanced compared with that seen in control cells, although it was significantly decreased in σ1SR-mCh cells (blue line). Interestingly, decreased [Ca2+]mt mobilization seen in σ1SR-mCh cells was significantly rescued to levels comparable with those seen in control cells by σ1R co-expression (Fig. 5B, green line). Peak amplitudes of [Ca2+]mt mobilization are summarized (0.18 ± 0.02 in control, 0.3 ± 0.01 in σ1R-mCh, 0.09 ± 0.02 in σ1SR-mCh, and 0.14 ± 0.01 in σ1SR-mCh plus σ1R expression).
To confirm that IP3Rs function in [Ca2+]mt mobilization, saponin-permeabilized cells were treated with IP3, and [Ca2+]mt was monitored in the presence of ICM buffer. Consistent with results seen after ATP application, IP3-mediated [Ca2+]mt mobilization was significantly enhanced in σ1R-mCh cells and decreased in σ1SR-mCh cells relative to control cells. In addition, [Ca2+]mt decreases seen in σ1SR-mCh cells were rescued by σ1R co-expression (Fig. 5C; peak amplitude 0.17 ± 0.04 in control cells, 0.22 ± 0.04 in σ1R-mCh cells, 0.13 ± 0.03 in σ1SR-mCh cells, and 0.19 ± 0.06 in σ1SR-mCh plus σ1R expression).
We next examined intracellular mobilization [Ca2+]c following ATP stimulation using the ratiometric indicator Fura-2/AM in Ca2+-free Krebs buffer. Interestingly, ATP-induced [Ca2+]c mobilization in σ1R-mCh cells was significantly reduced compared with controls. By contrast, [Ca2+]c mobilization in σ1SR-mCh cells was greater than that seen in control cells and that increase was suppressed by σ1R co-expression (Fig. 5D; peak amplitude 0.13 ± 0.02 in control cells, 0.12 ± 0.08 in σ1R-mCh cells, 0.16 ± 0.06 in σ1SR-mCh cells, and 0.1 ± 0.03 in σ1SR-mCh plus σ1R cells). This evidence strongly suggests that increased IP3-mediated [Ca2+]mt mobilization stimulated by σ1R overexpression decreases [Ca2+]c mobilization, although IP3-mediated [Ca2+]mt mobilization due to σ1SR overexpression enhances it.
We also confirmed that ATP-induced [Ca2+]mt mobilization was mediated by σ1Rs using siRNA knockdown of both σ1R isoforms. Expression levels of both proteins were down-regulated ∼70% by siRNA treatment (supplemental Fig. 1B). σ1R isoform knockdown significantly decreased ATP-induced [Ca2+]mt mobilization (Fig. 5E; peak amplitude 0.18 ± 0.03 in control cells and 0.1 ± 0.02 in σ1R isoform siRNA cells); conversely, knockdown of both σ1R and σ1SR protein elevated ATP-induced [Ca2+]c mobilization (Fig. 5F; peak amplitude 0.15 ± 0.05 in control cells and 0.19 ± 0.08 in σ1R isoform siRNA cells).
Overexpression of σ1R-mCh and σ1SR-mCh may alter the capacity of ER Ca2+ stores. To assess this possibility, we stimulated cells with thapsigargin, an inhibitor of the sarcoplasmic reticulum calcium ATPase. Thapsigargin treatment promoted a transient increase in [Ca2+]c by depleting Ca2+ stores in the absence of extracellular Ca2+, and subsequent addition of extracellular Ca2+ increased [Ca2+]c through capacitative Ca2+ entry. Both the capacity of ER Ca2+ stores and capacitative Ca2+ entry were comparable in cells with or without σ1R and σ1SR overexpression (supplemental Fig. 2). These results suggest that both σ1R isoforms affect the efficiency of mitochondrial Ca2+ uptake in response to IP3R-driven stimuli without changing the capacity of ER Ca2+ stores.
σ1SR Acts Antagonistically to σ1R following ER Stress
Because σ1SR expression suppresses IP3-mediated [Ca2+]mt mobilization and enhances receptor-mediated [Ca2+]c mobilization through the ER, we hypothesized that σ1SR overexpression increases the vulnerability of a cell to ER stress. To test this, we evaluated the effect of the ER stressor tunicamycin on overexpression of σ1R isoforms. Tunicamycin treatment promoted expression of both endogenous σ1R and σ1SR protein in σ1R-mCh but not control cells. Conversely, tunicamycin treatment reduced expression of both endogenous σ1R and σ1SR in σ1SR-mCh cells (Fig. 6B).
FIGURE 6.

σ1SR enhances IP3R destabilization following ER stress. A, representative immunoblots probed with various antibodies are shown in control, σ1R-mCh, and σ1SR-mCh cells at 0, 4, and 24 h after 2 μg/ml tunicamycin treatment. B, quantitative densitometry analyses are shown. n = 6 per experiment. Each bar represents the mean ± S.E. *, p < 0.05; **, p < 0.01 versus control cells.
