

Background: Androgen receptor (AR) is a major therapeutic target for inhibiting proliferation of prostate cancer cells.
Results: A newly identified AR inhibitor, CPIC, works by reducing AR binding to regulated genes.
Conclusion: CPIC has a different mode of action from classical AR antagonists such as bicalutamide.
Significance: CPIC has therapeutic potential in the treatment of prostate cancer and is a new tool for studying AR.
Keywords: Androgen Receptor, Cell Proliferation, Chromatin Immunoprecipitation (ChIP), Drug Screening, Gene Expression, Prostate Cancer, Small Molecules, AR Inhibitor, High-throughput Screening
Abstract
The androgen receptor (AR) has a critical role in the growth and progression of androgen-dependent and castration-resistant prostate cancers. To identify novel inhibitors of AR transactivation that block growth of prostate cancer cells, a luciferase-based high-throughput screen of ∼160,000 small molecules was performed in cells stably expressing AR and a prostate-specific antigen (PSA)-luciferase reporter. CPIC (1-(3-(2-chlorophenoxy) propyl)-1H-indole-3-carbonitrile) was identified as a small molecule that blocks AR transactivation to a greater extent than other steroid receptors. CPIC inhibited AR-mediated proliferation of androgen-sensitive prostate cancer cell lines, with minimal toxicity in AR-negative cell lines. CPIC treatment also reduced the anchorage-independent growth of LAPC-4 prostate cancer cells. CPIC functioned as a pure antagonist by inhibiting the expression of AR-regulated genes in LAPC-4 cells that express wild-type AR and exhibited weak agonist activity in LNCaP cells that express the mutant AR-T877A. CPIC treatment did not reduce AR levels or alter its nuclear localization. We used chromatin immunoprecipitation to identify the site of action of CPIC. CPIC inhibited recruitment of androgen-bound AR to the PSA promoter and enhancer sites to a greater extent than bicalutamide. CPIC is a new therapeutic inhibitor that targets AR-mediated gene activation with potential to arrest the growth of prostate cancer.
Introduction
Androgens such as testosterone and dihydrotestosterone (DHT),2 mediate their biological effects through the nuclear androgen receptor (AR). Binding of high affinity androgens to the ligand binding domain of AR induces the interdomain amino- and carboxyl-terminal (N/C) interaction (1–3). Activated AR translocates and accumulates in the nucleus, where it binds to specific androgen response elements (AREs) in the promoter and enhancer regions of androgen-regulated genes and initiates transcription by recruiting multiple coregulators and the basal transcription machinery in a sequential and cyclic fashion (4, 5). Inhibitors of AR-mediated transcription include selective AR modulators such as the prostate cancer therapeutic, bicalutamide (Casodex), and the experimental drug MDV3100. Bicalutamide (Bic) and MDV3100 inhibit AR transcriptional activity by competing with androgens for binding to the ligand binding domain of AR (6, 7). Bicalutamide can exhibit some agonist activity in cells containing mutant AR or expressing high AR levels (8, 9).
Androgens play a central role in the growth and development of the normal prostate gland and in the proliferation and progression of prostate cancers (10, 11). Patients with low grade tumors benefit from primary therapies for prostate cancer, including radical prostatectomy and androgen deprivation or anti-androgen therapy. However, these treatment strategies are much less effective for long term treatment of high grade tumors (Gleason score ≥7) with elevated recurrence rates after primary therapy. In castration-resistant (or castration-recurrent) prostate cancer (CRPC), there may be higher levels of AR (12), increased expression of AR-regulated genes (13–15), and AR coregulators such as MAGE-A11 and SRC/p160 coactivators (16–19), suggesting that these cancers remain dependent on AR. Several types of evidence support the continuing role of AR in CRPC. Many of the genes induced by androgens in androgen-dependent prostate cancer xenografts become elevated in CRPC (20). Recent studies show that many advanced prostate cancers fuel their growth by synthesizing their own androgens (21). AR continues to be a focus for new drug development, and it remains important to identify antagonists that block AR transcriptional activity (22). The importance of new approaches to targeting androgens and AR in CRPC is illustrated by Abiraterone acetate/Zytiga, an androgen synthesis inhibitor that prolongs survival in patients with advanced prostate cancer (23).
To search for new AR antagonists, we developed and implemented a cell-based high-throughput screen of ∼160,000 small molecules using HeLa cells that stably express AR and an androgen-inducible PSA-luciferase reporter. The relatively high levels of AR in these cells and the high concentration of androgen we used in the screens rendered bicalutamide largely ineffective as an AR antagonist. Here we describe 1-(3-(2-chlorophenoxy)propyl)-1H-indole-3-carbonitrile (CPIC), a lead compound that emerged from our screen. CPIC is a potent and selective AR antagonist that inhibits expression of endogenous AR-regulated genes and the androgen-dependent growth of prostate cancer cells. Chromatin immunoprecipitation (ChIP) assays using the synthetic androgen methyltrienolone (R1881) showed that CPIC decreased AR occupancy at AREs of two prominent AR-regulated genes, PSA and TMPRSS2, without affecting AR protein levels or AR nuclear translocation. In LNCaP cells that express a mutant AR-T877A and in LAPC-4 cells with wild-type AR, CPIC inhibited binding of R1881-AR to the PSA promoter and enhancer. The ability of CPIC to reduce recruitment of AR to multiple regulatory regions of androgen-responsive genes to a greater extent than bicalutamide suggests a new mode of action.
EXPERIMENTAL PROCEDURES
Chemical Libraries
The libraries screened were the ChemBridge MicroFormat small molecule library obtained from ChemBridgeTM containing ∼150,000 small molecules, the Marvel library developed at the University of Illinois by K. Putt and Hergenrother (24) containing ∼9,700 small molecules, and the NCI Diversity Set from NIH with ∼1,990 small molecules.
Plasmids
Expression vectors used have been previously described (25). pCMV-AR-(507–919) codes for the human AR DNA and ligand binding domains, and pCMV-AR-(1–503) codes for the AR N-terminal domain. PSA-Enh-Luc containing the PSA upstream enhancer region was generously provided by Michael Carey (University of California, Los Angeles).
Cell Culture
AR-positive cell lines included LNCaP and LAPC-4 human prostate cancer cells maintained in phenol-red free RPMI 1640 with 10% fetal bovine serum (FBS, Atlanta Biological, GA). LAPC-4 growth medium was routinely supplemented with 1 nm synthetic androgen R1881. Cells were transferred to RPMI 1640 containing 5% charcoal-dextran-stripped FBS (CD-FBS) at least 3 days before plating for an experiment. CWR-R1 cells were grown in modified iMEM (GIBCO#10488-001) containing 2.5 g/liter glucose, 1.2 g/liter niacinamide, 0.5 ml of insulin-transferrin-selenium (ITS, Roche Applied Science #11074547001), 10 ng/ml epidermal growth factor (EGF), and 2% FBS. The cells were transferred to medium containing 2% CD-FBS without EGF 3–4 days before the experiment.
HeLa-AR1C-PSA-ARE-Luc-A6 (HeLaA6) cells selected with hygromycin and Geneticin (G418) stably express human AR and a PSA-luciferase reporter gene containing the 5.8-kb PSA upstream enhancer and promoter region linked to the luciferase gene (26). HeLa-AR3A-PSA-ARE4-Luc-13 (HeLa13) cells selected with hygromycin and G418 stably express human AR and a PSA-luciferase reporter gene containing a 4× multimerized PSA upstream enhancer ARE1 linked to the E4 TATA box and luciferase gene. Both HeLa-AR cell lines were maintained in MEM supplemented with 10% FBS. HeLaA6 cells were maintained under selection with 0.1 mg/ml hygromycin B and 0.5 mg/ml Geneticin. HeLa13 cells were maintained under selection with 0.05 mg/ml hygromycin B and 0.5 mg/ml G418. Cells were transferred to medium containing 5% CD-FBS 3–4 days before the experiment. AR-negative cell lines included PC-3 human prostate cancer cells maintained in RPMI 1640 with 10% FBS and DU145 human prostate cancer cells and MDA-MB-231 human breast cancer cells grown in MEM with 10% FBS.
Other cell lines included estrogen receptor α (ERα)-containing T47D-KBluc breast cancer cells expressing an estrogen-responsive element (ERE)3-luciferase reporter gene (27), maintained in RPMI 1640 containing 10% FBS. Three days before induction with 17β-estradiol, cells were transferred to medium containing 5% CD-FBS. T47D/(A1–2) cells stably express the human glucocorticoid receptor (GR) and contain a mouse mammary tumor virus-luciferase reporter (28) and were maintained in MEM, 5% FBS, and 0.2 mg/ml G418. Mouse mammary tumor virus-Luc reporter is inducible by AR, progesterone receptor (PR), and GR depending on the activating ligand used. Before the experiment, cells were transferred to the above medium containing 5% CD-FBS.
The RPMI base medium was supplemented with 2 mm l-glutamine, 1.5 g/liter sodium bicarbonate, 4.5 g/liter glucose, 10 mm Hepes, pH 7.5, and 1 mm sodium pyruvate. MEM was supplemented with 10 mm HEPES, pH 7.4, and 2 mm l-glutamine. All cells were maintained at 37 °C in 5% CO2 in growth medium containing 1% penicillin and streptomycin and 2–10% FBS (Atlanta Biological, Atlanta, GA) without phenol red.
Soft-agar Colony Formation Assay
To assay anchorage-independent cell growth in soft agar, 1 and 0.7% Select Agar (Invitrogen) was prepared in water and warmed at 40 °C before use. 1.5 ml of 0.5% bottom agar diluted in 2× RPMI 1640 medium was added to each well of a 6-well cell culture plate and allowed to solidify at room temperature. Top agar was prepared by dilution in warm medium. LAPC-4 cells were resuspended in 1.5 ml of 0.35% top agar at 5000 cells/well and plated in 3 wells for each condition. The plates were kept at room temperature for 30 min until the top agar solidified, then 0.5 ml of medium containing the respective treatments was added on top of the agar. Culture medium containing the various treatments was changed every 3–4 days. Colonies were visible after 2 weeks in the hormone-treated wells and counted at day 28 using a dissecting microscope. Photographs of colonies were taken using a Zeiss AxioImager2 imaging system at 5× magnification.
Reporter Gene Assays
At least 3 days before each experiment cells were transferred to medium containing CD-FBS as described above. HeLaA6 and HeLa13 cells (50,000 cells/well) and T47DA/1-2 or T47D-KBluc cells (200,000 cells/well) were plated in 1 ml of medium in 24-well plates. After 24 h the indicated concentrations of 17β-estradiol, DHT, or dexamethasone were added along with each inhibitor or DMSO (vehicle). After 24 h of treatment, cells were lysed in 100 μl of Passive Lysis Buffer (Promega, Madison, WI), and luciferase activity was determined using BrightGlo firefly luciferase reagent (Promega). Unless otherwise mentioned, total DMSO (vehicle) concentration in all assays was maintained at or below 0.1%.
Cell Growth and Viability Assays
To assay cell growth, 2000 cells/well were plated in 96-well plates. LNCaP and LAPC-4 cells were maintained in CD-treated serum for at least 2–4 days before each experiment. All AR negative cell lines, MDA-MB-231, DU145, and PC-3, were plated in growth medium 24 h before treatment. Treatment medium containing vehicle or the indicated concentrations of R1881 with or without inhibitor compounds was added to the cells and incubated for the indicated number of days. Cell viability was determined using Promega CellTiter 96 Aqueous One Solution Cell Proliferation Assay (MTS).
Transient Transfection
LAPC-4 cells were plated (200,000 cells/well in 1 ml) in 24-well plates in 5% CD-FBS 3 days before transfection. On the day of transfection, the medium was changed to 0.2 ml of Opti-MEM. DNA and Lipofectamine2000 (Invitrogen) were diluted in Opti-MEM and incubated together for 20 min before adding to the well. A total of 500 ng of DNA (400 ng of PSA-Luc and 100 ng Renilla-Luc) was transfected into cells in each well at a DNA:Lipofectamine2000 ratio of 1:3. 24 h after transfection, 1 nm R1881 and the indicated concentrations of each inhibitor in DMSO were added to the cells and incubated for 48 h. Cells were lysed in 100 μl of passive lysis buffer (Promega), and luciferase activity was determined using Dual-Luciferase Reporter Assay (Promega #E1910). Transfections in HeLa cells were performed as described (29).
Radioligand Binding Assay
Competitive radioligand binding assays were performed by expressing pCMV-AR in monkey kidney COS cells and incubating cell cultures for 2 h at 37 °C with [3H]R1881 (17α-methyl-[3H] methyltrienolone, 82 Ci/mmol) in the absence and presence of competitor ligands (30, 31).
Endogenous Gene Expression
LNCaP and LAPC-4 cells were seeded into 6-well plates and grown for 3–4 days in medium containing 5% CD-FBS. Cells were then treated with ethanol or R1881 along with the indicated concentrations of the inhibitor for 24 h. RNA was extracted and purified using the Qiagen RNeasy kit. cDNA was prepared from 1 μg of RNA with M-MuLV reverse transcriptase from New England Biolabs. Diluted cDNA was used to perform quantitative RT-PCR using SYBR Green (ABI Thermocycler) with actin as the internal standard. Primers for quantitative RT-PCR were: β-actin forward primer (5′-TGT CAC CAA CTG GGA CGA CA) and reverse primer 5′-GGG GTG TTG AAG GTC TCA AA); PSA (kallikrein 3) forward primer (5′-GGT GAC CAA GTT CAT GCT GTG) and reverse primer (5′-GTG TCC TTG ATC CAC TTC CG); TMPRSS2 forward primer (5′-TAG TGA AAC CAG TGT GTC TGC) and reverse primer (5′-AGC GTT CAG CAC TTC TGA GGT CTT) (6).
Western Blot
Cells were plated at 300,000 cells/well in 6-well plates in medium containing 5% CD-FBS. The medium was changed on day 2, and on day 4 the cells received fresh medium containing the indicated treatments. Whole cell extracts were prepared after 24 h of treatment using 1× radioimmune precipitation assay buffer (Millipore) containing complete mini protease inhibitor mixture (Roche Applied Science). 30 μg of protein per lane was analyzed on 10% SDS-PAGE gels and transferred to nitrocellulose membranes (GE Healthcare). AR protein was detected using AR antibody AR (441) (sc-7305, Santa Cruz, CA), internal control α-tubulin was detected using monoclonal antibody T1699 (Sigma), and β-actin was detected using antibody A1978 (Sigma).
ChIP
LNCaP or LAPC-4 cells were grown in 5% CD-FBS for 4 days and pretreated with 10 μm CPIC, Bic, or DMSO as control for 1 h before treatment with 1 nm R1881 or vehicle (ethanol) for 4 h. Proteins were cross-linked with 1% formaldehyde for 10 min. Cell extracts were digested for 10 min with 50 units of micrococcal nuclease (New England Biolabs) at 37 °C and further sonicated to yield sheared DNA fragments with an average length of 200–1000 base pairs. The sonicated samples were pelleted by centrifugation, and the supernatant was diluted 5-fold with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl, pH 8.1, 167 mm NaCl, and protease inhibitor mixture). 50 μl of diluted supernatant was reserved as input (10%) for each treatment. The samples were precleared with 50 μl of protein A-Sepharose™ 4 Fast Flow (GE Healthcare) in ChIP dilution buffer (1:1) preblocked with 200 μg/ml sheared herring sperm DNA and 500 μg/ml BSA. The samples were then divided, and the remaining proteins were incubated with either 2 μg of anti-AR (C19), 2 μg of anti-RNA polymerase II (clone CTD4H8, Millipore), or control mouse IgG overnight at 4 °C. The antibody-protein-DNA complex was precipitated by incubating with 50 μl of Protein A-SepharoseTM beads for 2 h at 4 °C. The protein-DNA complex was eluted from the beads with elution buffer (1% SDS, 0.1 m NaHCO3). Cross-links were reversed, and DNA was eluted from the protein-DNA complexes by adding 200 mm NaCl and incubating overnight at 65 °C. Protein was digested using Proteinase K and incubating at 45 °C for 2 h. DNA was recovered and purified. Quantitative RT-PCR was performed to determine the change in AR and RNA polymerase II occupancy at various sites of AR binding. The double negative controls were nonspecific antibody (normal mouse IgG) and primers coding for intergenic regions that do not interact with AR. Thermal cycling conditions were 95 °C for 10 min followed by 50 cycles of 25 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C. Primers used were: PSA enhancer ARE forward primer (5′-ACC TGC TCA GCC TTT GTC TCT GAT) and reverse primer (5′-AGA TCC AGG CTT GCT TAC TGT CCT); PSA promoter ARE forward primer (5′-CCT AGA TGA AGT CTC CAT GAG CTA CA) and reverse primer (5′-GGG AGG GAG AGC TAG CAC TTG); middle region forward primer (5′-CTG TGC TTG GAG TTT ACC TGA) and reverse primer 5′-GCA GAG GTT GCA GTG AGC C) (32–34).