Tunicamycin treatment also promoted significant degradation of IP3R proteins in control but not σ1R-mCh cells, suggesting a chaperone activity for σ1R. More importantly, σ1SR-mCh overexpression enhanced degradation of IP3R proteins relative to that seen in control cells (Fig. 6B; 50.8 ± 14.1% in control cells, 77 ± 24.1% in σ1R-mCh cells, and 16.3 ± 8.8% in σ1SR-mCh cells at 24 h after tunicamycin treatment). Because σ1SR-mCh overexpression promoted protein degradation, we examined the effects of σ1SR-mCh overexpression on ER stress using stress markers, such as phosphorylation of PERK and its downstream target C/EBP homologous protein. As unexpected, neither PERK phosphorylation nor C/EBP homologous protein levels were enhanced by σ1SR-mCh overexpression (Fig. 6B). However, when we evaluated autophagic responses based on production of the autophagy marker LC3-II, the ratio of LC3-II to total (LC3-I plus LC3-II) LC3 was markedly increased by 24 h after tunicamycin treatment (Fig. 6B).
σ1SR Enhances Autophagosome Formation following ER Stress
Given that LC3-II production was enhanced in σ1SR-mCh cells (Fig. 6B), we determined the diameters of vacuoles containing LC3-II (34). Twenty four hours after tunicamycin treatment, diameters of ∼80% of LC3-positive vacuoles were smaller than the 1.0 μm seen in control cells (Fig. 7, A and D). σ1R-mCh overexpression decreased the number of LC3-positive vacuoles compared with control without changing the size distribution (Fig. 7, B, D and E). By contrast, σ1SR-mCh overexpression induced formation of larger autophagosomes than those seen in control cells, significantly shifting the size distribution profile. Over 50% of vacuoles were between 1.0 and 1.5 μm at 24 h after tunicamycin treatment (Fig. 7,C and D). The total number of LC3-positive particles also markedly increased in σ1SR-mCh cells following tunicamycin treatment (Fig. 7E; 98.3 ± 11.4 in controls, 66.3 ± 7.2 in σ1R-mCh cells, and 195 ± 16.7 in σ1SR-mCh cells). The size of LC3-positive vacuoles did not change in control, σ1R-mCh, and σ1SR-mCh cells not treated with tunicamycin. Taken together, σ1R overexpression inhibited formation of tunicamycin-induced autophagosomes, although σ1SR overexpression promoted autophagosome formation under ER stress conditions.
FIGURE 7.

σ1SR enhances autophagosome formation following ER stress. A–C, LC3 immunostaining in control (A), σ1R-mCh (B), and σ1SR-mCh (C) cells 24 h after 2 μg/ml tunicamycin treatment. Enlarged images indicate the boxed areas. Scale bars, 30 μm in low magnification and 10 μm in high magnification images. D and E, quantitative analysis of particle size (D) and total number (E) of LC3-immunoreactive autophagosomes 24 h after tunicamycin treatment. Ten fields (30 cells per field) in each condition were chosen randomly and photographed. Each bar represents the mean ± S.E. *, p < 0.05; **, p < 0.01 versus control cells.
σ1SR Suppresses Mitochondrial ATP Production and Enhances Apoptosis following ER Stress
Finally, we asked how σ1SR overexpression enhances the autophagic response. Because IP3R-mediated Ca2+ transport into mitochondrial Ca2+ promotes oxidative phosphorylation, respiration, and ATP production by activating the tricarboxylic acid cycle (2, 35), we speculated that ATP production would be suppressed by σ1SR overexpression. Therefore, we measured changes in cellular ATP production with or without tunicamycin-induced ER stress. Unexpectedly, overexpression of σ1R or σ1SR markedly enhanced ATP production without tunicamycin treatment. In control cells, tunicamycin treatment significantly increased ATP production at 24 h but markedly suppressed it after 48 h of treatment. σ1R overexpression significantly promoted ATP production both at 24 and 48 h after tunicamycin treatment (Fig. 8A). ATP production in σ1SR-mCh cells was significantly suppressed at both the 24- and 48-h time points compared with control cells (Fig. 8A; 41.9 ± 1.1% in controls, 86.4 ± 5.3% in σ1R-mCh cells, and 14.5 ± 0.24% in σ1SR-mCh cells at 48 h compared with control cells at a resting state). To confirm that changes in ATP production are related to mitochondrial Ca2+ mobilization, we examined cellular ATP levels with or without tunicamycin treatment in the presence or absence of Ru360, a mitochondrial Ca2+ uptake blocker. ATP production enhanced by σ1R or σ1SR overexpression or by tunicamycin treatment was completely inhibited by Ru360 treatment (supplemental Fig. 3).
FIGURE 8.