Statistical Analysis
Results are expressed as the mean ± S.E. of at least three independent experiments. Significance was established when p < 0.05. Student's t test was used for comparison of the means between two groups.
RESULTS
Establishment of Stable HeLa Cell Lines and Cell-based High-throughput Screening
For this work HeLaA6 (26) and HeLa13 cells were established to stably express PSA-ARE-Luc reporter genes and AR at levels similar to or greater than LNCaP and LAPC-4 cells. HeLaA6 cells express considerably more AR protein than prostate cancer cell lines like LNCaP or LAPC-4 (35) (Fig. 1A). The higher level of AR results in 10–40 μm bicalutamide acting as an agonist in HeLaA6 cells (supplemental Fig. S1A), and in this regard they resemble CRPC. To more nearly mimic the environment of early-stage prostate cancer, we established the HeLa13 cell line that stably expresses AR protein at levels similar to LNCaP and LAPC-4 cells (Fig. 1A). There is a 20-fold difference in the dose-response curves between the two cell lines. The half-maximal effective DHT concentration (EC50) was ∼50 pm in the HeLaA6 cells compared with ∼1 nm in the HeLa13 cells (Fig. 1B).
FIGURE 1.
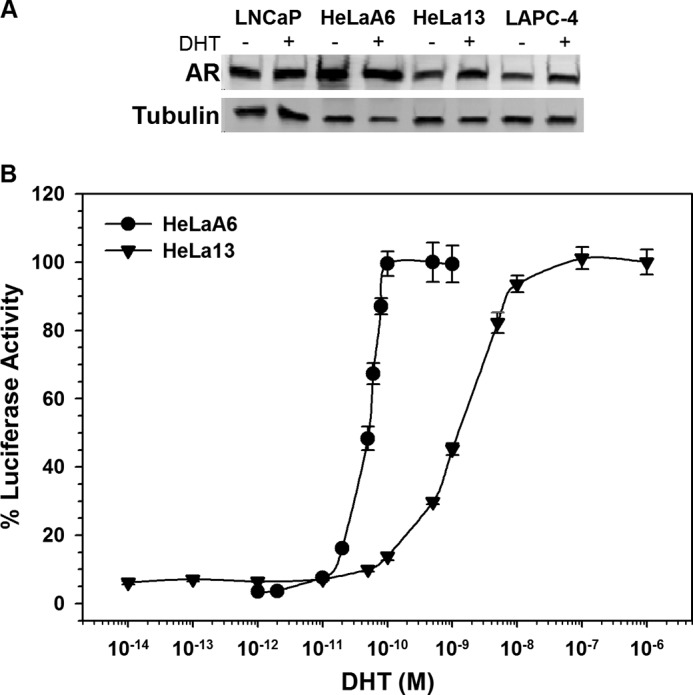
Comparison of AR level and activity in HeLaA6 and HeLa13 cells. A, a Western blot shows AR levels in HeLaA6 and HeLa13 cells. After at least 3 days in medium containing 5% CD-FBS, ethanol (−) or 10 nm DHT (+) was added. After 24 h, the cells were harvested, protein was extracted, and equal amounts of protein were fractionated on 10% polyacrylamide gels and analyzed by Western blotting. Tubulin served as a loading control. B, shown are the DHT dose-response curves for HeLaA6 and HeLa13 cells. HeLaA6 or HeLa13 cells were plated 1 day before treatment with the indicated concentrations of DHT or ethanol (vehicle) control. After 24 h of incubation, lysates were prepared, and PSA luciferase activity was measured in extracts from three wells of cells (mean ± S.E.). Activity in the presence of saturating DHT was set to 100%.
To identify new small molecule inhibitors of AR action, we performed a luciferase-reporter based high-throughput screen using HeLaA6 cells and ∼160,000 small molecules in several libraries at the University of Illinois High-throughput Screening Facility. Feasibility for the cell-based high-throughput screen was established using a combination of manual and robotic steps. In preliminary experiments we tested two screening methods. The more classical method of first plating the cells, waiting 24 h, and adding the small-molecule test compounds and an alternative approach of adding the inhibitor and cells simultaneously to the wells (supplemental Methods 1 and 2). Although we were concerned that many small molecules might interfere with cell attachment, we found that the two methods yielded similar outcomes (supplemental Fig. S2). Because Method 2 was easier to automate, it was used in the large-scale screen.
Identification of CPIC as Lead Compound
To minimize the detection of moderate affinity competitor ligands, HeLaA6 cells were assayed in the presence of 5 nm DHT to fully saturate AR (see Fig. 1B). The lead inhibitor to emerge from our studies, CPIC (Fig. 2A), was subsequently shown to be a competitive inhibitor of androgen binding to AR. We evaluated the potency of CPIC in cells expressing high (HeLaA6) and moderate (HeLa13) levels of AR. The potency of CPIC in HeLaA6 and HeLa13 cells was compared with bicalutamide, a competitive AR antagonist that inhibits androgen binding to AR. CPIC elicited a concentration-dependent inhibition of AR-induced luciferase activity in the HeLaA6 cells with an IC50 of 5 μm and was much more effective in HeLa13 cells with an IC50 of 0.09 μm (Fig. 2B). In contrast to bicalutamide, which had substantial agonist activity in HeLaA6 cells in the absence of DHT, CPIC lacked agonist activity in these cells (supplemental Fig. S1) and thus functioned as a pure antagonist.
FIGURE 2.

CPIC inhibits androgen induction of PSA-luciferase activity. A, shown is the structure of CPIC. B, dose-response studies of CPIC inhibition of DHT-AR induced PSA-luciferase in HeLaA6 and HeLa13 cells. Cells were seeded in 24-well plates and treated with medium ± 10 nm DHT containing DMSO (Veh), the indicated concentrations of CPIC, or 10 μm bicalutamide (Bic) for 24 h and assayed for PSA luciferase activity. Activity of the reporter in the presence of 10 nm DHT and DMSO vehicle was set to 100%. Data represent the mean of three experiments ±S.E. C, LAPC-4 cells transiently transfected with 400 ng of PSA-Enh-luciferase plasmid and 100 ng of CMV-Renilla luciferase were treated with the indicated concentrations of CPIC or DMSO in the presence of 2 nm R1881 for 48 h. Cell lysates were assayed for firefly luciferase and normalized with Renilla-luciferase activity. Bars represent the mean of three experiments ± S.E.
To evaluate the potency of CPIC in prostate cancer cells containing wild-type AR, LAPC-4 human prostate cancer cells were transfected with PSA-luciferase reporter. CPIC effectively inhibited luciferase activity induced by R1881-AR with an IC50 of 1–5 μm (Fig. 2C).