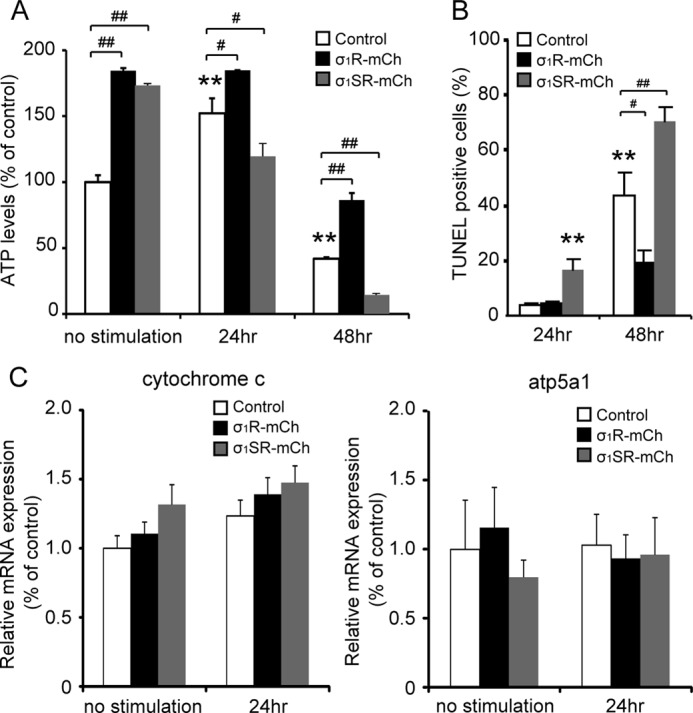
σ1SR suppresses mitochondrial ATP production and enhances apoptosis following ER stress. A, intracellular ATP levels were measured in untreated cells or cells treated with 2 μg/ml tunicamycin at 24 and 48 h. B, TUNEL-positive cells were counted in treated cells at 24 and 48 h. One hundred cells from 13 randomly selected fields were counted in each experiment. C, real time PCR analysis showed that cytochrome c and ATP5A1 mRNA expression showed no significant differences between each group. n = 6 per experiment. Each bar represents the mean ± S.E. **, p < 0.01 versus control at no stimulation. #, p < 0.05; ##, p < 0.01 versus control at each time point.
Because ATP production was suppressed 48 h after tunicamycin treatment and treatment could promote apoptosis, we evaluated apoptosis using TUNEL staining. σ1SR-mCh overexpression significantly enhanced the number of TUNEL-positive cells 24 h after tunicamycin treatment, although σ1R-mCh overexpression significantly suppressed tunicamycin-induced apoptosis. By contrast, σ1SR-mCh overexpression markedly enhanced tunicamycin-induced apoptosis at 48 h (Fig. 8B; percentages of TUNEL-positive cells were 43.5 ± 8.4% in control, 19.1 ± 4.4% in σ1R-mCh, and 70.4 ± 5.4% in σ1SR-mCh).
To confirm that σ1R-mCh and σ1SR-mCh overexpression does not alter mitochondrial protein levels, we examined the expression of cytochrome c and the α subunit of ATP synthase (ATP5A1), subunits of the respiratory chain complex. σ1R-mCh and σ1SR-mCh cells showed no change in mRNA levels of these factors (Fig. 8C), indicating that σ1R-mCh and σ1SR-mCh overexpression does not interfere with mitochondrial structure or directly regulate IP3R-mediated ATP production.
DISCUSSION
Here, we identified σ1SR, a novel truncated isoform of the σ1R. Using Neuro-2a cells, we showed that under physiological conditions the σ1SR overexpression slightly stimulates ATP-induced cytosolic Ca2+ mobilization, in contrast with a small reduction promoted by σ1R overexpression (Fig. 5D). These changes are likely due to increased and decreased Ca2+ mobilization into mitochondria induced by σ1SR and σ1R overexpression, respectively. Unexpectedly, σ1SR overexpression increased ATP production in the absence of stress stimuli, such as ER stress (Fig. 8A), an activity not associated with reduction in ATP-induced mitochondrial Ca2+ mobilization. Thus, although ATP-induced mitochondrial Ca2+ mobilization is significantly reduced by σ1SR overexpression, mitochondrial Ca2+ levels may be moderately increased with mitochondrial elongation. In support of this idea, increased ATP production seen following σ1SR overexpression was eliminated by treating cells with the mitochondrial Ca2+ transport blocker Ru360 (supplemental Fig. 3).
Because basal ATP levels increase following overexpression of either isoform (Fig. 8A) and σ1R overexpression in PC12 cells reportedly stimulates neurite sprouting in response to nerve growth factor (36), we hypothesized that σ1SR overexpression might stimulate Neuro-2a cell differentiation. To evaluate this possibility, we measured morphological changes in both σ1R- and σ1SR-overexpressing Neuro-2a cells. Consistent with the findings of Ref. 36, σ1R overexpression significantly stimulated neurite extension as compared with control cells. Similarly, like σ1R overexpression, σ1SR overexpression significantly stimulated neurite extension (supplemental Fig. 4; 3.2 ± 1.3% in control, 45.5 ± 4.3% in σ1R, and 46 ± 7.4% in σ1SR-transfected cells).