CPIC Competes with Androgen for Binding to AR and Disrupts AR N/C Interaction
The ability of CPIC to compete with R1881 binding to AR was evaluated in cell-based assays. In competitive radiometric binding assays performed using 5 nm [3H]R1881, CPIC competed for binding to AR (Fig. 3A). If CPIC is a competitive inhibitor of AR, increasing the hormone concentration should reduce the ability of CPIC to bind AR and block its action. To test this, we varied the DHT concentration by 100-fold and tested the ability of CPIC to inhibit PSA-Luc in HeLaA6 cells. Increasing the concentration of DHT from 0.1 to 10 nm increased the IC50 for inhibiting DHT-AR induced PSA-Luc transcription by ∼20-fold (Fig. 3B). These data demonstrate that one mechanism for CPIC inhibition of AR-mediated transcription is by competing with androgen binding to AR.
FIGURE 3.

CPIC competes with androgens for binding to AR and reduces the AR N/C interaction. A, shown is a competitive radioligand binding assay. Relative binding affinity of CPIC for AR was determined using 5 nm [3H]R1881 and a range of CPIC concentrations as described (31). Data are the average of duplicate experiments. B, shown is the effect of DHT concentration on CPIC inhibition of AR-induced luciferase activity. HeLaA6 cells were seeded in 24-well plates and maintained for 24 h in medium containing 0.1 nm DHT (circle) or 10 nm DHT (triangle) and the indicated concentrations of CPIC. Data represent the average of triplicate experiments ± S.E. C, HeLa cells were transfected with 100 ng of PSA-Enh-Luc reporter plasmid, 50 ng of pCMV-AR-(1–503), and 50 ng of pCMV-AR-(507–919). Cells were incubated in the absence or presence of 10 nm DHT and the indicated concentrations of CPIC or Bic. Luciferase units of DHT-treated wells were set at 100%. The luciferase activity is representative of two independent experiments.
To evaluate the effect of CPIC on the interdomain AR N/C interaction, we performed mammalian two-hybrid assays using constructs containing the N- and C-terminal regions of AR. Transfections were performed in HeLa cells using pCMV-AR-(1–503) (the AR N-terminal region), pCMV-AR-(507–919) (N-terminal deletion of AR), and PSA-Enh-Luc reporter plasmids in the absence and presence of 10 nm DHT. Bicalutamide and other antagonists that bind to the ligand binding pocket of AR disrupt this crucial interaction and alter receptor structure and function (36, 37). CPIC was substantially more effective than bicalutamide in inhibiting the androgen-induced AR N/C interdomain interaction (Fig. 3C).
CPIC Is Relatively Specific Inhibitor of AR-mediated Transactivation
The AR ligand binding domain shares >50% sequence homology with the GR ligand-binding domain (38). We, therefore, evaluated the effect of CPIC on transcription by GR and ERα in cell lines stably expressing reporter genes. Inhibition of ERα was evaluated in T47D-KBluc cells that express endogenous ERα and are stably transfected to express an (ERE)3-luciferase reporter. Inhibition of GR was evaluated in T47D/(A1–2) cells that stably express GR and a mouse mammary tumor virus-luciferase reporter gene. Fig. 4 shows that CPIC did not inhibit GR transcriptional activity and modestly inhibited ERα activity at 20 μm CPIC. The results suggest that CPIC is a relatively specific inhibitor of AR-mediated transcription.
FIGURE 4.

Partial inhibition of ERα activity at high concentrations of CPIC. T47D-KBluc and T47D/(A1–2) cells were seeded in 24-well plates in medium containing 5% CD-FBS. After the cells attached, treatment medium was added with 5 nm 17β-estradiol for ER in T47D-KBluc cells and 5 nm dexamethasone for GR in T47D/(A1–2) cells and the indicated concentrations of CPIC (or DMSO vehicle). After 24 h cell lysates were analyzed for luciferase activity. Data are the average of triplicate experiments ± S.E.
CPIC Inhibits AR-dependent Proliferation of Prostate Cancer Cells
We evaluated the effect of CPIC on the proliferation of AR positive prostate cancer cell lines. LAPC-4 cells contain wild-type AR. LNCaP cells, the most widely used androgen-sensitive cell line, contain high levels of the mutant AR-T877A, and CWR-R1 cells contain mutant AR-H874Y, both of which are highly inducible by androgens and other steroids (9, 39). CPIC inhibited androgen-mediated AR-dependent growth of all 3 cell lines, with an IC50 of ∼2 μm in LNCaP cells (Fig. 5A), ∼0.3 μm in LAPC-4 cells (Fig. 5B), and ∼5 μm in CWR-R1 cells (Fig. 5C).
FIGURE 5.

Effects of CPIC on prostate cancer cell growth. LNCaP cells (A), LAPC-4 cells (B), and CWR-R1 cells (C) were treated with the indicated concentrations of CPIC in the presence or absence of 1 nm R1881. After 4 days of treatment for LNCaP and CWR-R1 cells and 8 days for slow-growing LAPC-4, cell proliferation was measured using MTS. Cell growth in R1881+DMSO was set to 100%. D, AR-negative PC-3, MDA-MB-231, and DU145 cells were inoculated at 2000 cells/well in 96-well plates and treated with 100 μl of medium containing the indicated concentrations of CPIC or DMSO (vehicle). Growth of the cells was evaluated after 3 days. DMSO wells were set to 100%. Data points represent the mean of 8 wells ± S.E.
The specificity of CPIC inhibition of AR-mediated cell growth was tested using AR-negative PC-3 and DU145 prostate cancer cells and MDA-MB-231 breast cancer cells. CPIC had little effect on the growth of PC-3 or MDA-MB-231 cells (Fig. 5D). However, at high concentrations, CPIC slowed but did not arrest the proliferation of DU145 cells. Thus, functional concentrations of <5 μm CPIC were relatively specific for inhibition of AR-mediated cell growth. We also evaluated several small molecules structurally related to CPIC. These small molecules lacked the combined potency and specificity of CPIC. One member of this structural family, PIC19.7 (2-methyl-1-(3-(o-tolyloxy)propyl)-1H-indole-3-carbonitrile), exhibited an excellent toxicity profile (supplemental Fig. S3) but had substantially lower potency than CPIC and inhibited ER-mediated transactivation at high concentrations.
CPIC Inhibits Anchorage-independent Growth of LAPC-4 Cells
Anchorage-independent growth is a hallmark of cancer cells. Growth in soft agar is often used to evaluate anchorage independence of human prostate cancer cells. We tested the ability of CPIC to inhibit colony formation of wild-type AR containing LAPC-4 cells grown in soft agar. LAPC-4 cells supplemented with medium containing 1 nm R1881 formed large colonies (>0.5 mm) after 4 weeks (Fig. 6, R1881). The growth of these cells was completely inhibited in the presence of 10 μm CPIC as well as 10 μm Bic (Fig. 6). When colonies from all the wells of each treatment condition were counted, the R1881-treated plate contained 77 colonies/well mostly >0.5 mm in diameter. In comparison, there were no colonies >0.5 mm in diameter in the R1881+ CPIC-treated wells and on average 2 small colonies/well less than 0.5 mm in diameter. A similar effect was seen in the R1881+ Bic-treated wells. The data indicate that CPIC inhibits androgen stimulation of anchorage-dependent (Fig. 5) and anchorage-independent (Fig. 6) growth of prostate cancer cells.
FIGURE 6.

CPIC inhibits anchorage-independent growth of LAPC-4 prostate cancer cells in soft agar. 5000 LAPC-4 cells were plated into top agar. Cells were treated with medium containing DMSO (vehicle), 1 nm R1881, 1 nm R1881 + 10 μm CPIC, or 1 nm R1881 + 10 μm Bic and replenished every 3–4 days. After 28 days, colonies were counted and photographed at 5× magnification. Inset, the bar graph represents the average of the total number of colonies counted in each well of the treatments. Photographs are representative of the entire well and of triplicate experiments.