Mitochondrial elongation and extension of gap junctions between ER and mitochondria promoted by σ1SR and σ1R overexpression likely account for enhanced mitochondrial ATP production. Elongated mitochondria express higher levels of the dimeric form of ATPase, which is associated with more efficient ATP production (37). Increased contact space between the ER and mitochondria also likely enhances mitochondrial Ca2+ transport (38, 39). An ER-associated sorting protein, phosphofurin acidic cluster sorting protein 2 (PACS-2), is required for association of mitochondria with the ER. PACS-2 depletion induces mitochondrial fragmentation and uncoupling with the ER, resulting in aggravated ER stress (40). Mitofusin-2 (Mfn2) in the ER is also required for adhesion between mitochondrial and ER membranes. Depletion of Mfn2 impairs ER-mitochondrial Ca2+ transport (25) and induces cell death in cerebellar granule neurons (41). Taken together, σ1R overexpression causes elongation of mitochondria and enhances IP3R-induced Ca2+ transport, thereby promoting mitochondrial ATP production. Thus, elevated mitochondrial energy production likely promotes cell survival in the presence of ER stress.
σ1SR Expression Promotes Mitochondrial ATP Depletion and Autophagy Only under ER Stress Conditions
The σ1R reportedly stabilizes IP3Rs to maintain Ca2+ transport from the ER into mitochondria in CHO cells (11). We confirmed that σ1R overexpression enhances IP3-induced mitochondrial Ca2+ transport and ATP production, whereas σ1SR did not bind to IP3Rs, and its overexpression did not enhance IP3-induced mitochondrial Ca2+ transport. Under ER stress conditions, σ1SR expression had detrimental effects on mitochondrial Ca2+ transport through the ER (Fig. 5B), promoting mitochondrial ATP depletion (Fig. 8). σ1SR overexpression also destabilized IP3Rs (Fig. 6B), which may account for decreased mitochondrial Ca2+ transport. Finally, σ1SR overexpression promoted an autophagic response to ER stress following tunicamycin treatment without altering PERK activity (Figs. 6B and 7). Autophagy functions to recycle energy and nutrients in nutrient starvation and ER stress conditions (42), but the balance between autophagy and cell death is highly dependent on intracellular Ca2+ levels (43). IP3R protein levels and activity are critical to inhibit autophagy (44). Indeed, autophagy is inhibited and promoted by IP3R agonists (such as IP3) and antagonists (such as xestospongins), respectively (45–47). In addition, IP3R is required to inhibit autophagy under physiological conditions (6). The lack of mitochondrial Ca2+ transport by IP3R depletion inhibits pyruvate dehydrogenase and increases the AMP/ATP ratio, thereby aggravating autophagy via AMP-activated protein kinase (6). IP3Rs also inhibit autophagy through binding with Bcl-2 and Beclin-1 (known as autophagy-related gene 6) in HeLa cells (48). Inhibition of IP3Rs by xestospongin B promotes disruption of complexes formed by IP3R, Bcl-2, and Beclin-1, activating autophagy. Taken together, IP3R dysfunction through σ1R down-regulation accounts for autophagic mechanisms in σ1SR-expressed cells.
Physiological Relevance of Interaction between σ1R and σ1SR and Their Ligands
As shown in Fig. 1, the σ1R is composed two transmembrane domains (TM1, amino acids 11–29; TM2, amino acids 91–109), an extracellular loop, and intracellular N and C termini with the C-terminal region including a large soluble domain (49, 50). Pharmacological studies indicate that numerous compounds, including benzomorphans (SKF-10047, pentazocine), antipsychotics (haloperidol), antidepressants (fluvoxamine), steroids (dehydroepiandrosterone, progesterone), and drugs of abuse (methamphetamine, cocaine) can bind to the σ1R, primarily through the TM2 and C-terminal regions (50). The σ1R agonist (+)-pentazocine positively modulates σ1R/IP3R association and stabilizes the complex at ER-mitochondria contact sites under ER stress. As a result, IP3 binding to the IP3R increases, and Ca2+ efflux is enhanced (11). In addition, ligand-induced regulation of this function apparently resides largely in the N terminus, contributing to functional coupling of C- and N-terminal σ1R fragments. Wu and Bowen (51) reported that agonist binding to the σ1R may change its conformation such that the N-terminal segment dissociates from the C terminus, which in turn can interact more avidly with IP3R. Here, we showed that σ1R binds to the IP3R via its C terminus (Fig. 4). σ1SR displays the opposite function, suppressing mitochondrial ATP production and promoting cell death following tunicamycin-induced ER stress. We suggest that σ1SR, whose sequence is almost identical to the σ1R N terminus (Fig. 1), interacts with the σ1R C terminus, inhibiting σ1R-mediated IP3R-derived ER-mitochondrial Ca2+ transport.