CPIC Inhibits Expression of Endogenous AR-regulated Genes
To evaluate the effect of CPIC on endogenous gene expression, the levels of mRNAs for several well characterized androgen-regulated genes were measured in LNCaP and LAPC-4 cells. The PSA and TMPRSS2 genes are highly induced by androgens acting through AR. In LNCaP cells, 10 μm CPIC exhibited weak agonist activity and blocked AR-mediated transcription of PSA and TMPRSS2 mRNAs in a dose-dependent manner with IC50 values of ∼0.5 and 0.3 μm, respectively (Fig. 7A). In contrast to LNCaP cells, in LAPC-4 cells that express wild-type AR, CPIC was a pure antagonist and was more effective than bicalutamide in inhibiting induction of PSA and other genes (Fig. 7B and supplemental Fig. S4B). TMPRSS2 mRNA was minimally induced by androgen in LAPC-4 cells, and thus the effect of CPIC could not be evaluated (data not shown). CPIC also inhibited androgen-regulated expression of other genes including kallikrein 2 and TMEPAI in LNCaP and LAPC-4 cells (supplemental Fig. S4).
FIGURE 7.

CPIC inhibits expression of endogenous AR-regulated genes. LNCaP (A) and LAPC-4 (B) cells were seeded in 6-well plates at 300,000 cells/well in medium containing 5% CD-FBS for at least 3 days. Cells were treated with or without 1 nm R1881 and the indicated concentrations of CPIC or Bic for 24 h before RNA extraction. mRNA was quantitated using quantitative RT-PCR and normalized to β-actin. Data represent the mean of three independent experiments ±S.E.
The AR-T877A mutant in LNCaP cells is activated by adrenal androgens, estrogens, and progestins as well as many anti-androgens (39), which may explain the weak agonist activity of CPIC. Alternatively, higher levels of AR in LNCaP cells compared with LAPC-4 cells (Fig. 1A) or cell type specificity might be responsible for the weak agonist activity of CPIC. To test the effect of the AR-T877A mutation, transient transfections were performed in HeLa cells. For both wild-type AR and AR-T877A, CPIC did not induce luciferase activity above the no hormone control (supplemental Fig. S5). Because CPIC did not exhibit weak agonist activity in HeLaA6 cells containing high levels of wild-type AR (supplemental Fig. S1B) or in HeLa cells transfected with AR-T877A (supplemental Fig. S5), it seems likely that cell context contributes to the weak agonist activity of CPIC in LNCaP cells.
CPIC Does Not Decrease AR Levels or Reduce Nuclear Translocation
CPIC could inhibit AR-mediated gene expression through multiple mechanisms (40) that include (a) increased AR degradation, (b) reduced nuclear localization of liganded-AR, (c) inhibition of AR recruitment to response elements on DNA, and (d) altered coregulator recruitment. Western blot analysis showed that CPIC had little or no effect on intracellular levels of AR (Fig. 8A).
FIGURE 8.

CPIC does not decrease AR levels or alter nuclear localization of AR. A, shown is a Western blot of AR levels in LNCaP cells treated with CPIC. LNCaP cells were plated in medium containing 5% CD-FBS and maintained for at least 3 days. Treatment medium with (+) or without (−) 10 nm DHT containing DMSO (vehicle) or the indicated inhibitor was added to the cells. After 24 h, cell lysates were prepared, and equal amounts of protein were analyzed by Western blotting and tubulin used as a loading control. B, visualization of intracellular AR in LNCaP cells is shown. LNCaP cells were incubated with 10 nm R1881, 10 μm Bic, or 10 μm CPIC for 24 h, and intracellular AR was visualized by fluorescent microscopy using AR polyclonal antibody (Abcam, ab3510). Objective magnification, 40×.
We used fluorescent polyclonal antibody to visualize intracellular AR in LNCaP and LAPC-4 cells. AR was predominantly nuclear in the presence of 10 nm R1881. In LNCaP cells, when 10 μm CPIC or Bic was present for either 4 or 24 h, AR was predominantly localized in the nucleus (Fig. 8B and supplemental Fig. S6A). Staining DNA with DAPI showed nuclear co-localization of CPIC-bound AR in LAPC-4 cells (supplemental Fig. S6B). These data suggest that CPIC promotes nuclear localization of AR and might influence AR association with DNA.
CPIC Inhibits AR Binding to Androgen-responsive Genes
ChIP was used to evaluate the effect of CPIC on AR recruitment to regulatory regions of androgen-responsive genes in LNCaP and LAPC-4 cells. PSA and TMPRSS2 are two well characterized androgen-regulated genes with defined AREs in their promoter and enhancer regions (41, 42). In LNCaP cells, R1881 increased AR recruitment to the PSA enhancer and promoter regions (Fig. 9, A and B) and the TMPRSS2 enhancer (supplemental Fig. S7A). RNA polymerase II was also recruited to these AREs in the presence of androgen-bound AR. Consistent with its weak agonist activity in LNCaP cells, with 10 μm CPIC alone there was a small increase in association of AR with the PSA enhancer and promoter (Fig. 9, A and B). Although there is diversity in the reported effects of bicalutamide on AR binding to the PSA enhancer in LNCaP cells (6, 43, 44), most reports suggest that bicalutamide interferes with androgen action at the PSA promoter by altering AR interaction with coregulators rather than inhibiting AR DNA binding (34, 45, 46). We found that bicalutamide inhibited recruitment of R1881-bound AR to the PSA enhancer to a greater extent than the PSA-promoter (Fig. 9, A and B). CPIC strongly inhibited recruitment of R1881-AR to the PSA-promoter, PSA-enhancer, and the TMPRSS2 enhancer (Fig. 9, A and B, and supplemental Fig. S7A).
FIGURE 9.

CPIC inhibits R1881-AR occupancy at AREs in LNCaP cells. Before ChIP, LNCaP cells were maintained for 3 days in medium containing 5% CD-FBS. After 1 h of incubation with DMSO, 10 μm CPIC, or 10 μm Bic, the cells were treated with 1 nm R1881 or ethanol and incubated for 4 h. Protein complexes were cross-linked, and AR (A, B) or RNA polymerase II (RNAPoIII; C, D) or control IgG antibody was used to pull down protein-bound chromatin fragments. Occupancy at the PSA enhancer, PSA promoter, and a control ARE-free region (middle region) between the two sites was determined using quantitative RT-PCR. -Fold enrichment over IgG control was plotted. Data represent the mean of three PCRs ± S.E. and are representative of other experiments. Significance of the differences between CPIC and the DMSO control was tested using Student's t test. p < 0.01 when compared with the respective controls. IP, immunoprecipitate.
Because CPIC has no effect on AR levels or nuclear localization (Fig. 8), the decrease in AR occupancy is likely an effect of CPIC on AR binding to AREs and not a result of reduced levels of nuclear AR. Consistent with its ability to reduce AR binding to the promoter and enhancer regions of PSA and TMPRSS2, CPIC also reduced recruitment of RNA polymerase II (Fig. 9, C and D, and supplemental Fig. S7B). The results suggest that CPIC inhibits AR binding to AREs.
In LNCaP cells, 10 μm CPIC exhibits weak agonist activity at the PSA gene (Figs. 7A and 9). To evaluate the effect of CPIC in cells in which it acts as a pure antagonist, we performed ChIP in LAPC-4 cells. Because LAPC-4 cells contain lower levels of AR relative to LNCaP cells, there are few instances of ChIP performed using these cells. In LAPC-4 cells, 10 μm CPIC in the absence of R1881 did not increase AR occupancy at the PSA enhancer or promoter (Fig. 10, A and B) or the kallikrein 2-enhancer (supplemental Fig. S7C). Bicalutamide had no effect on R1881-AR recruitment at the PSA promoter, whereas CPIC strongly inhibited R1881-AR recruitment to the PSA promoter (Fig. 10B). CPIC was somewhat more effective than Bic in reducing AR recruitment to the PSA enhancer (Fig. 10A). Consistent with its inhibition of AR binding at the PSA regulatory regions, CPIC also prevents recruitment of RNA polymerase II (Fig. 10, C and D, and supplemental Fig. S7D).
FIGURE 10.