Lymphocytes also express another σ1R splice variant, which is replaced at Ala-13, Leu-28, and Ala-86 to Thr-13, Pro-28, and Val-86, respectively, and lacks 31 amino acids corresponding to residues 119–149 in the σ1R protein (52). The σ1R isoform lacks ligand binding sites. In NG108-15 cells, the full-length σ1R forms a trimeric complex with the cytoskeletal adaptor protein ankyrin B and IP3R in the ER membrane, and σ1R agonists cause dissociation of ankyrin B from the IP3R, promoting IP3R activation (53). Indeed, the C-terminal σ1R peptides (amino acids 102–223) transfected into MCF-7 breast tumor cells promote decreased levels of ankyrin B associated with the IP3R compared with untransfected cells, enhancing IP3R activation (51). In addition, the N-terminal σ1R peptides (amino acids 1–100) expressed in MCF-7 cells weakly associate with ankyrin B and IP3R complexes, but have little capacity to enhance IP3R activity (51). In addition, a glutathione S-transferase (GST) fusion form of σ1R (amino acids 116–223) has chaperone activity, blocking aggregation of denatured citrate synthase in vitro. However, GST-σ1R (amino acids 29–92) lacks chaperone activity (11). Taken together with these studies, σ1SR as defined here likely lacks chaperone activity and ligand binding ability and acts instead as a dominant negative form of σ1R by blocking C-terminal chaperone activity under ER stress conditions or disrupting IP3R/σ1R interaction. However, under normal conditions, overexpression of either isoform promotes similar phenotypes relevant to ATP production, mitochondrial elongation, and neurite extension, suggesting that endogenous σ1SR has an alternate unique function in mitochondrial elongation and adhesion with the ER.
We showed that ATP-induced [Ca2+]c mobilization in σ1SR-mCh cells was greater than that seen in control cells in Ca2+-free Krebs buffer (Fig. 5D). σ1SR expression possibly destabilizes IP3Rs by inhibiting chaperone activity of endogenous σ1R, thereby reducing IP3R-derived ER-mitochondrial Ca2+ transport (Fig. 9). This activity may elicit enhanced [Ca2+]c mobilization following ATP stimulation of σ1SR-mCh cells. However, extensive studies are required to define how IP3R/σ1R interaction is disrupted by σ1SR overexpression.
FIGURE 9.

Schematic representation of altered mitochondrial ATP production and cell death in the presence of two σ1R isoforms following ER stress. Upper, in ER stress conditions, σ1R stabilizes IP3Rs and sustains mitochondrial Ca2+ uptake-derived ATP production, enhancing survival. Lower, by contrast, σ1SR expression destabilizes IP3Rs and promotes dysfunction of IP3R-derived ER-mitochondrial Ca2+ transfer through functional loss of σ1R, resulting in ATP depletion and autophagic apoptosis. In resting conditions, both σ1R isoforms positively regulate mitochondrial biogenesis.
Among the three IP3R isoforms, type-3 IP3R plays a critical role in induction of apoptosis by preferentially transmitting Ca2+ signals into mitochondria (54). siRNA-based knockdown of type-3 IP3R significantly decreases IP3-induced mitochondrial Ca2+ concentrations in CHO cells, whereas knockdown of type-1 IP3R reduces cytosolic Ca2+ concentration (54). In addition, type-3 IP3Rs are particularly enriched at the MAM, whereas type-1 IP3R is homogeneously expressed in ER membranes in neurons (9). σ1Rs form a trimeric complex with ankyrin-B and the type-3 IP3Rs but not with type-1 IP3Rs in NG108-15 cells (53). Taken together, σ1R acts as a specific chaperone for type-3 IP3R at the MAM, regulating [Ca2+]mt mobilization in neurons. Notably, σ1R agonists, including (+)-pentazocine, increase [Ca2+]c from the ER following bradykinin stimulation of NG108-15 cells (55). In addition, σ1R (amino acids 102–223)-transfected MCF-7 cells also show increased [Ca2+]c induced by bradykinin stimulation (51), suggesting that other σ1R-binding proteins regulate intracellular Ca2+ mobilization. In the future, we will investigate whether these binding proteins regulate [Ca2+]c mobilization after stimulation with σ1R ligands in neurons and define σ1SR function in [Ca2+] mobilization.
Pathophysiological Relevance of σ1SR
Autophagy is activated in several neurodegenerative disorders, although its significance in neuronal death and survival remains to be defined (56). Al-Saif et al. (19) reported a SIGMAR1 missense mutation in exon 2 associated with juvenile amyotrophic lateral sclerosis (c.304G→C) that substitutes glutamine for glutamic acid at residue 102 (E102Q), which occurs at the σ1SR splice site (Fig. 1). Expression of the E102Q mutant enhances apoptosis in the mouse motor neuron-like cell line NSC34 (19). Further studies are needed to reveal functions of σ1SR in neurodegenerative disorders.
In conclusion, we have identified a novel σ1R isoform, σ1SR, as a key molecule in ER stress-related cellular apoptosis through regulation of ER-mediated mitochondrial biogenesis. Importantly, σ1SR does not bind IP3Rs but inhibits the ability of σ1R to promote mitochondrial ATP production through IP3R-mediated Ca2+ transport. Future studies should address pathological conditions in which σ1SR is up-regulated and potential interference with σ1R and mitochondrial function.
Acknowledgments
We thank Dr. Teruo Hayashi of the National Institute on Drug Abuse, Department of Health and Human Services, for kindly providing antibodies against the N and C termini of σ1R; Dr. Akihiko Tanimura and Dr. Yosuke Tojyo in the Department of Pharmacology, School of Dentistry, Health Sciences University of Hokkaido, for helpful advice on Ca2+ imaging analysis; and Dr. Atsushi Miyawaki at the Brain Science Institute, RIKEN, for kindly providing ratiometric pericam-mt/pcDNA3.
This work was supported by a grant-in-aid for scientific research on innovative areas (Foundation of Synapse and Neurocircuit Pathology), Grants-in-aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan and Core Research for Evolutional Science and Technology, Japan Science and Technology Agency, 22390109 and 24659024 (to K. F.) and 23790072 and 23110501 (to N. S.), and from the Smoking Research Foundation (to K. F.).

This article contains supplemental Figs. 1–4 and Table 1.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) AB721301.
- ER
- endoplasmic reticulum
- ALS
- amyotrophic lateral sclerosis
- ATP5A1
- ATP synthase
- IP3
- inositol 1,4,5-trisphosphate
- IP3R
- inositol 1,4,5-trisphosphate receptor
- MAM
- mitochondrion-associated ER membrane
- Mfn2
- Mitofusin-2
- PACS-2
- phosphofurin acidic cluster sorting protein 2
- PERK
- RNA-dependent protein kinase-like ER kinase
- σ1SR
- short form σ1 receptor
- σ1R
- σ1 receptor
- mt
- mitochondrial
- ICM
- intracellular like medium
- eGFP
- enhanced GFP
- erRFP
- ER-targeted RFP.
REFERENCES
- 1. Robb-Gaspers L. D., Burnett P., Rutter G. A., Denton R. M., Rizzuto R., Thomas A. P. (1998) Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 17, 4987–5000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Csordás G., Renken C., Várnai P., Walter L., Weaver D., Buttle K. F., Balla T., Mannella C. A., Hajnóczky G. (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rizzuto R., Brini M., Murgia M., Pozzan T. (1993) Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262, 744–747 [DOI] [PubMed] [Google Scholar]
- 4. Rizzuto R., Pinton P., Carrington W., Fay F. S., Fogarty K. E., Lifshitz L. M., Tuft R. A., Pozzan T. (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766 [DOI] [PubMed] [Google Scholar]
- 5. Hanson C. J., Bootman M. D., Roderick H. L. (2004) Cell signaling. IP3 receptors channel calcium into cell death. Curr. Biol. 14, R933-R935 [DOI] [PubMed] [Google Scholar]
- 6. Cárdenas C., Miller R. A., Smith I., Bui T., Molgó J., Müller M., Vais H., Cheung K. H., Yang J., Parker I., Thompson C. B., Birnbaum M. J., Hallows K. R., Foskett J. K. (2010) Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vance J. E. (1990) Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256 [PubMed] [Google Scholar]
- 8. Giorgi C., De Stefani D., Bononi A., Rizzuto R., Pinton P. (2009) Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int. J. Biochem. Cell Biol. 41, 1817–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hayashi T., Rizzuto R., Hajnoczky G., Su T. P. (2009) MAM. More than just a housekeeper. Trends Cell Biol. 19, 81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hanner M., Moebius F. F., Flandorfer A., Knaus H. G., Striessnig J., Kempner E., Glossmann H. (1996) Purification, molecular cloning, and expression of the mammalian σ1-binding site. Proc. Natl. Acad. Sci. U.S.A. 93, 8072–8077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hayashi T., Su T. P. (2007) σ1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 131, 596–610 [DOI] [PubMed] [Google Scholar]
- 12. Hayashi T., Su T. P. (2003) σ1 receptors (σ1-binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum. Roles in endoplasmic reticulum lipid compartmentalization and export. J. Pharmacol. Exp. Ther. 306, 718–725 [DOI] [PubMed] [Google Scholar]
- 13. Martina M., Turcotte M. E., Halman S., Bergeron R. (2007) The σ1 receptor modulates NMDA receptor synaptic transmission and plasticity via SK channels in rat hippocampus. J. Physiol. 578, 143–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Celsi F., Pizzo P., Brini M., Leo S., Fotino C., Pinton P., Rizzuto R. (2009) Mitochondria, calcium, and cell death. A deadly triad in neurodegeneration. Biochim. Biophys. Acta 1787, 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maurice T., Lockhart B. P. (1997) Neuroprotective and anti-amnesic potentials of σ receptor ligands. Prog. Neuropsychopharmacol. Biol. Psychiatry 21, 69–102 [DOI] [PubMed] [Google Scholar]