CPIC inhibits R1881-AR occupancy at AREs in LAPC-4 cells. LAPC-4 cells were maintained for 3 days in medium containing 5% CD-FBS, and ChIP was carried out as described in Fig. 9 and under “Experimental Procedures.” AR and RNA polymerase II occupancy at the PSA enhancer, PSA promoter, and a control ARE-free region (middle region) between the two sites was determined using quantitative RT-PCR. -Fold enrichment over IgG control was plotted. Data represent the mean of three PCRs ± S.E. Significance of the differences between CPIC and the DMSO control was tested using Student's t test and p < 0.05 when compared with the respective controls. For AR recruitment to the PSA promoter, R1881 + 10 μm Bic was not significantly different from R1881 + DMSO. IP, immunoprecipitate.
To investigate the effect of CPIC on interaction of coactivators to AR, we used mammalian 2-hybrid assays. Because this assay uses GAL4 DNA binding domain, we investigated the effects of CPIC on coactivator recruitment independent of its effects on AR binding to AREs in responsive genes. Previous work showed that TIF2 (GRIP1/SRC2) interacts with the AR ligand binding domain through its LXXLL motifs (1). In mammalian two-hybrid assays in HeLa cells, CPIC moderately reduces the interaction between the AR ligand binding domain and the TIF2 fragment required for interaction with AR (supplemental Fig. S8). There was essentially no effect on TIF2 interaction at 5 μm CPIC and 50% inhibition at 10 μm CPIC.
We evaluated the relationship between the weak agonist activity of CPIC in LNCaP cells that induced PSA and TMPRSS2 mRNAs and the extent to which CPIC enhances AR occupancy at regulatory sites in PSA and TMPRSS2. CPIC alone induced TMPRSS2 mRNA to ∼24% of maximum and AR binding to the TMPRSS2 enhancer to ∼26% of maximum (Fig. 7 and supplemental Fig. S7). There was also a correlation between the ability of CPIC to inhibit R1881 induction of PSA and TMPRSS2 mRNAs and its ability to inhibit binding of AR to the regulatory regions of these genes. These results suggest that CPIC acts by decreasing AR binding to AREs in androgen-responsive genes.
DISCUSSION
The importance of AR in the development and growth of prostate cancer make AR an important therapeutic target. AR has properties unique among the steroid receptor family that influence androgen-dependent gene regulation. The AR interdomain interactions and a more limited role of coactivator LXXLL motifs (1, 3) highlight significant differences between AR and other steroid receptors. One way to probe the mechanism of AR action is by identification and characterization of novel small molecule inhibitors. The identification of a new coactivator binding surface on AR using low potency small molecule inhibitors, unrelated to CPIC, identified by screening (47) supports the utility of using small molecules as probes of AR actions. New small molecule inhibitors also have the potential to be therapeutically relevant.
Molecular mechanisms leading to the development of prostate cancer resistance to antagonists are not fully understood. In a limited number of cases, AR mutations such as AR-T877A can confer resistance to anti-androgens that function as weak agonists (39). To identify new small molecule AR inhibitors, we developed and implemented a cell-based high-throughput screen. Because LNCaP cells contain mutant AR-T877A and LAPC-4 cells contain wild-type AR but have a doubling time of ∼3 days, these cell lines were unsuitable for a luciferase-based screen. Although assays for androgen-stimulated proliferation of LNCaP and LAPC-4 cells work well in a 96-well format, they did not reach the requisite level of precision and reproducibility needed for a high-throughput screen in 384-well plates. For both the primary screen and preliminary analysis of hits, HeLa cells stably expressing wild-type AR and a PSA-luciferase reporter were used. The classical approach to cell-based screening involves adding the test compounds to cells that are previously plated. We found a simpler approach in which the inhibitor and cells are added at the same time, produced equivalent results, and could be readily automated. Consistent with its properties in LNCaP cells stably transfected to express elevated levels of wild-type AR (6), bicalutamide was an agonist in the HeLaA6 cells that express high levels of AR compared with minimal agonist activity in HeLa13 cells with moderate levels of AR similar to prostate cancer cells. In contrast to bicalutamide, which did not inhibit the androgen-induced AR N/C interaction at the submicromolar concentrations tested, CPIC potently disrupted the androgen-dependent interdomain AR N/C interaction.
Based on our assays, CPIC was selected for detailed evaluation. An effective small molecule inhibitor of AR should exhibit high potency and specificity. Our studies show that CPIC inhibits AR transcriptional activity, with little or no effect on GR or ERα under the same conditions where other small molecules robustly inhibited ERα and GR (26, 48). CPIC effectively inhibited androgen-AR-dependent proliferation of LNCaP, LAPC-4, and CWR-R1 prostate cancer cells. At relevant concentrations, CPIC had little or no effect on the proliferation of several AR-negative cell lines. Anchorage-independent colony formation is a characteristic of many cancers, and CPIC nearly abolished the AR-dependent proliferation of LAPC-4 prostate cancer cells in soft agar. Our studies, therefore, suggest that CPIC is a potent and selective inhibitor of AR action in prostate cancer cells.
Radioligand binding assays and gene expression studies using stably transfected cell lines indicate that high levels of CPIC compete with androgen binding. By far the most straightforward explanation of these data is that CPIC is a competitive inhibitor of androgen binding to AR. However, our data do not exclude the possibility that CPIC binds to AR outside of the ligand binding pocket and induces an inactive AR conformation opposed to androgen binding. When very high concentrations of androgens are present, AR is predominantly in the active conformation, which suggests a competitor phenotype.
Although radioligand binding and cell-based assays show that CPIC competes with androgen for binding to AR, the weak agonist activity of CPIC in LNCaP cells did not result from the AR-T877A mutation. Cell context and high AR-T877A are likely responsible for the weak agonist activity of CPIC.
Interestingly, CPIC has a different site of action than two experimental competitive inhibitors of AR, MDV3100 (6) and ARN-509 (49). In part because MDV3100 required a relatively high 100 mg/kg/day dose to elicit a maximum in vivo response, ARN-509 was produced that elicited a maximum response at 30 mg/kg/day. A major site of action of both MDV3100 and ARN-509 is inhibition of AR nuclear localization (6, 49). In contrast, CPIC has no effect on nuclear localization and acts at the level of AR binding to regulatory regions in responsive genes.
CPIC exhibited weak agonist activity in LNCaP cells and modestly induced PSA and TMPRSS2 mRNAs in the absence of androgen. However, 10 μm CPIC and 10 μm bicalutamide alone had similar minimal effects on the proliferation of LNCaP cells. Tamoxifen, which competes with estrogens for binding to ER, exhibits partial agonist activity in stimulating gene expression in MCF-7 human breast cancer cells (50, 51) but is widely used in breast cancer therapy. Because the weak agonist activity of CPIC in LNCaP cells does not stimulate of LNCaP cell proliferation, CPIC has therapeutic potential.
The inability of CPIC to influence AR levels or block agonist-induced AR nuclear localization together with the results from ChIP assays indicates that the inhibitory effects of CPIC occur at the gene level. R1881 induced an ∼27-fold increase in AR occupancy at the PSA enhancer, an ∼11-fold increase in occupancy at the PSA promoter, and an ∼18-fold increase in occupancy at the TMPRSS2 regulatory region. The extent to which CPIC acts as a weak agonist in LNCaP cells and induced PSA and TMPRSS2 mRNAs correlated with the extent to which CPIC enhanced AR occupancy at regulatory sites in PSA and TMPRSS2. These data suggest that CPIC-bound AR at the PSA and TMPRSS2 genes is transcriptionally competent.
Consistent with the mRNA data, CPIC does not exhibit any weak agonist activity in LAPC-4 cells. In these cells CPIC significantly reduced androgen-induced AR recruitment and consequently reduced RNA polymerase II recruitment to the PSA promoter and enhancer regions. In contrast, bicalutamide in the presence of androgen did not inhibit AR occupancy at the PSA promoter in LNCaP cells (5) and has been reported to recruit corepressors to the promoter region (34, 45). Our data indicate that CPIC functions as an AR inhibitor by decreasing the interaction of AR with regulatory regions of androgen-responsive genes.
AR and other steroid receptors exhibit a high level of conformational flexibility. Small molecules such as CPIC and bicalutamide may elicit different AR conformations. Our analysis suggests that although CPIC is similar to bicalutamide in its ability to inhibit androgen-induced transcription at the PSA gene locus, they may evoke different conformational changes when bound to AR and have different mechanisms of action. CPIC was much more effective than bicalutamide in the inhibition of R1881-AR binding to the PSA promoter. The weak agonist activity of CPIC in LNCaP cells could be due to: an AR conformation in which binding to AREs is reduced but not eliminated; an altered coactivator population that stabilizes weak binding of CPIC-AR to AREs; or the existence of multiple CPIC-AR conformations, one of which binds DNA and one of which is unable to bind DNA. With its unique mode of action, CPIC is a new potential inhibitor of prostate cancer growth.
Acknowledgments
We are most grateful to Dr. Chen Zhang (Director, High Throughput Screening Facility, University of Illinois) for advice and help in performing the primary high-throughput screen and to Amanda J. Blackwelder and John T. Minges for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants DK-071909 (to D. J. S.), HD16910 (to E. M. W.), and P01-CA77739 (NCI; to E. M. W.). This work was also supported by Department of Defense Prostate Cancer Research Program Grant W81XWH-09-1-0309 (to D. J. S.).

This article contains supplemental Figs. S1–S8 and Methods 1 and 2.
- DHT
- dihydrotestosterone
- AR
- androgen receptor
- ARE
- androgen response element
- Bic
- bicalutamide
- CD-FBS
- charcoal-dextran stripped FBS
- CPIC
- 1-(3-(2-chlorophenoxy)propyl)-1H-indole-3-carbonitrile
- CRPC
- castration-resistant prostate cancer
- ER
- estrogen receptor
- GR
- glucocorticoid receptor
- PSA
- prostate-specific antigen
- R1881
- synthetic androgen methyltrienolone
- R1881-AR
- AR activated by the binding of R1881
- MEM
- minimum essential medium
- Luc
- luciferase
- N/C
- amino- and carboxyl-terminal interaction.
REFERENCES
- 1. He B., Kemppainen J. A., Voegel J. J., Gronemeyer H., Wilson E. M. (1999) Activation function 2 in the human androgen receptor ligand binding domain mediates interdomain communication with the NH2-terminal domain. J. Biol. Chem. 274, 37219–37225 [DOI] [PubMed] [Google Scholar]
- 2. He B., Kemppainen J. A., Wilson E. M. (2000) FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J. Biol. Chem. 275, 22986–22994 [DOI] [PubMed] [Google Scholar]
- 3. He B., Minges J. T., Lee L. W., Wilson E. M. (2002) The FXXLF motif mediates androgen receptor-specific interactions with coregulators. J. Biol. Chem. 277, 10226–10235 [DOI] [PubMed] [Google Scholar]
- 4. Shang Y., Hu X., DiRenzo J., Lazar M. A., Brown M. (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103, 843–852 [DOI] [PubMed] [Google Scholar]
- 5. Kang Z., Pirskanen A., Jänne O. A., Palvimo J. J. (2002) Involvement of proteasome in the dynamic assembly of the androgen receptor transcription complex. J. Biol. Chem. 277, 48366–48371 [DOI] [PubMed] [Google Scholar]
- 6. Tran C., Ouk S., Clegg N. J., Chen Y., Watson P. A., Arora V., Wongvipat J., Smith-Jones P. M., Yoo D., Kwon A., Wasielewska T., Welsbie D., Chen C. D., Higano C. S., Beer T. M., Hung D. T., Scher H. I., Jung M. E., Sawyers C. L. (2009) Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324, 787–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scher H. I., Beer T. M., Higano C. S., Anand A., Taplin M. E., Efstathiou E., Rathkopf D., Shelkey J., Yu E. Y., Alumkal J., Hung D., Hirmand M., Seely L., Morris M. J., Danila D. C., Humm J., Larson S., Fleisher M., Sawyers C. L. (2010) Antitumor activity of MDV3100 in castration-resistant prostate cancer. A phase 1–2 study. Lancet 375, 1437–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wilding G., Chen M., Gelmann E. P. (1989) Aberrant response in vitro of hormone-responsive prostate cancer cells to antiandrogens. Prostate 14, 103–115 [DOI] [PubMed] [Google Scholar]
- 9. Tan J., Sharief Y., Hamil K. G., Gregory C. W., Zang D. Y., Sar M., Gumerlock P. H., DeVere White R. W., Pretlow T. G., Harris S. E., Wilson E. M., Mohler J. L., French F. S. (1997) Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol. Endocrinol. 11, 450–459 [DOI] [PubMed] [Google Scholar]
- 10. Debes J. D., Tindall D. J. (2004) Mechanisms of androgen-refractory prostate cancer. N. Engl. J. Med. 351, 1488–1490 [DOI] [PubMed] [Google Scholar]
- 11. Lamont K. R., Tindall D. J. (2011) Minireview. Alternative activation pathways for the androgen receptor in prostate cancer. Mol. Endocrinol. 25, 897–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Linja M. J., Savinainen K. J., Saramäki O. R., Tammela T. L., Vessella R. L., Visakorpi T. (2001) Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 61, 3550–3555 [PubMed] [Google Scholar]
- 13. Chen C. D., Welsbie D. S., Tran C., Baek S. H., Chen R., Vessella R., Rosenfeld M. G., Sawyers C. L. (2004) Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 10, 33–39 [DOI] [PubMed] [Google Scholar]
- 14. Mohler J. L., Gregory C. W., Ford O. H., 3rd, Kim D., Weaver C. M., Petrusz P., Wilson E. M., French F. S. (2004) The androgen axis in recurrent prostate cancer. Clin. Cancer Res. 10, 440–448 [DOI] [PubMed] [Google Scholar]
- 15. Taplin M. E., Balk S. P. (2004) Androgen receptor. A key molecule in the progression of prostate cancer to hormone independence. J. Cell Biochem. 91, 483–490 [DOI] [PubMed] [Google Scholar]
- 16. Gregory C. W., He B., Johnson R. T., Ford O. H., Mohler J. L., French F. S., Wilson E. M. (2001) A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 61, 4315–4319 [PubMed] [Google Scholar]
- 17. Bai S., He B., Wilson E. M. (2005) Melanoma antigen gene protein MAGE-11 regulates androgen receptor function by modulating the interdomain interaction. Mol. Cell. Biol. 25, 1238–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wilson E. M. (2010) Androgen receptor molecular biology and potential targets in prostate cancer. Ther. Adv. Urol. 2, 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karpf A. R., Bai S., James S. R., Mohler J. L., Wilson E. M. (2009) Increased expression of androgen receptor coregulator MAGE-11 in prostate cancer by DNA hypomethylation and cyclic AMP. Mol. Cancer Res. 7, 523–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gregory C. W., Hamil K. G., Kim D., Hall S. H., Pretlow T. G., Mohler J. L., French F. S. (1998) Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res. 58, 5718–5724 [PubMed] [Google Scholar]
- 21. Mohler J. L., Titus M. A., Bai S., Kennerley B. J., Lih F. B., Tomer K. B., Wilson E. M. (2011) Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res. 71, 1486–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scher H. I., Sawyers C. L. (2005) Biology of progressive, castration-resistant prostate cancer. Directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol. 23, 8253–8261 [DOI] [PubMed] [Google Scholar]
- 23. O'Donnell A., Judson I., Dowsett M., Raynaud F., Dearnaley D., Mason M., Harland S., Robbins A., Halbert G., Nutley B., Jarman M. (2004) Hormonal impact of the 17α-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br J. Cancer 90, 2317–2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Putt K. S., Hergenrother P. J. (2004) A nonradiometric, high-throughput assay for poly(ADP-ribose) glycohydrolase (PARG). Application to inhibitor identification and evaluation. Anal. Biochem. 333, 256–264 [DOI] [PubMed] [Google Scholar]