- 16. Urani A., Romieu P., Roman F. J., Yamada K., Noda Y., Kamei H., Manh Tran H., Nagai T., Nabeshima T., Maurice T. (2004) Enhanced antidepressant efficacy of σ1 receptor agonists in rats after chronic intracerebroventricular infusion of β-amyloid-(1–40) protein. Eur. J. Pharmacol. 486, 151–161 [DOI] [PubMed] [Google Scholar]
- 17. Schetz J. A., Perez E., Liu R., Chen S., Lee I., Simpkins J. W. (2007) A prototypical σ1 receptor antagonist protects against brain ischemia. Brain Res. 1181, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luty A. A., Kwok J. B., Dobson-Stone C., Loy C. T., Coupland K. G., Karlström H., Sobow T., Tchorzewska J., Maruszak A., Barcikowska M., Panegyres P. K., Zekanowski C., Brooks W. S., Williams K. L., Blair I. P., Mather K. A., Sachdev P. S., Halliday G. M., Schofield P. R. (2010) σ nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron disease. Ann. Neurol. 68, 639–649 [DOI] [PubMed] [Google Scholar]
- 19. Al-Saif A., Al-Mohanna F., Bohlega S. (2011) A mutation in σ1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 70, 913–919 [DOI] [PubMed] [Google Scholar]
- 20. Shioda N., Yamamoto Y., Watanabe M., Binas B., Owada Y., Fukunaga K. (2010) Heart-type fatty acid-binding protein regulates dopamine D2 receptor function in mouse brain. J. Neurosci. 30, 3146–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tagashira H., Bhuiyan S., Shioda N., Hasegawa H., Kanai H., Fukunaga K. (2010) σ1 receptor stimulation with fluvoxamine ameliorates transverse aortic constriction-induced myocardial hypertrophy and dysfunction in mice. Am. J. Physiol. Heart. Circ. Physiol. 299, H1535–H1545 [DOI] [PubMed] [Google Scholar]
- 22. Takeuchi Y., Fukunaga K. (2003) Differential subcellular localization of two dopamine D2 receptor isoforms in transfected NG108-15 cells. J. Neurochem. 85, 1064–1074 [DOI] [PubMed] [Google Scholar]
- 23. Shioda N., Moriguchi S., Shirasaki Y., Fukunaga K. (2006) Generation of constitutively active calcineurin by calpain contributes to delayed neuronal death following mouse brain ischemia. J. Neurochem. 98, 310–320 [DOI] [PubMed] [Google Scholar]
- 24. Shioda N., Beppu H., Fukuda T., Li E., Kitajima I., Fukunaga K. (2011) Aberrant calcium/calmodulin-dependent protein kinase II (CaMKII) activity is associated with abnormal dendritic spine morphology in the ATRX mutant mouse brain. J. Neurosci. 31, 346–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Brito O. M., Scorrano L. (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610 [DOI] [PubMed] [Google Scholar]
- 26. Manders E. M., Verbeek F. J., Aten J. A. (1993) Measurement of co-localization of objects in dual color confocal images. J. Microsc. 169, 375–382 [DOI] [PubMed] [Google Scholar]
- 27. Tanimura A., Turner R. J. (1996) Inositol 1,4,5-trisphosphate-dependent oscillations of luminal [Ca2+] in permeabilized HSY cells. J. Biol. Chem. 271, 30904–30908 [DOI] [PubMed] [Google Scholar]
- 28. Shioda N., Han F., Morioka M., Fukunaga K. (2008) Bis(1-oxy-2-pyridinethiolato)oxovanadium(IV) enhances neurogenesis via phosphatidylinositol 3-kinase/Akt and extracellular signal-regulated kinase activation in the hippocampal subgranular zone after mouse focal cerebral ischemia. Neuroscience 155, 876–887 [DOI] [PubMed] [Google Scholar]
- 29. Su T. P., London E. D., Jaffe J. H. (1988) Steroid binding at σ receptors suggests a link between endocrine, nervous, and immune systems. Science 240, 219–221 [DOI] [PubMed] [Google Scholar]
- 30. Hellewell S. B., Bruce A., Feinstein G., Orringer J., Williams W., Bowen W. D. (1994) Rat liver and kidney contain high densities of σ1 and σ2 receptors. Characterization by ligand binding and photoaffinity labeling. Eur. J. Pharmacol. 268, 9–18 [DOI] [PubMed] [Google Scholar]
- 31. Kekuda R., Prasad P. D., Fei Y. J., Leibach F. H., Ganapathy V. (1996) Cloning and functional expression of the human type 1 σ receptor (hσR1). Biochem. Biophys. Res. Commun. 229, 553–558 [DOI] [PubMed] [Google Scholar]
- 32. Seth P., Leibach F. H., Ganapathy V. (1997) Cloning and structural analysis of the cDNA and the gene encoding the murine type 1 σ receptor. Biochem. Biophys. Res. Commun. 241, 535–540 [DOI] [PubMed] [Google Scholar]
- 33. Nagai T., Sawano A., Park E. S., Miyawaki A. (2001) Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. U.S.A. 98, 3197–3202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hajnóczky G., Csordás G., Krishnamurthy R., Szalai G. (2000) Mitochondrial calcium signaling driven by the IP3 receptor. J. Bioenerg. Biomembr. 32, 15–25 [DOI] [PubMed] [Google Scholar]
- 36. Takebayashi M., Hayashi T., Su T. P. (2002) Nerve growth factor-induced neurite sprouting in PC12 cells involves σ1 receptors. Implications for antidepressants. J. Pharmacol. Exp. Ther. 303, 1227–1237 [DOI] [PubMed] [Google Scholar]
- 37. Strauss M., Hofhaus G., Schröder R. R., Kühlbrandt W. (2008) Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 27, 1154–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bernardi P. (1999) Mitochondrial transport of cations. Channels, exchangers, and permeability transition. Physiol. Rev. 79, 1127–1155 [DOI] [PubMed] [Google Scholar]
- 39. Green D. R., Kroemer G. (2004) The pathophysiology of mitochondrial cell death. Science 305, 626–629 [DOI] [PubMed] [Google Scholar]
- 40. Simmen T., Aslan J. E., Blagoveshchenskaya A. D., Thomas L., Wan L., Xiang Y., Feliciangeli S. F., Hung C. H., Crump C. M., Thomas G. (2005) PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 24, 717–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jahani-Asl A., Cheung E. C., Neuspiel M., MacLaurin J. G., Fortin A., Park D. S., McBride H. M., Slack R. S. (2007) Mitofusin 2 protects cerebellar granule neurons against injury-induced cell death. J. Biol. Chem. 282, 23788–23798 [DOI] [PubMed] [Google Scholar]
- 42. Klionsky D. J. (2007) Autophagy. From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 8, 931–937 [DOI] [PubMed] [Google Scholar]
- 43. Debnath J., Baehrecke E. H., Kroemer G. (2005) Does autophagy contribute to cell death? Autophagy 1, 66–74 [DOI] [PubMed] [Google Scholar]
- 44. Decuypere J. P., Monaco G., Bultynck G., Missiaen L., De Smedt H., Parys J. B. (2011) The IP3 receptor-mitochondria connection in apoptosis and autophagy. Biochim. Biophys. Acta 1813, 1003–1013 [DOI] [PubMed] [Google Scholar]
- 45. Sarkar S., Floto R. A., Berger Z., Imarisio S., Cordenier A., Pasco M., Cook L. J., Rubinsztein D. C. (2005) Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 170, 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Criollo A., Maiuri M. C., Tasdemir E., Vitale I., Fiebig A. A., Andrews D., Molgó J., Díaz J., Lavandero S., Harper F., Pierron G., di Stefano D., Rizzuto R., Szabadkai G., Kroemer G. (2007) Regulation of autophagy by the inositol trisphosphate receptor. Cell Death. Differ. 14, 1029–1039 [DOI] [PubMed] [Google Scholar]
- 47. Criollo A., Vicencio J. M., Tasdemir E., Maiuri M. C., Lavandero S., Kroemer G. (2007) The inositol trisphosphate receptor in the control of autophagy. Autophagy 3, 350–353 [DOI] [PubMed] [Google Scholar]
- 48. Vicencio J. M., Ortiz C., Criollo A., Jones A. W., Kepp O., Galluzzi L., Joza N., Vitale I., Morselli E., Tailler M., Castedo M., Maiuri M. C., Molgó J., Szabadkai G., Lavandero S., Kroemer G. (2009) The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin-1. Cell Death. Differ. 16, 1006–1017 [DOI] [PubMed] [Google Scholar]
- 49. Aydar E., Palmer C. P., Klyachko V. A., Jackson M. B. (2002) The σ receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron 34, 399–410 [DOI] [PubMed] [Google Scholar]
- 50. Su T. P., Hayashi T., Maurice T., Buch S., Ruoho A. E. (2010) The σ1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol. Sci. 31, 557–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wu Z., Bowen W. D. (2008) Role of σ1 receptor C-terminal segment in inositol 1,4,5-trisphosphate receptor activation. Constitutive enhancement of calcium signaling in MCF-7 tumor cells. J. Biol. Chem. 283, 28198–28215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ganapathy M. E., Prasad P. D., Huang W., Seth P., Leibach F. H., Ganapathy V. (1999) Molecular and ligand-binding characterization of the σ receptor in the Jurkat human T lymphocyte cell line. J. Pharmacol. Exp. Ther. 289, 251–260 [PubMed] [Google Scholar]
- 53. Hayashi T., Su T. P. (2001) Regulating ankyrin dynamics. Roles of σ1 receptors. Proc. Natl. Acad. Sci. U.S.A. 98, 491–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mendes C. C., Gomes D. A., Thompson M., Souto N. C., Goes T. S., Goes A. M., Rodrigues M. A., Gomez M. V., Nathanson M. H., Leite M. F. (2005) The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J. Biol. Chem. 280, 40892–40900 [DOI] [PubMed] [Google Scholar]
- 55. Hayashi T., Maurice T., Su T. P. (2000) Ca2+ signaling via σ1-receptors. Novel regulatory mechanism affecting intracellular Ca2+ concentration. J. Pharmacol. Exp. Ther. 293, 788–798 [PubMed] [Google Scholar]
- 56. Nixon R. A. (2006) Autophagy in neurodegenerative disease. Friend, foe, or turncoat? Trends Neurosci. 29, 528–535 [DOI] [PubMed] [Google Scholar]