- 25. Lagarde W. H., Blackwelder A. J., Minges J. T., Hnat A. T., French F. S., Wilson E. M. (2012) Androgen receptor exon 1 mutation causes androgen insensitivity by creating a phosphorylation site and inhibiting melanoma antigen-A11 activation of N/C interaction-dependent transactivation. J. Biol. Chem. 287, 10905–10915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kretzer N. M., Cherian M. T., Mao C., Aninye I. O., Reynolds P. D., Schiff R., Hergenrother P. J., Nordeen S. K., Wilson E. M., Shapiro D. J. (2010) A noncompetitive small molecule inhibitor of estrogen-regulated gene expression and breast cancer cell growth that enhances proteasome-dependent degradation of estrogen receptor α. J. Biol. Chem. 285, 41863–41873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wilson V. S., Bobseine K., Gray L. E., Jr. (2004) Development and characterization of a cell line that stably expresses an estrogen-responsive luciferase reporter for the detection of estrogen receptor agonist and antagonists. Toxicol. Sci. 81, 69–77 [DOI] [PubMed] [Google Scholar]
- 28. Nordeen S. K., Kühnel B., Lawler-Heavner J., Barber D. A., Edwards D. P. (1989) A quantitative comparison of dual control of a hormone response element by progestins and glucocorticoids in the same cell line. Mol. Endocrinol. 3, 1270–1278 [DOI] [PubMed] [Google Scholar]
- 29. Askew E. B., Gampe R. T., Jr., Stanley T. B., Faggart J. L., Wilson E. M. (2007) Modulation of androgen receptor activation function 2 by testosterone and dihydrotestosterone. J. Biol. Chem. 282, 25801–25816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. He B., Gampe R. T., Jr., Hnat A. T., Faggart J. L., Minges J. T., French F. S., Wilson E. M. (2006) Probing the functional link between androgen receptor coactivator and ligand-binding sites in prostate cancer and androgen insensitivity. J. Biol. Chem. 281, 6648–6663 [DOI] [PubMed] [Google Scholar]
- 31. Wilson E. M. (2009) Methods for measuring ligand dissociation and nuclear receptor turnover in whole cells. Methods Mol. Biol. 505, 21–33 [DOI] [PubMed] [Google Scholar]
- 32. Andersen R. J., Mawji N. R., Wang J., Wang G., Haile S., Myung J. K., Watt K., Tam T., Yang Y. C., Bañuelos C. A., Williams D. E., McEwan I. J., Wang Y., Sadar M. D. (2010) Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino terminus domain of the androgen receptor. Cancer Cell 17, 535–546 [DOI] [PubMed] [Google Scholar]
- 33. Joseph J. D., Wittmann B. M., Dwyer M. A., Cui H., Dye D. A., McDonnell D. P., Norris J. D. (2009) Inhibition of prostate cancer cell growth by second-site androgen receptor antagonists. Proc. Natl. Acad. Sci. U.S.A. 106, 12178–12183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shang Y., Myers M., Brown M. (2002) Formation of the androgen receptor transcription complex. Mol. Cell 9, 601–610 [DOI] [PubMed] [Google Scholar]
- 35. Sobel R. E., Sadar M. D. (2005) Cell lines used in prostate cancer research. A compendium of old and new lines. Part 1. J. Urol. 173, 342–359 [DOI] [PubMed] [Google Scholar]
- 36. Wilson E. M. (2011) Analysis of interdomain interactions of the androgen receptor. Methods Mol. Biol. 776, 113–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Langley E., Zhou Z. X., Wilson E. M. (1995) Evidence for an anti-parallel orientation of the ligand-activated human androgen receptor dimer. J. Biol. Chem. 270, 29983–29990 [DOI] [PubMed] [Google Scholar]
- 38. Chang C. S., Kokontis J., Liao S. T. (1988) Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science 240, 324–326 [DOI] [PubMed] [Google Scholar]
- 39. Veldscholte J., Berrevoets C. A., Ris-Stalpers C., Kuiper G. G., Jenster G., Trapman J., Brinkmann A. O., Mulder E. (1992) The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J. Steroid Biochem. Mol. Biol. 41, 665–669 [DOI] [PubMed] [Google Scholar]
- 40. Shapiro D. J., Mao C., Cherian M. T. (2011) Small molecule inhibitors as probes for estrogen and androgen receptor action. J. Biol. Chem. 286, 4043–4048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Q., Li W., Liu X. S., Carroll J. S., Jänne O. A., Keeton E. K., Chinnaiyan A. M., Pienta K. J., Brown M. (2007) A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol. Cell 27, 380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cleutjens K. B., van Eekelen C. C., van der Korput H. A., Brinkmann A. O., Trapman J. (1996) Two androgen response regions cooperate in steroid hormone regulated activity of the prostate-specific antigen promoter. J. Biol. Chem. 271, 6379–6388 [DOI] [PubMed] [Google Scholar]
- 43. Jia L., Choong C. S., Ricciardelli C., Kim J., Tilley W. D., Coetzee G. A. (2004) Androgen receptor signaling: mechanism of interleukin-6 inhibition. Cancer Res. 64, 2619–2626 [DOI] [PubMed] [Google Scholar]
- 44. Masiello D., Cheng S., Bubley G. J., Lu M. L., Balk S. P. (2002) Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J. Biol. Chem. 277, 26321–26326 [DOI] [PubMed] [Google Scholar]
- 45. Kang Z., Janne O. A., Palvimo J. J. (2004) Coregulator recruitment and histone modifications in transcriptional regulation by the androgen receptor. Mol. Endocrinol. 18, 2633–2648 [DOI] [PubMed] [Google Scholar]
- 46. Hodgson M. C., Astapova I., Hollenberg A. N., Balk S. P. (2007) Activity of androgen receptor antagonist bicalutamide in prostate cancer cells is independent of NCoR and SMRT corepressors. Cancer Res. 67, 8388–8395 [DOI] [PubMed] [Google Scholar]
- 47. Estébanez-Perpiñá E., Arnold L. A., Arnold A. A., Nguyen P., Rodrigues E. D., Mar E., Bateman R., Pallai P., Shokat K. M., Baxter J. D., Guy R. K., Webb P., Fletterick R. J. (2007) A surface on the androgen receptor that allosterically regulates coactivator binding. Proc. Natl. Acad. Sci. U.S.A. 104, 16074–16079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mao C., Patterson N. M., Cherian M. T., Aninye I. O., Zhang C., Montoya J. B., Cheng J., Putt K. S., Hergenrother P. J., Wilson E. M., Nardulli A. M., Nordeen S. K., Shapiro D. J. (2008) A new small molecule inhibitor of estrogen receptor α binding to estrogen response elements blocks estrogen-dependent growth of cancer cells. J. Biol. Chem. 283, 12819–12830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Clegg N. J., Wongvipat J., Joseph J. D., Tran C., Ouk S., Dilhas A., Chen Y., Grillot K., Bischoff E. D., Cai L., Aparicio A., Dorow S., Arora V., Shao G., Qian J., Zhao H., Yang G., Cao C., Sensintaffar J., Wasielewska T., Herbert M. R., Bonnefous C., Darimont B., Scher H. I., Smith-Jones P., Klang M., Smith N. D., De Stanchina E., Wu N., Ouerfelli O., Rix P. J., Heyman R. A., Jung M. E., Sawyers C. L., Hager J. H. (2012) ARN-509. A novel antiandrogen for prostate cancer treatment. Cancer Res. 72, 1494–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Frasor J., Chang E. C., Komm B., Lin C. Y., Vega V. B., Liu E. T., Miller L. D., Smeds J., Bergh J., Katzenellenbogen B. S. (2006) Gene expression preferentially regulated by tamoxifen in breast cancer cells and correlations with clinical outcome. Cancer Res. 66, 7334–7340 [DOI] [PubMed] [Google Scholar]
- 51. Jiang X., Ellison S. J., Alarid E. T., Shapiro D. J. (2007) Interplay between the levels of estrogen and estrogen receptor controls the level of the granzyme inhibitor, proteinase inhibitor 9, and susceptibility to immune surveillance by natural killer cells. Oncogene 26, 4106–4114 [DOI] [PubMed] [Google Scholar]